
Abstract
In recent years, research has focused on the immunoreactive components of the Sporothrix schenckii cell wall that can be relevant targets for preventive and therapeutic vaccines against sporotrichosis, an emergent worldwide mycosis. In a previous study, we identified a 47-kDa enolase as an immunodominant antigen in mice vaccinated with an adjuvanted mixture of S. schenckii cell wall proteins. Here, we sought to assess the protective potential of a Sporothrix spp. recombinant enolase (rSsEno) formulated with or without the adjuvant Montanide Pet-GelA (PGA) against the S. brasiliensis infection in mice. Mice that were immunized with rSsEno plus PGA showed increased antibody titters against rSsEno and increased median survival time when challenged with S. brasiliensis as compared with mice that had not been immunized or that were immunized with rSsEno alone. Immunization with rSsEno plus PGA induced a predominantly T-helper 1 cytokine pattern after in vitro stimulation of splenic cells with rSsEno: elevated levels of IFN-γ and IL-2, as well as of other cytokines involved in host defense against sporotrichosis, such as TNF-alpha, IL-6, and IL-4. Furthermore, we show for the first time the presence of enolase in the cell wall of both S. schenckii and S. brasiliensis. As a whole, our results suggest that enolase could be used as a potential antigenic target for vaccinal purposes against sporotrichosis.
Similar content being viewed by others


Introduction
Sporotrichosis is a subcutaneous mycosis of subacute or chronic evolution mainly caused by traumatic inoculation of different species of the Sporothrix genus affecting both humans and animals1. The disease has a universal geographical distribution, although it is endemic in Latin America, including in Peru, México, Colombia, Guatemala and, especially, Brazil, where in the last 20 years, it became an important zoonosis, with the infected cat being the main source of transmission2,3,4. Species of the Sporothrix genus are thermodymorphic fungi with a saprophytic life at 25 °C and a filamentous form. The parasitic form at 35–37 °C is a yeast1,5. The human infection is acquired in two ways: traumatic inoculation through the skin with Sporothrix species (spp.) or inhalation. Zoonotic transmission principally occurs from infected cats to humans6.
The genus Sporothrix is currently classified into two clades: i) the clinical clade, which includes S. brasiliensis, S. globosa, S. luriei and S. schenckii sensu stricto and ii) the environmental clade, composed mainly of species less pathogenic to man and animals, such as S. mexicana, S. pallida and S. chilensis7,8. Brazil is the only country that has reported all species of the clinical clade, and S. brasiliensis is the most virulent species9,10. This species is also the most prevalent during zoonotic transmission through scratches and bites from infected cats8. In this country, though sporotrichosis has been reported in most states, the disease is a neglected disease, particularly in the state of Rio de Janeiro, where the largest number of cases has been reported, representing a serious public health problem3. The Oswaldo Cruz Foundation (Fiocruz), Rio de Janeiro, a referral center for the diagnosis and treatment of this mycosis, diagnosed over 4000 humans and feline sporotrichosis cases between 1998 and 201211. More recently, according to data from the epidemiological bulletin of 001/2018 of the sanitary vigilance service of the state of Rio de Janeiro, from January 2015 through May 2018, more 3510 human cases were confirmed12, which shows a progressive increase in the incidence and prevalence of this mycosis.
Sporotrichosis is usually controlled through the use of itraconazole – in combination with potassium iodide in cats –, or terbinafine in immunocompetent patients who exhibit the less severe clinical forms of the disease (lymphocutaneous and fixed cutaneous lesions)13,14. However, in immunocompromised patients with neoplastic diseases, transplantation or AIDS, the conventional treatment with classical antifungals is generally ineffective15,16. The lack of a veterinary and/or human vaccine against this disease has awakened interest in the identification of S. schenckii cell wall immunoreactive components involved in fungal pathogenesis17 and the induction of the immune response18 that can be used for immunoprophylaxis and immunotherapy against sporotrichosis.
In previous studies, our group showed that sera obtained from mice immunized with an S. schenckii- cell wall protein (CWP) formulated with the adjuvant aluminum hydroxide (AH) showed reactivity against two proteins, one of 71 kDa and another of 47 kDa. The latter was functionally identified as enolase and predicted to be an adhesin by the Fungal RV database19. These immune sera showed opsonizing properties, enhancing the phagocytosis of S. schenckii, and they inhibited the fungal adhesion to fibroblasts in vitro. Passive transfer of immune serum to nonimmunized mice conferred protection against challenges with the fungus. These findings indicated the induction of protective immunity from the vaccine formulation against experimental sporotrichosis and the potential use of both antigens for an antifungal vaccine. More recently, we showed that serum from mice vaccinated with AH-adsorbed CWPs, and serum obtained from mice immunized with the same antigenic source but formulated with Montanide Gel Pet A adjuvant (PGA), reacted with the S. brasiliensis yeast cell wall20. Such cross-reactivity, as well as the fact that both formulations confer protection in mice challenged either with S. schenckii or S. brasileinsis, suggested the existence of shared immunodominant antigens that could prove beneficial for the simultaneous protection against these species, which are the more virulent of the genus Sporothrix.
Enolase (2-phospho-D-glycerate hydrolase, EC 4.2.1.11) is a metalloenzyme that requires the metal ion magnesium (Mg2+) to catalyze the dehydration of 2-phosphoglycerate (2-PG) to phosphoenolpyruvate (PEP), a product that is used to produce energy (ATP) in eukaryotic and prokaryotic cells21. In mammals, there are at least 4 isoforms of enolase: α‐enolase (eno1), expressed in almost all tissues; β‐enolase (eno3), predominantly expressed in adult skeletal muscle; γ‐enolase (eno2), found in neurons and neuroendocrine tissues22; and eno4, expressed in human and mouse sperm23. Enolase has been identified on the cell surface of C. albicans24, Plasmodium falciparum25, Ascaris suum26, Streptococcus sobrinus27, S. suis serotipo II28, S. iniae29, Plasmodium spp.30 and Clonorchis sinensis31. In addition, the immunogenicity and protective properties of anti-enolase immune response have been reported for diverse pathogens24,27,32. Furthermore, enolase is probably the only immunogenic antigen shared by eukaryotic and prokaryotic pathogens that has been proposed as an antigenic target for diagnostic, therapeutic, and prophylactic purposes against different diseases.
In this study, we report for the first time the presence of enolase in the cell wall of Sporothrix spp. and also that a PGA-adjuvanted vaccine formulation using a recombinant enolase was able to confer protection either actively to a subsequent challenge with S. brasiliensis or passively through the serum of vaccinated mice. Our results thus suggest that enolase could be used as a potential antigenic target for vaccinal purposes against sporotrichosis.
Results
Production, purification, and characterization of rSsEno
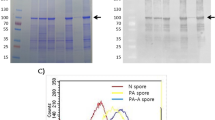
Figure 1A shows that rSsEno expressed in the IPTG-induced pET28a::SsEno-transformed E. coli BL21 cells was produced both in the pellet, as well as in soluble fraction of the lysed cells (Fig. 1A, lane 3). Based on this result, the rSsEno containing the soluble fraction (after filtration, Fig. 1, lane) was purified by Ni2+-affinity (Fig. 1A, lane 6) and preparative SEC (Fig. 1, lane 7), respectively, resulting in an apparent purity 95% on SDS- PAGE with Coomassie blue staining. The final yield was approximately of 15 mg of pure rSsEno per L.

SDS-PAGE and structure analysis of rSsEno expressed in E. coli BL21. The recombinant plasmid pET28a::SsEno-transformed E. coli BL21 cells were induced in the presence of 0,2 mM IPTG for 4 h at 30 °C. The cells were lysed by sonication, and the supernatant containing the recombinant protein was purified by affinity and preparative SEC, respectively. All the samples were analyzed by SDS-PAGE 12%, and the protein was stained with Coomassie Blue R250 in the gel. (A) Expression and purification of rSsEno. Molecular mass markers in kDa (lane 1), non-IPTG-induced pET28a::SsEno-transformed E. coli BL21 cells lysate (lane 2), IPTG-induced pET28a::SsEno-transformed E. coli BL21 cells lysate (lane 3); supernatant of lysed IPTG-induced pET28a::SsEno-transformedE. coli BL21 cells (lane 4), supernatant of lysed cells filtered through Hydrophlic Durapore Membrane, 0.45 µm cutoff, 47 mm diameter (lane 5), rSsEno purified by Ni2+ affinity chromatography (lane 6) and by preparative SEC (lane 7). (B) The CD spectrum shows that rSsEno was obtained mainly with a secondary structure composed by α-helices and β-sheets. (C) Intrinsic emission fluorescence spectra for enolase in the folded (black curve) and unfolded (red curve) states induced by 6 M Gnd-HCl. (D) Analytical SEC performed for rSsEno (red line). The MW standard protein mix elution pattern is represented by the black line: 1) Apoferritin (480 kDa); 2) γ-Globulin (160 kDa); 3) BSA (67 kDa); 4) carbonic anhydrase (29 kDa); 5) Cytochrome C (12 kDa). The column void is identified by blue dextran (blue line).
The rSsEno far UV-CD spectrum shows two bands at ~209 and 218 nm, which indicate the presence α-helices and β-sheets folded structures (Fig. 1B). Deconvolution analysis of the rSsEno far UV CD spectra revealed that the secondary structure of the proteins in the preparation contains 24% α-helices, 22% β-sheets, 18% of turns and 34% of random coils. Intrinsic emission fluorescence also indicated that the rSsEno was purified in a folded state. The mean maximum emission emission wavelength (λmax) of about 338 nm indicates that the 6 tryptophan residues present in the rSsEno structure were partially protected from the solvent (Fig. 1C). On the other hand, treatment of rSsEno with a chemical denaturant caused a red shift and fluorescence quenching suggesting at least a partial exposition of the tryptophan residues to the solvent33. Analytical SEC analysis showed that rSsEno elutes near the column void (identified by blue dextran in Fig. 1D) leaving a tail after the main peak, which indicates the protein sample as a heterogeneous solution probably due to the presence of oligomers of smaller size in equilibrium; this suggests that the recombinant protein should be organized by various oligomeric forms. Small angle x-ray scattering (SAXS) data (not shown) indicated that rSsEno behaved as an oligomeric particle with a weight-average molecular weight of 580 ± 60 kDa. Taken together, our biophysical data suggest that rSsEno was purified folded and as a mixture of different oligomeric forms.
Sequence alignment of the S. schenckii enolase
The sequence alignment analysis among the S. schenckii-, F. catus- and H. sapiens-enolase revealed an expected result; the enolase from humans and cats showed a degree of identify of 95% (Fig. 2). However, both enolases showed an identity of 62% with the enolase of S. schenckii. Moreover, although we don’t present it in alignment form, the S. schenckii enolase has 100% sequence identity with the ones from S. schenckii 1099–18 and S. brasiliensis 5110, belonging together to the Cluster identity UniRef100 U7PSS1. Specifically, according to the uniprot database (https://www.uniprot.org/), there are five enolase genes in the Sporothrix spp. genome, of which three, namely SPSK_03292, HMPREF1624_06143, and SPBR_00513, code for the same 438 a.a. protein in S. schenckii ATCC58251, S. schenckii 1099–18, and S. brasiliensis 5110, respectively; the two other genes, namely SPI_04152 and SPI_06529, encode the 439 and 465 a.a. proteins found in S. insectorum RCEF 264. Unlike S. schenckii and S. brasiliensis, S. insectorum, is not clinically associated with human and feline sporotrichosis.

Multiple sequence alignments of S. schenckii. The deduced amino acid sequence of S. schenckii (ERS97971.1), Felis catus (M3WCP0_FELCA) and Homo sapiens (P06733) were aligned by the Clustal Omega server. The conserved amino acids in all sequences are labeled with asterisks; the conservative and semi-conservative substitutions are labeled with two and one points, respectively. The percentage of amino acid sequence identity between all enolases is indicated.
Specificity of the anti-rSsEno serum
The specificity of the antibodies raised in rSsEno immunized mice was examined by immunoblotting against recombinant enolase or CWPs isolated from S. schenckii ATCC 16345. As shown in Fig. 3 (A), the anti-rSsEno sera reacted with the recombinant protein and against a single reactive band present in S. schenckii ATCC 16345 CWPs with the expected 47 kDa molecular mass, corresponding to the native enolase19. Interestingly, the serum obtained from infected cats with sporotrichosis confirmed a specific high reactivity against the recombinant protein (Fig. 3B,C), indicating that, during natural infection, the fungal enolase can induce anti-enolase antibodies. Sera from uninfected control cats exhibited no immunoreactivity with the rSsEno (Fig. 3B).

Western blot analysis showing the specificity of the anti-rSsEno sera and the reactivity of the sera from cats with sporothricosis against rSsEno. Samples of S. schenckii ATCC 16345 CWPs and rSsEno were tested by 12% SDS-PAGE under nonreducing conditions and after immobilization on a nitrocellulose membrane. The strips were incubated at 37 °C for 1 h with anti-rSsEno serum or naïve mouse serum, and the immunoblots were visualized by adding 3,3′-diaminobenzidine substrates after being treated with goat anti-mouse IgG-HRP. Panel A, column 1: molecular weight marker; column 2: S. schenckii ATCC 16345 CWPs resolved by SDS-PAGE 12%; columns 3 and 4: nitrocellulose strips containing the S. schenckii ATCC 16345 CWPs treated with NS and anti-rSsEno serum, respectively; columns 5 and 6: nitrocellulose strips containing rSsEno treated with naïve mice-serum and anti-rSsEno serum, respectively. Panel B, strips containing rSsEno were incubated with sera from cats with or without sporotrichosis (NS) and immunoblots were incubated with goat anti-feline IgG-HRP. Each cat serum is identified by the admission number of the Laboratory of Clinical Research in Dermatozoonoses in Domestic Animals of the National Institute of Infectology Evandro Chagas (FIOCRUZ).
Enolase is present in the cell wall of S. schenckii spp
After confirming their specificity, the anti-rSsEno serum was used to detect enolase in the S. schenckii ATCC 16345, S. schenckii 1099–18, S. brasiliensis Ss250 and S. brasiliensis Ss256 cell wall. Figure 4(A,C,E,G) shows an intense and significant (p < 0.05) median fluorescence intensity (MFI) in yeasts treated with the anti-rSsEno serum compared to yeast treated with serum from nonimmunized mice (NIS), evidencing enolase on the cell surface of these strains. The MFI shift was greater for S. brasiliensis Ss250 and Ss256 (Fig. 4F–H) compared to S. schenckii ATCC 16345 (Fig. 4B) and S. schenckii 1099–18 (Fig. 4D), suggesting that this protein is expressed more on the cell wall of S. brasiliensis, the more virulent species. The presence of enolase on the cell surface of the studied fungi was also confirmed by transmission microscopy using the immunogold stain. Figure 4(I–N) showed that enolase appears distributed along the cell wall of S. schenckii ATCC 16345, S. schenckii 1099–18, S. brasiliensis Ss250 and S. brasiliensis Ss256, which might facilitate its recognition by the host’s immune system, although it also appears, as expected, in the cellular cytoplasm of these species, since its classical function is to catalyze the reversible conversion of 2-phosphoglycerate to phosphoenolpyruvate22,34.

Demonstration of the enolase on the cell surface of Sporothrix spp.-yeasts by flow cytometry and electron microscopy. A S. schenckii ATCC 16345 (Ss16345), S. schenckii 1099–18 (Ss1099–18), S. brasiliensis Ss250 (Ss250), or S. brasiliensis Ss256 (Ss256) yeasts suspension was previously incubated with anti-rSsEno serum (SAE) or serum from nonimmunized mice (NIS) for 1 h at 37 °C. After washing, the cells were exposed to FITC-conjugated rabbit anti-mouse IgG and examined using a flow cytometer. (A,C,E,G) Representative histograms from one of three independent experiments for the indicated Sporothrix spp. yeasts treated with NIS or SAE. Bar graphs show the median fluorescence intensity (MFI) of the FITC staining for S. schenckii ATCC 16345 (B), S. schenckii 1099–18 (D), S. brasiliensis Ss250 (F) and S. brasiliensis Ss256 (H). The results are presented as the mean ± SD of three independent experiments, and statistical significance was determined by Student’s paired t test (I-N). *P < 0.05; **P < 0.01 ***P < 0.001. Ultrathin sections of each fungus were incubated overnight with SAE or NIS and then treated overnight with an Au-conjugated secondary antibody, at 4 °C. Grids were observed with in a transmission electron microscope after being stained with uranyl acetate and lead citrate. The micrographs show enolase (Eno) on the cell wall (CW) or cytoplasm (C) of S. schenckii ATCC 16345-(I,J), S. schenckii 1099–18 (K), or S. brasiliensis Ss250 (L) yeasts treated with SAE. (M,N) show Eno on the CW of S. brasiliensis Ss256-yeasts treated with SAE.
Antibody response
To assess the immunogenic potential of S. schenckii-enolase, sera from the experimental group obtained seven days after the last boost was subjected to ELISA using rSsEno as an antigen. Our results showed that animals immunized only with enolase stimulated high IgG specific antibody production (Fig. 5A) compared to the PBS group. However, as expected, the specific antibody production was significantly higher (p < 0.05) when enolase was formulated with the PGA adjuvant. We also determined the rSsEno-specific IgG1, IgG2a and IgG3 antibodies induced by each formulation. Mice immunized with rSsEno100 and PGA + rSsEno100 induced higher IgG1 and IgG3 antibody levels against rSsEno compared to the PBS control group, but the level of both subclasses was higher in the mice immunized with the PGA-adjuvanted formulation (Fig. 5B,C). The PGA + rSsEno100 formulation was the only formulation that induced the production of IgG2a (Fig. 5C).

Immunization with rSsEno with or without PGA conjugation enhanced the antibody response. BALB/c mice were s.c. immunized three times with rSsEno100, PGA + rSsEno100 or PBS as a negative control. Sera collected seven days after the last boost was used to determine antigen-specific IgG (A), IgG1 (B), IgG2a (C), and IgG3 (D) titers by ELISA. The results are presented as the mean ± SD of 5 mice from one of three independent experiments, and statistical significance was determined by one-way ANOVA using Tukey’s multiple comparisons test and a 95% confidence interval. *(p < 0.05), **(p < 0.01), ***(p < 0.001) and ****(p < 0.0001) for comparison with the control group or as indicated.
Cytokine profile analysis
The effect of anti-enolase vaccination on the pattern of cytokines was evaluated in the supernatant of splenocyte cultures from nonimmunized and immunized mice after in vitro stimulation with rSsEno100. A higher production of IL-2 and IFN-ɣ from the Th1 profile, IL-4 and IL-6, which are involved in the production of antibodies, and TNF-α, which is released during the innate immune response (also with IL-6) in mice vaccinated with PGA + rSsEno100, was observed (Fig. 6). All of these cytokines are involved in defense against S. schenckii, which is additional evidence of protective immunogenicity induced by the vaccine formulation.

Vaccinated mice with rSsEno100 and PGA + rSsEno100 showed differences in Th1, Th2 and Th17 cytokine profiles. BALB/c mice were s.c. immunized three times with rSsEno100, PGA + rSsEno100 or PBS as a negative control. Total splenocytes of each animal were obtaining seven days after the last immunization and stimulated in vitro with rSsEno. After 24 h of incubation, supernatant-accumulated cytokines (IL-2, IL-4, IL-6, IL17A, IFN-γ, TNF and IL-10) were measured by cytokine cytometric bead array kit ELISA. The results are presented as the mean ± SD of 5 mice from one of three independent experiments, and statistical significance was determined by one-way ANOVA using Tukey’s multiple comparisons test and a 95% confidence interval. *(p < 0.05), **(p < 0.01), ***(p < 0.001) and ****(p < 0.0001) for comparison the control group or as indicated.
Challenge studies
To test whether rSsEno in the formulation with PGA adjuvant protects against systemic sporotrichosis in mice, seven days after booster immunization, mice from each group were challenged intravenously with 105 S. brasiliensis Ss250 yeasts, a highly virulent strain. The mortality of nonimmunized mice was of 100% before 40 days postinfection, while the rSsEno-immunized mice showed over 50% survival, and those immunized with the PGA-adjuvanted formulation exhibited the highest percentage of survival (over 90%) at the end of the experiment (45 days postinfection) (Fig. 7A). Furthermore, passively immunizing mice with anti-rSsEno serum led to a significant decrease in the number of CFUs in the spleen (P < 0.01) and liver (p < 0.001) following infection with S. brasiliensis Ss250 via the i.p. route as compared to those previously treated with NIS or PBS. Our results thus indicate that enolase has potential use as an immunogen for both therapeutic and prophylactic purposes.

rSsEno immunization induces protection against disseminated sporotrichosis. BALB/c mice were immunized (s.c.) three times with rSsEno100, PGA + rSsEno100, or PBS and after seven days and after seven days were challenged i.v. with 1 × 105 S. brasiliensis Ss250 yeast cells. Mice survival was monitored daily for 45 days post-challenge (n = 10 in all groups). In another study, two hours prior to infection with 106 S. brasiliensis Ss250 yeasts, BALB/c mice were passively immunized (i.p.) with a pool of sera from PGA + rSsEno100-, NIS- (serum from non-immunized mice), or PBS-injected mice; five days post-infection the number of CFUs was determined in the spleen (B) and liver (C) of each animal. Differences in survival were determined by the log-rank test. Results of the passive immunization study are presented as the mean ± SD of 7 mice from one of two separate experiments. Statistical significance was determined by one-way ANOVA using Tukey’s multiple comparisons test and a 95% confidence interval. **(p < 0.01), ***(p < 0.001), and ****(p < 0.0001) for comparisons with the control group or as indicated.
Discussion
In the last two decades, sporotrichosis has been a hyperendemic zoonosis in Brazil transmitted by infected cats. The high incidence of sporotrichosis, together with the low effectiveness of the treatment, especially in immunocompromised individuals, has reinforced the need to identify antigenic targets on the cell surface of species of clinical interest of the genus Sporothrix for immunological prevention and therapeutic intervention17,35.
In this study, the enolase of S. schenckii was obtained by expression in E. coli, then purified and partially characterized. Our results showed that the His-tagged rSsEno was successfully produced with a molecular weight of 50 kDa and with a native-like structure as suggested by signals of the presence of secondary and local tertiary structures, obtained in the CD and tryptophan fluorescence experiments. We expected that rSsEno would be assembled in the form of dimers, as reported for yeast enolase36,37. However, analytical SEC indicated the presence of various oligomeric species of a weight-average molecular weight of about 580 kDa as indicated by SAXS data.
Enolase has been described as a moonlighting protein that exhibits multiple nonglycolytic functions, probably because of its different multimeric structures32. Ehinger et al.38 reported that α-enolase of Streptococcus pneumonia forms an octamer in solution and that due to its binding to human plasminogen, it probably resides on the cellular surface of this pathogen and can be involved in virulence. Wu et al.39 also reported that Staphylococcus aureus recombinant enolase is organized in dimers and octamers and that the latter probably exist in vivo since it showed enzymatic activity in vitro. Whether the complex oligomeric state of rSsEno in solution is the same as its native form in S. schenckii, and its functional role in vivo, is a subject for future studies.
The reactivity of sera from cats with sporotrichosis against rSsEno and the lack of reactivity with sera from uninfected control cats evidenced the antigenic role and probable immunogenicity of the S. schenckii enolase during the infectious process in these animals. Coupled with a 38% difference in sequence homology, this indicates that S. schenckii enolase may contain conserved regions distinct from its cat and human orthologs, suggesting that S. schenckii enolase can be used for vaccine and/or therapeutic strategies against or as a diagnostic tool for sporotrichosis in cats. Although α-enolase autoantibodies have been associated with a wide variety of human autoimmune diseases including systemic lupus erythematosus, autoimmune-mediated retinopathy, autoimmune hepatitis, severe asthma, and Hashimoto’s encephalopathy22, as far as we know, the association of these antibodies with clinical autoimmune disorders in cats is unknown. In any case, we are currently assessing the immunogenic potential of synthetic peptides derived from non-homologous regions (to its feline ortholog) of the S. schenckii enolase, which should eliminate any safety problems regarding the development of auto-immunity in the feline host.
Different studies have shown that enolase on the cell surface of bacteria, fungi and parasites acts as a virulence factor that facilitates the colonization and dissemination of these pathogens in the host25,40,41. Although there are no studies assessing the effects of the absence of enolase in S. schenckii, Ko et al.42 showed that C. albicans ENO1 (enolase) null mutants exhibit reduced hyphal growth, decreased virulence in BALB/c mice and increased susceptibility to amphotericin B, miconazole and other antifungal drugs. We also know, from experiments of a yet to be published study from our group, that rSsEno is able to bind extracellular matrix constituents such as fibronectin and plasminogen and that an anti-rSsEno polyclonal serum generated in BALB/c mice inhibits the adhesion of S. schenckii ATCC 16345, S. brasiliensis Ss250, and S. brasiliensis Ss256 yeasts to fibroblasts, suggesting that enolase may be a virulence factor within the Sporothrix genus (data not shown). In this study, we show for the first time that enolase is present on the cellular surface of S. schenckii and S. brasiliensis species, and interestingly, this expression was higher on yeast cell walls from S. brasiliensis, suggesting that the level of enolase expression on the cell surface of species of the genus Sporothrix can be related to the invasiveness and virulence of these pathogens in the host. In this way, Roth et al.43 showed that the level of expression of enolase is 15-fold higher in red blood cells infected with P. falciparum compared to uninfected cells. More recently, Marcos et al.44 observed a considerable increase of this protein in the cell wall of Paracoccidiodes brasiliensis when the fungus was cultivated in BHI medium enriched with sheep blood or during fungal infection in mice, suggesting a role for enolase as a virulence factor of these fungi in host cells.
The generation of a Th1 and Th17 response is necessary for protective immunity against Staphylococcus aureus and C. albicans45. Ferreira et al.46 demonstrated in a model of S. schenckii infection in BALB/c mice that the Th1 and Th17 immune response were able to control the infection. Recently, our group reported a similar result in a model of C57BL6 mice subcutaneously infected with either S. schenckii or S. brasiliensis. However, the higher virulence of S. brasiliensis caused a long-lasting infection associated with severe tissue lesions that stimulated a regulatory T cell (Tregs) response with deleterious effects on the Th1 and Th1/Th17 response, although a compensatory Th17 response was induced47. We also demonstrated in an immunoprophylaxis study in BALB/c mice that either aluminum hydroxide adjuvant or PGA, both formulated with the S. schenckii ATCC 16345 CWPs containing the immunoreactive enolase, induced a Th1, Th2 and Th17 profile, in addition to high stimulation of specific antibodies that conferred protection in these animals after challenge with S. schenckii ATCC 16345 or S. brasiliensis Ss25020.
To verify whether rSsEno could be used as an antigenic target for a sporotrichosis vaccine, we performed a survival study in immunized mice after intravenous infection with the highly virulent strain S. brasiliensis Ss250. The survival above 90% seen in mice immunized with PGA + rSsEno100 is strong evidence of the protective capacity of our vaccine candidate. In addition, probably the Th1, and not Th1/Th2cytokine profile observed ex vivo in PGA + rSsEno100-immunized mice played a significant role in vivo in favoring protection, since rSsEno100-immunized mice showed ex vivo a stimulation of Th2 cytokines, which may be associated to decreased survival of those animals (~ 48%) postchallenge. Li et al.24 showed that a Th1 and Th2 immune response pattern induced by recombinant enolase of C. albicans emulsified with Freund’s adjuvant (AF) was enough to confer protection on C57BL/6 mice challenged with a lethal dose of C. albicans strains SC5314 and 3630. In addition, passive immune serum transfer, characterized by the prevalence of IgG2a- and IgG1-specific antigen isotypes, also demonstrated effective protection against both fungal C. albicans lineages, showing that antibodies against enolase could be useful to treat of candidiasis. Zhang et al.48 also showed that the enolase of Streptococcus suis serotype 2 plus AF formulation induced a mixed Th1 (IgG2a) and Th2 (IgG1) response that also conferred protection in challenged animals with two pathogenic strains of S. suis. This same immune response profile and protective efficacy were observed in mice immunized with the Ascaris suum enolase after infection with infective larvae of this parasite49.
Several studies have shown that the IgG antibody response50,51 and especially IgG152,53, IgG2a and IgG3 isotypes19,20 against S. schenckii and S. brasiliensis cell wall proteins is associated with in vivo protection through neutralization (IgG1 and IgG3) and Fc-mediated phagocytosis by macrophages (IgG2a). Our results showed that rSsEno100 and PGA + rSsEno100 stimulated a Th2 (IgG1 and IgG3) and Th1/Th2 (IgG1, IgG2a and IgG3) immune response, respectively. Thus, the anti-S. brasiliensis protection conferred by the active or passive immunization of mice with anti-rSsEno may be related to the IgG profile. Moreover, Almeida et al.54 showed that opsonization with a humanized anti-gp70 (Sporothrix spp. immunogenic protein) IgG1 antibody (P6E7) increased the phagocytosis of S. schenckii yeasts by human monocyte-derived macrophages and that the passive transference of P6E7 to BALB/c mice three days post-infection decreased the fungal burden in the spleen a week later compared with the control. Therefore, a serotherapy-based strategy with anti-rSsEno holds great promise for the treatment and/or prevention of sporotrichosis.
In summary, for the first time, a recombinant form of S. schenckii (rSsEno) enolase was obtained folded and partially characterized. The weight-average molecular mass of rSsEno determined by SAXS was of about 580 kDa, while the aSEC showed a tail after the main peak indicating the presence of smaller oligomers. This organization is different from the enolases from other fungi, at least in the absence of magnesium which are enolase co-factors. The identification of enolase on the cell wall of S. brasiliensis and S. schenckii and its recognition by serum from cats affected with sporotrichosis are reported in this study. A vaccine formulation of rSsEno plus PGA adjuvant induced a Th1/Th2 response and high titers of specific antibodies that favored the protection to mice challenged with a highly virulent S. brasiliensis isolate. In addition, the anti-enolase serum induced by the vaccine candidate conferred protection to naïve mice. All these results show that the enolase of Sporothrix spp. may be a vaccine antigen candidate for feline sporotrichosis prevention.
Materials and Methods
Animals
For this study, male 5–7-week-old BALB/c mice were purchased from “Centro Multidisciplinar para Investigação Biológica na Área da Ciência de Animais de Laboratório” (CEMIB), Universidade de Campinas (UNICAMP), São Paulo, Brasil. Animals were housed in individually ventilated cages in an ambient controlled temperature and 12-h light/dark cycles. All animals were acclimatized to the conditions for 1 week before the experiments, and water and food was offered ad libitum. This study was carried out in strict accordance with the recommendations for the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health, and the protocols were approved by the Institutional Ethics Committee for Animal Use in Research of the Faculty of Pharmaceutical Sciences of Araraquara – UNESP (Proc. CEUA/FCF/CAR no. 57/2015).
Microorganisms
The strains S. schenckii ATCC 16345, S. schenckii 1099–18, S. brasiliensis Ss250 (GenBank: KC693883.1) and S. brasiliensis Ss256 (KC693889.1), both S. brasiliensis strains isolated from feline sporotrichosis, and S. schenckii ATCC 16345 were kindly provided by the Oswaldo Cruz Foundation, Rio de Janeiro, Brazil. S. schenckii 1099–18 was provided by Dr. Celuta Sales Alviano at the Institute of Microbiology, Federal University of Rio de Janeiro (Brazil). Mycelial-to-yeast phase conversion was accomplished as previously described by Ferreira and collaborators46.
Expression and purification of recombinant S. schenckii enolase (rSsEno)
The encoding DNA for S. schenckii 58251 enolase containing 438 amino acids and a molecular mass of 47 kDa (Accession Code: ERS97971.1 of the GenBank database) was synthesized by Epoch Life Science Inc. optimized for production in E. coli. It was subcloned into the pET28a plasmid between the Nde I and Eco RI restriction enzymes in fusion with a His-tag at the N-terminus. The resultant plasmid (pET28a::SsEno) is capable of expressing a His-tagged rSsEno of about 50 kDa.
Escherichia coli DH5α was used as the cloning host for the propagation of pET28a::SsEno on lysogeny broth (LB) agar medium containing 30 μg/mL of kanamycin, and the authenticity of the cloning procedure was confirmed by sequencing. For recombinant protein expression, E. coli BL21 cells transformed with pET28a::SsEno were grown at 37 °C in LB medium containing 30 μg/mL of kanamycin until they reached an OD600 in the range of 0.5–0.7. The expression of rSsEno was induced by 0.2 mmol/L of isopropyl β-D-1-thiogalactopyranoside (IPTG) at 30 °C for 4 h. The cells were separated by centrifugation for 20 min at 8000 rpm, and the pellet was resuspended in 20 mL buffer A (NaPO4 20 mM, NaCl 500 mM and imidazole 20 mM, pH 7,4) containing 5 U of DNAse (Promega) and 30 ug/mL lysozyme (Sigma) for 30 min on ice. The cell homogenate was sonicated, filtrate and then centrifuged at 19,000 rpm for 20 min at 4 °C. The rSsEno-containing supernatant was filtered through a Hydrophlic Durapore Membrane, 0.45 µm cutoff, 47 mm diameter (Millipore) and further subjected to Ni2+-affinity chromatography in buffer A. The rSsEno was then eluted in buffer B (NaPO4 20 mM, NaCl 500 mM and imidazole 500 mM, pH 7,4). After elution, the material obtained was subjected to a preparative SEC with a Superdex 200 pg 16/60 column (GE Healthcare Life Sciences) in Tris-HCl 25 mM, NaCl 100 mM and β-mercaptoethanol 2 mM at pH 7.5, and subjected to biophysical tests. The eluted protein was also concentrated using the Amicon Ultra 15 mL 3k device (Millipore) after being dialyzed for 24 h at 4 °C against phosphate buffer saline (PBS, pH 7, 2–7, 4). The rSsEno concentration was measured by the BCA assay (Pierce) (for immunological tests) or by absorbance at 280 nm (for biophysical tests) using an extinction coefficient of 54,620 M−1 cm−1, obtained from the rSsEno primary structure through Protparam Tool (web.expasy.org/protparam/). The efficacy of the expression and purification processes was assessed by 12% SDS-polyacrylamide gel electrophoresis (SDS-PAGE).
Biophysical characterization
Secondary structure analysis for rSsEno was performed by far-UV (195–260 nm) CD in a J-815 spectropolarimeter (Jasco Inc.) coupled to a Peltier PFD 425 S for the temperature control system. rSsEno was tested in Tris-HCl buffer (pH 7.5), 100 mM NaCl and 2 mM β-mercaptoethanol, at 20 °C and the secondary structure content was estimated using the CDNN Deconvolution program55. The folded structure for rSsEno was also investigated by intrinsic fluorescence emission using a Fluorescence Spectrophotometer Hitachi F-4500 with excitation wavelength at 295 nm and recording the emission fluorescence spectra from 305 nm to 420 nm. The recombinant protein was prepared at 1 µM in Tris-HCl buffer (pH 7.5), 100 mM NaCl and 2 mM β-mercaptoethanol, at 20 °C. The effect of a chemical denaturant, 6 M guanidinium-HCl (Gnd-HCl), on the rSsEno structure was also investigated by intrinsic fluorescence.
In addition, the rSsEno oligomeric state was analyzed by analytical size exclusion chromatography on a Superdex 200 GL 10/30 column (GE Healthcare LifeSciences) coupled to a ÄKTA Prime Plus (GE Healthcare LifeSciences) and equilibrated with the same buffer described above, at room temperature. Blue dextran was used in order to identify the column void. The column was calibrated using a pool of protein markers of known size.
The rSsEno oligomeric state was also investigated by small angle X-ray scattering (at SAXS1 beamline, at the Brazilian Synchrotron Light Laboratory, Campinas, Brazil). Measurements were done using a monochromatic X-ray beam (λ = 1.488 Å), and rSsEno at 0.5 and 0.9 mg.mL−1 (prepared in Tris-HCl buffer pH 7.5, 100 mM NaCl and 2 mM β-mercaptoethanol) were applied into a 1 mm path length capillary mica. Data were recorded using a Pilatus 300 K detector, with a ~900 mm sample-to-detector distance. Scattering curves were normalized by concentration, extrapolated to infinite dilution and used to yield the Guinier curve in order to estimate-average molecular mass and the average size of the scattering particle56,57.
Extraction of S. schenckii ATCC 16345 CWPs
Extraction of the S. schenckii ATCC 16345 CWPs was performed per Portuondo et al.19. S. schenckii ATCC 16345 yeast cells collected from logarithmically growing cultures were incubated with a protein extraction buffer containing 2 mM dithiothreitol, 1 mM phenylmethylsulfonyl fluoride, and 5 mM EDTA in This-HCl buffer for 2 h at 4 °C under mild agitation. The S. schenckii ATCC 16345 CWPs-containing supernatant was collected, dialyzed against PBS, and then concentrated using the Amicon Ultra 15 mL 3 k device concentrator (Millipore). The proteins were then precipitated by overnight incubation with 10% (w/v) trichloroacetic acid in acetone at 4 °C, and the resulting pellets were washed in ice-cold acetone, dried in a SpeedVac and reconstituted PBS. The protein concentration was measured by the BCA assay (Pierce).
SDS-PAGE, western blot analysis
Samples containing 20 µg of protein S. schenckii ATCC 16345 CWPs and purified rSsEno (5 µg) were resolved on an SDS-PAGE 12% as described by Laemmli58. Two gels were stained with Coomassie brilliant blue R250, and the other gels were transferred to 0.45-μm-nitrocellulose membranes (GE Healthcare) using a mini Tank VEP-2 electroblotting system (Owl Separation Systems, Thermo Scientific) at 50 mM for 3 h. The membrane-cut strips were saturated with 5% dried skim milk in PBS for 4 h at 37 °C, and the strips containing rSsEno were incubated overnight at room temperature (RT) with anti-rSsEno serum (obtained from BALB/c mice seven days after being immunized subcutaneously twice at 14 day intervals with 100 µg rSsEno emulsified with Freund´s adjuvant) or sera from cats with confirmed sporotrichosis* (n = 34) obtained from the Laboratório de Pesquisa Clínica em Dermatozoonoses em Animais Domésticos (Lapclin-Dermzoo)/Instituto Nacional de Infectologia Evandro Chagas (INI)/Fundação Oswaldo Cruz, Rio de Janeiro, Brazil. One strip containing S. schenckii ATCC 16345 CWPs was incubated with anti-rSsEno. Sera from naïve mice or sera from cats with no evidence of sporotrichosis (n = 3) were utilized as negative controls. All sera were diluted 1:100 in PBS. After three washes with PBS, the strips were further incubated for 2 h with goat anti-mouse IgG (Sigma-Aldrich) diluted 1:500 or goat anti-feline IgG (Southern Biotech) diluted 1:1000. Both antibodies were conjugated with horseradish peroxidase (HRP). Protein signals were visualized by adding 3,3′-diaminobenzidine plus hydrogen peroxide. *The sporotrichosis diagnosis was carried out as follow: Samples from skin or nasal mucosa lesions were collected using a sterile swab and seeded onto Sabouraud Dextrose Agar and Mycobiotic Agar (Difco), incubated at 25 °C for four weeks. Microscopic and macroscopic characteristics of the mycelia cultures were evaluated on Potato Dextrose Agar. Dimorphism was confirmed by conversion to the yeast phase on BHI Agar at 37 °C.
Alignment of enolase sequences
We compared conservation (similarity) between the enolase of S. schenckii and the cat and human enolase. The enolase amino acid sequences of S. schenckii (GenBank Accession No. ERS97971.1 and Felis catus (UniProt Accession: M3 WCP0_FELCA, Homo sapiens (UniProt Accession: P06733) were aligned through the default settings within Clustal Omega59.
Flow cytometry
To demonstrate the enolase on the cell wall of S. schenckii ATCC 16345, S. schenckii 1099–18, S. brasiliensis Ss250 and S. brasiliensis Ss256, 106 yeasts were incubated for 1 h at 37 °C with anti-rSsEno serum. Serum from naïve mice was used as a nonspecific binding control at a 1:50 dilution. After incubation, cells were washed twice with PBS for 1 h at 37 °C and then incubated with a FITC-conjugated rabbit anti-mouse IgG antibody (Sigma-Aldrich) at a 1:500 dilution. After washing, samples were acquired with the BD Accuri C6 flow cytometer (BD Biosciences). The acquisition threshold was set to 50,000 on FSC-H for debris exclusion, and at least 50,000 events were effectively included in each analysis. Binding of serum antibodies to the yeast cell surface was assessed through the median fluorescence intensity (MFI) on the FL1 channel using the flow cytometer’s proprietary software.
Electron microscopy
To visualize enolase on the S. schenckii ATCC 16345, S. schenckii 1099–18, S. brasiliensis Ss250, and S. brasiliensis Ss256 cell surface, we performed pre-embedding immunogold experiments using intact yeast cells this fungus, as described previously43. Briefly, the yeast cells were fixed with 2.5 glutaraldehyde v/v in 0.1 M cacodylate buffer, pH 7.2, for 24 h at 4 °C. Ultrathin sections of each fungus were prepared and treated overnight with the primary antibody (polyclonal anti-rSsEno) diluted 1:100 in PBS at 4 °C. The grids were then incubated overnight with the labeled Au-conjugated secondary antibody rabbit IgG (10 nm average particle size, 1:20) at 4 °C. The grids were stained with 4% uranyl acetate and lead citrate and observed with a Jeol 1011 transmission electron microscope (Jeol, Tokyo, Japan). Controls were obtained by incubating the ultrathin sections with NS.
Immunization schedule
BALB/c mice (n = 5) were injected subcutaneously (s.c.) three times in the back of the neck, with 2-week intervening period, with one of the following formulations diluted in 100 μl of PBS: rSsEno100 alone (100 μg), PGA + rSsEno100 [10% Montanide Pet Gel A (PGA), SEPPIC, France plus 100 μgrSsEno] or PBS alone as a negative control. One week after the lastimmunization, mice were euthanized in a CO2 chamber and bled by heart puncture to obtain serum, which was aliquoted and stored at −20 °C until use.
Quantification of the rSsEno-specific antibody response by ELISA
rSsEno IgG, IgG1, IgG2a and IgG3 antibody titration was conducted as described by Portuondo et al.19 with some modifications. Briefly, a 96-well ELISA plate (Costar) was coated with 5 μg rSsEno/mL in PBS and incubated overnight at 4 °C. The plate was washed with washing buffer (0.1% Tween 20 in PBS) and then saturated for 1 h at RT with blocking buffer (5% dried skim milk in washing buffer). Next, dilutions (1:100 in blocking buffer) of the serum samples were added to each well and incubated for 2 h at RT. After washing, 100 µl/well of peroxidase-conjugated anti-mouse IgG (1/500) (Sigma) in blocking buffer was added and incubated at 37 °C for 1 h. For determination of the IgG1, IgG2a and IgG3 subclasses, ELISA plates coated as before were first incubated with an unconjugated rabbit anti-mouse IgG1, IgG2a or IgG3 (Bio-Rad) at 37 °C for 1 h and then with a peroxidase-conjugated goat anti-rabbit IgG (Sigma) overnight at 4 °C. After exhaustive washing, immune complexes were revealed by incubation with tetramethylbenzidine for 30 min at RT. The reaction was stopped by the addition of 50 µL/well 1 M H2SO4, and the absorbance was read with an ELISA reader (Multiskan ascent, Labsystem) at 450 nm.
Cytokine production
To evaluate the cytokine production induced by rSsEno-stimulated spleen cells, splenocytes isolated from each group of animals were harvested seven days after the third immunization. Collected cells were washed, suspended in complete RPMI−1640 medium (cRPMI; RPMI−1640 medium containing 0.02 mM 2-mercaptoethanol, 100 U/mL penicillin, 100 U/mL streptomycin, 2 mM l-glutamine, and 5% fetal bovine serum) and then plated in triplicate in 96-well plates (Costar, USA) to final concentration 2.5 × 106 cells/mL with 20 µg of rSsEno/mL in cRPMI for 24 h at 37 °C with 5% CO2. Concanavalin A (0.25 l g/ml) or cRPMI alone were used as positive and negative controls, respectively. Supernatant-accumulated cytokine concentrations (IL-2, IL−10, IL-4, IL-6, IFN-γ, TNF-α, and IL-17A) were simultaneously measured using the mouse Th1/Th2/Th17 cytokine cytometric bead array (CBA) kit (BD Biosciences). Briefly, 50 μL of each standard or supernatant sample was incubated for 2 h at RT with an equal volume of PhycoErythrin (detection reagent) and the mixed capture beads. After incubation, the samples were centrifuged at 200 × g for 5 min, and the pellet was resuspended in 300 μL of wash buffer and analyzed using a flow cytometer (BD Accuri C6, BD Biosciences).
Protection assay
BALB/c mice (n = 10) were immunized according to the immunization schedule described previously. Seven days after the final boost, mice were challenged intravenously with 105 of the highly virulent S. brasiliensis Ss250 yeasts in 0.1 mL of PBS via the tail vein, as described by Ishida et al.60. Animals were monitored daily for 45 days postinfection to determine the survival curve and efficacy of each vaccine formulation. In another experiment, a 200 µL of a 1:2 dilution of a pooled immune serum from PGA + rSsEno100-immunized mice was passively transferred via the i.p. route to BALB/c mice (n = 7) two hours prior to infection with 106 S. brasiliensis Ss250 yeasts also via the i.p; mice pre-treated with NIS or PBS were used as controls. Protection was assessed by determining the number of CFUs recovered from the spleen and liver on day 5 post-infection. This was done by plating an adequate dilution of the organs’ macerate on Mycosel agar plates.
Statistical analysis
All statistics were performed using Graph Pad Prism version 6.01 by applying Student’s t-test or one-way analysis of variance (ANOVA) followed by Tukey’s post-test. Survival data were compared using the log-rank test. In this study, a p value of < 0.05 was considered significant. The results are expressed as the mean ± SD.
References
Barros, M. B., de Almeida, Paes., R. & Schubach, A. O. Sporothrix schenckii and sporotrichosis. Clin Microbiol Rev 24, 633–654 (2011).
Chakrabarti, A., Bonifaz, A., Gutierrez-Galhardo, M. C., Mochizuki, T. & Li, S. Global epidemiology of sporotrichosis. Medical mycology 53, 3–14 (2015).
Carlos, I. Z. & Batista-Duharte, A. Sporotrichosis: An Emergent Disease. In Sporotrichosis. Springer International Publishing 1–23 (2015).
Lopes-Bezerra, L. M. et al. Sporotrichosis between 1898 and 2017: The evolution of knowledge on a changeable disease and on emerging etiological agents. Medical mycology 56(suppl_1), S126–S143 (2018).
Lopes-Bezerra, L. M., Schubach, A. & Costa, R. O. Sporothrix schenckii and sporotrichosis. Anais da Academia Brasileira de Ciências 78(2), 293–308 (2006).
Zhou, X., Rodrigues, A. M., Feng, P. & Hoog, G. S. Global ITS diversity in the Sporothrix schenckii complex. Fungal Divers 66, 153–165 (2014).
Rodrigues, A. M., de Hoog, G. S. & de Camargo, Z. P. Sporothrix species causing outbreaks in animals and humans driven by animal-animal transmission. PLoS Pathog 12, e1005638 (2016).
Orofino-Costa, R., Macedo, P. M. D., Rodrigues, A. M. & Bernardes-Engemann, A. R. Sporotrichosis: an update on epidemiology, etiopathogenesis, laboratory and clinical therapeutics. Anais brasileiros de dermatologia 92(5), 606–620 (2017).
Arrillaga-Moncrieff, I. et al. Different virulence levels of the species of Sporothrix in a murine model. Clin Microbiol Infect Jul 15(7), 651–5 (2009).
Boechat, J. S. et al. Feline sporotrichosis: associations between clinical-epidemiological profiles and phenotypic-genotypic characteristics of the etiological agents in the Rio de Janeiro epizootic area. Mem Inst Oswaldo Cruz Mar 113(3), 185–196 (2018).
Gremião, I. D. et al. Feline sporotrichosis: epidemiological and clinical aspects. Medical mycology 53(1), 15–21 (2015).
Almeida, P. & Giordano, C. Sporotrichosis Epidemiological Bulletin 001/2018. Secretaria de Estado de Saude do Rio de Janeiro, Boletim Epidemiológico, [in Portuguese]. 2018
Batista-Duharte, A. et al. Therapeutic and prophylactic tools for sporotrichosis: current strategies and future tendencies. In: Sporotrichosis. Springer International Publishing p. 147–77 (2015).
Bonifaz, A. & Tirado-Sánchez, A. Cutaneous disseminated and extracutaneous sporotrichosis: Current status of a complex disease. Journal of Fungi 3(1), 6 (2017).
Aung, A. K., The, B. M., McGrath, C. & Thompson, P. J. Pulmonary sporotrichosis: case series and systematic analysis of literature on clinico-radiological patterns and management outcomes. Medical mycology 51(5), 534–544 (2013).
Mahajan, V. K. Sporotrichosis: an overview and therapeutic options. Dermatol Res Pract 272376 (2014).
Alba-Fierro, C. A. et al. Immune Response Induced by an Immunodominant 60 kDa Glycoprotein of the Cell Wall of Sporothrix schenckii in Two Mice Strains with Experimental Sporotrichosis. J Immunol Res 6, 52–5831 (2016).
Carlos, I. Z. et al. Current research on the immune response to experimental sporotrichosis. Mycopathologia 168(1), 1–10 (2009).
Portuondo, D. L. et al. A cell wall protein-based vaccine candidate induce protective immune response against Sporothrix schenckii infection. Immunobiology 221, 300–309 (2016).
Portuondo, D. L. et al. Comparative efficacy and toxicity of two vaccine candidates against Sporothrix schenckii using either Montanide™ Pet Gel A or aluminum hydroxide adjuvants in mice. Vaccine 35(34), 4430–4436 (2017).
Díaz-Ramos, A., Roig-Borrellas, A., García-Melero, A. & López-Alemany, R. α-Enolase, a multifunctional protein: its role on pathophysiological situations. BioMed Research International (2012).
Ji, H. W. et al. Progress in the biological function of alpha-enolase. Animal Nutrition 2(1), 12–17 (2016).
Nakamura, N. et al. Disruption of a spermatogenic cell-specific mouse enolase 4 (eno4) gene causes sperm structural defects and male infertility. Biology of reproduction 88(4) (2013).
Li, W. et al. Immunization with the glycolytic enzyme enolase confers effective protection against Candida albicans infection in mice. Vaccine 29(33), 5526–5533 8 (2011).
Pal-Bhowmick, I., Mehta, M., Coppens, I., Sharma, S. & Jarori, G. K. Protective properties and surface localization of Plasmodium falciparum enolase. Infection and immunity 75(11), 5500–5508 (2007).
Chen, N. et al. Ascaris suum enolase is a potential vaccine candidate against ascariasis. Vaccine 30(23), 3478–3482 (2012).
Dinis, M. et al. Oral therapeutic vaccination with Streptococcus sobrinus recombinant enolase confers protection against dental caries in rats. The Journal of infectious diseases 199(1), 116–123 (2009).
Feng, Y. et al. Streptococcus suis enolase functions as a protective antigen displayed on the bacterial cell surface. The Journal of infectious diseases 10, 1583–1592 (2009).
Wang, J. et al. Cloning and characterization of surface-localized α-Enolase of Streptococcus iniae, an effective protective antigen in mice. International journal of molecular sciences 16(7), 14490–14510 (2015).
Dutta, S., DasSarma, P., DasSarma, S. & Jarori, G. K. Immunogenicity and protective potential of a Plasmodium spp. enolase peptide displayed on archaeal gas vesicle nanoparticles. Malaria journal 14(1), 406 (2015).
Yu, J. et al. Oral delivery of Bacillus subtilis spore expressing enolase of Clonorchis sinensis in rat model: induce systemic and local mucosal immune responses and has no side effect on liver function. Parasitology research 114(7), 2499–2505 (2015).
Arce-Fonseca, M. et al. Recombinant Enolase of Trypanosoma cruzi as a Novel Vaccine Candidate against Chagas Disease in a Mouse Model of Acute Infection. Journal of immunology research (2018).
Batista, F. A., Gava, L. M., Pinheiro, G. M., Ramos, C. H. & Borges, j. C. From Conformation to Interaction: Techniques to Explore the Hsp70/Hsp90 Network. Curr. Protein Pept. Sci. 16(8), 735–753 https://doi.org/10.2174/1389203716666150505225744 (2015).
Pancholi, V. Multifunctional α-enolase: its role in diseases. Cellular and Molecular Life Sciences CMLS 58(7), 902–920 (2001).
Portuondo, D. L. F., Ferreira, L. S., Urbaczek, A. C., Batista-Duharte, A. & Carlos, I. Z. Adjuvants and delivery systems for antifungal vaccines: current state and future developments. Medical mycology 53, 69–89 (2015).
Brewer, J. M., Fairwell, T., Travis, J. & Lovins, R. E. Investigation of the subunit structure of yeast enolase. Biochemistry 9(4), 1011–1016 (1970).
Zhang, E., Brewer, J. M., Minor, W., Carreira, L. A. & Lebioda, L. Mechanism of Enolase: The Crystal Structure of Asymmetric Dimer Enolase-2-Phospho-d-glycerate/Enolase− Phosphoenolpyruvate at 2.0 Å Resolution. Biochemistry 36(41), 12526–12534 (1997).
Ehinger, S., Schubert, W. D., Bergmann, S., Hammers chmidt, S. & Heinz, D. W. Plasmin (ogen)-binding α-enolase from Streptococcus pneumoniae: crystal structure and evaluation of plasmin (ogen)-binding sites. Journal of molecular biology 343(4), 997–1005 (2004).
Wu, Y. et al. Octameric structure of Staphylococcus aureus enolase in complex with phosphoenolpyruvate. Acta Crystallographica Section D 71(12), 2457–2470 (2015).
Silva, R. C. et al. Extracellular enolase of Candida albicans is involved in colonization of mammalian intestinal epithelium. Frontiers in cellular and infection microbiology 4, 66 (2014).
Floden, A. M., Watt, J. A. & Brissette, C. A. Borrelia burgdorferi enolase is a surface-exposed plasminogen binding protein. PloS one 6(11), e27502 8 (2011).
Ko, H. C., Hsiao, T. Y., Chen, C. T. & Yang, Y. L. Candida albicans ENO1 null mutants exhibit altered drug susceptibility, hyphal formation, and virulence. Journal of Microbiology 51(3), 345–351 (2013).
Roth, E. J., Calvin, M. C., Max-Audit, I., Rosa, J. & Rosa, R. The enzymes of the glycolytic pathway in erythrocytes infected with Plasmodium falciparum malaria parasites. Blood 72(6), 1922–1925 (1988).
Marcos, C. M. et al. Surface-expressed enolase contributes to the adhesion of Paracoccidioides brasiliensis to host cells. FEMS yeast research 12(5), 557–570 (2012).
Lin, L. et al. Th1-Th17 cells mediate protective adaptive immunity against Staphylococcus aureus and Candida albicans infection in mice. PLoS pathogens 5(12), e1000703 (2009).
Ferreira, L. S. et al. Optimal clearance of Sporothrix schenckii requires an intact Th17 response in a mouse model of systemic infection. Immunobiology 220, 985–992 (2015).
Batista-Duharte, A. et al. Sporothrix brasiliensis induces a more severe disease associated with sustained Th17 and regulatory T cells responses than Sporothrix schenckii sensu stricto in mice. Fungal Biology (2018).
Zhang, A. et al. Identification and characterization of a novel protective antigen, Enolase of Streptococcus suis serotype 2. Vaccine 27(9), 1348–1353 (2009).
Chen, N. et al. Ascaris suum enolase is a potential vaccine candidate against ascariasis. Vaccine 30(23), 3478–3482. Clin Microbiol Infect 2009 Jul 15(7), 651–5 (2012).
Charoenvit, Y. N. & Taylor, R. L. Experimental sporotrichosis in Syrian hamsters. Infection and immunity 23(2), 366–372 (1979).
Nascimento, R. C. & Almeida, S. R. Humoral immune response against soluble and fractionate antigens in experimental sporotrichosis. FEMS Immunology & Medical Microbiology 43, 241–247 (2005).
Nascimento, R. C. et al. Passive immunization with monoclonal antibody against a 70‐kDa putative adhesin of Sporothrix schenckii induces protection in murine sporotrichosis. European journal of immunology 38, 3080–3089 (2008).
de Almeida., J. R., Kaihami, G. H., Jannuzzi, G. P. & De Almeida., S. R. Therapeutic vaccine using a monoclonal antibody against a 70-kDa glycoprotein in mice infected with highly virulent Sporothrix schenckii and Sporothrix brasiliensis. Med. Mycol. 53, 42–50 (2015).
de Almeida, J. R. et al. The efficacy of humanized antibody against the Sporothrix antigen, gp70, in promoting phagocytosis and reducing disease burden. Frontiers in microbiology 8, 345 (2017).
Bohm, G., Muhr, R. & Jaenicke, R. Quantitative-Analysis of Protein Far Uv Circular-Dichroism Spectra by Neural Networks. Protein Eng 5, 191–195 (1992).
Dores-Silva, P. R. D., Minari, K., Ramos, C. H. I., Barbosa, L. R. S. & Borges, J. C. Structural and stability studies of the human mtHsp70-escort protein 1: an essential mortalin co-chaperone. International journal of biological macromolecules 56, 140–148 (2013).
Borges, J. C., Seraphim, T. V., Dores-Silva, P. R. & Barbosa, L. R. A review of multi-domain and flexible molecular chaperones studies by small-angle X-ray scattering. Biophysical reviews 8(2), 107–120 (2016).
Laemmli, U. K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227(5259), 680 (1970).
Sievers, F. et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol 7, 539 (2011).
Ishida, K. et al. Efficacy of a poly-aggregated formulation of amphotericin B in treating systemic sporotrichosis caused by Sporothrix brasiliensis. Medical mycology 56(3), 288–296 (2017).
Acknowledgements
This work was supported by Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP, grant number 2015/09340-4, 2017/13228-0 and 2017/26774-3). S.A.P., J.C. B and I.Z.C are recipients of productivity fellowship from ‘Conselho Nacional de Pesquisa e Desenvolvimento Tecnológico’ (CNPq).
Author information
Authors and Affiliations
Contributions
Conception of the work: D.L.P.F., A.B.D., I.Z.C. Design of research: D.L.P.F., A.B.D., I.Z.C., P.R.D.S.; L.S.F. and D.T.M. Performed experiments: D.L.P.F., A.B.D., P.R.D.S.; C.S.O., L.S.F., D.T.M., M.L.A.L. and C.M.M., Data analysis: D.L.P.F., A.B.D., I.Z.C., P.R.D.S.; L.S.F., D.T.M., C.M.M., F.G., J.C.B. Interpreted results of experiments: D.L.P.F., A.B.D., S.A.P., I.Z.C. L.M.G. and J.C.B., Prepared figures: D.L.P.F., A.B.D., I.Z.C., Wrote the manuscript: D.L.P.F., A.B.D., I.Z.C. L.S.F. and J.C.B. All authors contributed to the final version of the paper and gave final approval for publication.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Portuondo, D.L., Dores-Silva, P.R., Ferreira, L.S. et al. Immunization with recombinant enolase of Sporothrix spp. (rSsEno) confers effective protection against sporotrichosis in mice. Sci Rep 9, 17179 (2019). https://doi.org/10.1038/s41598-019-53135-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-53135-z
- Springer Nature Limited
This article is cited by
-
Guideline for the management of feline sporotrichosis caused by Sporothrix brasiliensis and literature revision
Brazilian Journal of Microbiology (2021)
-
Extracellular Vesicles From Sporothrix brasiliensis Yeast Cells Increases Fungicidal Activity in Macrophages
Mycopathologia (2021)