
Abstract
Milk microbiomes significantly influence the pathophysiology of bovine mastitis. To assess the association between microbiome diversity and bovine mastitis, we compared the microbiome of clinical mastitis (CM, n = 14) and healthy (H, n = 7) milk samples through deep whole metagenome sequencing (WMS). A total of 483.38 million reads generated from both metagenomes were analyzed through PathoScope (PS) and MG-RAST (MR), and mapped to 380 bacterial, 56 archaeal, and 39 viral genomes. We observed distinct shifts and differences in abundance between the microbiome of CM and H milk in phyla Proteobacteria, Bacteroidetes, Firmicutes and Actinobacteria with an inclusion of 68.04% previously unreported and/or opportunistic strains in CM milk. PS identified 363 and 146 bacterial strains in CM and H milk samples respectively, and MR detected 356 and 251 bacterial genera respectively. Of the identified taxa, 29.51% of strains and 63.80% of genera were shared between both metagenomes. Additionally, 14 archaeal and 14 viral genera were found to be solely associated with CM. Functional annotation of metagenomic sequences identified several metabolic pathways related to bacterial colonization, proliferation, chemotaxis and invasion, immune-diseases, oxidative stress, regulation and cell signaling, phage and prophases, antibiotic and heavy metal resistance that might be associated with CM. Our WMS study provides conclusive data on milk microbiome diversity associated with bovine CM and its role in udder health.
Similar content being viewed by others

Introduction
Mastitis is one of the most prevalent diseases in the dairy industry with the highest clinical and economic significance worldwide1. The condition usually happens when pathogenic microbes enter the mammary gland, mostly by the disruption of the physical barriers of the mammary quarters, requiring prompt and appropriate host defenses to prevent colonization and subsequent disease pathology2. Diverse groups of microbes are known to colonize the mammary quarters of cows and have evolved novel mechanisms that facilitate their proliferation, leading to clinical mastitis (CM). Despite knowledge of a few of these invading microbial groups, the etiology of bovine mastitis is continuously changing, with new microbial species identified as causing disease frequently. Additionally, although bacteria are the main cause of mastitis3, other microbes like archaea, viruses, and fungi might be associated with the disease process4 and should therefore be investigated as well. During the progression of the mastitis, dysbiosis of the milk microbiome can occur with the increase of opportunistic pathogenic bacteria and reduction of healthy commensal bacteria5. Until recently, investigations of the microbiome associated with bovine mastitis have been mostly restricted to individual pathogen isolation and characterization. The disease is caused by epidemiologically diverse groups of microorganisms and categorized into contagious and environmental mastitis6. The udder of the dairy cows is the primary reservoir of contagious pathogens including Staphylococcus aureus, Streptococcus agalactiae, Streptococcus dysgalactiae, Mycoplasma spp., and Corynebacterium bovis1,6. The involvement of the bovine mammary gland microbiome in the host-pathogen interaction has infrequently been investigated except during the infectious episode7. Environmental pathogens such as Escherichia coli, Klebsiella pneumoniae, Klebsiella oxytoca, Enterobacter aerogenes, Streptococcus dysgalactiae, and Streptococcus uberis1,6 can also be implicated in disease.
Rapid advances in high-throughput NGS technology and bioinformatics tools8 during the last decade have initiated a transition from clinical microbiology to genomic characterization of the microbiome associated with infection, including mastitis which affects lactating women5 and animals9. However, until recently, 16S rRNA partial gene sequencing approach remained the most commonly used genomic survey tool in studying the bovine mastitis microbiome1,7. This technique is highly useful in resolving more than 90.0% of isolates at the genus level. However, it has a number of inherent limitations including the polymerase chain reaction (PCR) bias, inability to detect viruses, lower taxonomic resolution at the species or strain level, and limiting information on gene abundance and functional profiling10. These factors eventually limit the ability to fully explore the microbiome and its interaction with the host comprehensively. A complimentary, shotgun whole metagenome sequencing (WMS) approach reflecting the total microbial makeup of a sample (bacterial, archaeal, fungal, viral) has been used successfully to gain insights into the phylogenetic composition and species diversity of a variety of microbiomes11, including profiling of their functional attributes12. Thus, data such as the identity and abundance of genes related to microbial metabolism, virulence, and antibiotic resistance can be generated simultaneously enabling identification of unknown etiological agents that play a role in mammary gland pathogenesis.
The relatively overexpressed genes associated with immune suppression13, systemic oxidative stress3 and inflammatory processes14 coming from metabolic activities of the microbiome (bacteria, archaea and virus) are crucial factors for the development and progression of bovine CM. Surprisingly from the beginning of the twenty-first century, a rapid increase in antimicrobial resistance, particularly multidrug resistance (MDR), in bovine mastitis pathogens has been observed, which corresponds with the relatively higher abundance of genes coding for antibiotics and toxic compounds resistance in the CM milk microbiome15. Therefore, summarizing the variation in biota and protein functional diversity in clinical and healthy milk microbiomes using cutting-edge genomic technologies like WMS16 and associated bioinformatic tools is essential in understanding the pathophysiological conditions of bovine CM. Here we report the first study to apply high-throughput sequencing data (on an average 23.01 million reads per sample) to investigate the microbiome of bovine CM and H milk17. The results revealed that cows suffering from CM have a distinct microbial community with altered protein functions compared to their healthy counterparts, which leads to pathophysiological conditions.
Results
Structure and composition of the bovine milk microbiome
The rarefaction curves based on observed species metrics reached the plateau after on average 23.01 million reads (Supplementary Fig. 1a; Data 1) suggesting that the depth of coverage was sufficient to capture the entire microbial diversity within the samples. We found significant differences in alpha-diversity (Observed species and Shannon estimated) between the clinical mastitis (CM) samples and healthy controls (H) regardless of the method used to tabulate microbial abundances i.e., either PathoScope (PS) or MG-RAST (MR) (PS; p = 0.005, MR; p = 0.007, U test), showing higher diversity in the microbial ecosystem of CM milk (Supplementary Fig. 1b–d). Beta diversity (PCoA) also showed significant microbial disparity (p = 0.001) between CM and H sample groups (Supplementary Fig. 2a,b). At phylum level, NMDS also showed distinct differences (p = 0.001) between the sample categories (Supplementary Fig. 3a,b).
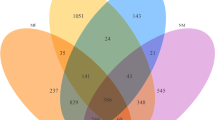
At the domain level, bacteria were the most abundant community, with an average abundance of 99.49%, followed by viruses (0.38%), and archaea (0.13%) (Supplementary Data 1). Though the relative abundance of microbes was higher in CM compared to H milk, the abundance fluctuated more (CV = 886.90 vs 511.80; PS, CV = 1521.41 vs 1221.92; MR). The unique and shared distribution of microbial taxa found in CM and H samples by the two analytic tools is represented in Venn diagrams (Fig. 1). A total of 363 bacterial strains in CM and 146 in H metagenomes were detected in PS analysis, of which 116 (29.51%) strains were present in the both sample sets (Fig. 1a). However, with the MR pipeline, 356 and 251 bacterial genera were detected in CM and H samples, respectively whereas 227 (63.8%) genera were common in both metagenomes (Fig. 1b). By comparing the detected bacterial genera between two analytic tools, 98 unique genera were identified, of them 62.24% genera were solely associated with the onset of bovine CM (Fig. 1c; Supplementary Data 2). In addition, MR detected 54 and 42 archaeal, and 35 and 25 viral genera in CM and H samples, respectively, and among them 25.00% and 35.00% archaeal and viral genera, respectively had sole association with CM (Fig. 1d,e). Unlike MR, PS detected only one archaeal genera (Methanobrevibacter) in CM and none in H samples.

Taxonomic composition of bovine milk microbiome. Venn diagrams representing the core unique and shared microbiomes of bovine clinical mastitis (CM) and healthy (H) milk. (a) Venn diagram comparison of bacteria at strain level by PathoScope (PS), (b) Venn diagram showing unique and shared bacterial genera by MG-RAST (MR), (c) Shared and unique bacterial genera distribution between PS and MR, (d,e) Venn diagrams representing unique and shared archaeal and viral genera, respectively found in bovine milk as analysed with MR pipeline. Microbiota shared between the conditions are indicated in bold.
CM-associated bacteria changes at the genus level
The current microbiome study demonstrated notable differences among the microbial community in CM and H milk using both bioinformatics tools. Proteobacteria, Bacteroidetes, Firmicutes, and Actinobacteria (contributing to 96.51% of the total sequences, U test, p = 0.001) were the four most abundant phyla in PS and MR analyses. The relative abundances of the top 40 bacterial genera were compared between the CM and H cohorts through PS and MR analyses (Fig. 2). Among the predominating phyla, Proteobacteria was the most diverse and included a wide variety of genera including Acinetobacter, Pseudomonas, Escherichia, Vibrio, Erwinia, and Pantoea. The phylum Firmicutes was dominated by Streptococcus, Enterococcus, Staphylococcus, and Bacillus while Chryseobacterium, Porphyromonas and Prevotella were predominating in Bacteroidetes phylum, and Corynebacterium was the most abundant genus in the phylum Actinobacteria. Among the detected genera, Acinetobacter (60.14%), Campylobacter (10.93%), Pantoea (0.66%), Klebsiella (0.63%), Kluyvera (0.42%), Salmonella (0.31%), Enterobacter (0.30%), Shewanella (0.30%), Escherichia (0.28%), Citrobacter (0.15%), and Bacillus (0.10%) had higher mean relative abundance in CM samples regardless of analytical tool, while the rest of the genera had relatively lower mean abundances (<0.10%). In contrast, the H milk metagenomes also had higher mean relative abundances of Acinetobacter (52.90%) in PS and MR pipelines followed by Pseudomonas (22.81%), Micromonospora (10.57%), Eubacterium (5.37%), Catenibacterium (2.12%), and Ralstonia (0.12%), and the rest of the genera had much lower abundances (<0.10%). In general, MR detected higher numbers of microbial genera than PS (Supplementary Tables 2 and 3), however results from the both tools were concordant, with 98.00% of the total microbial abundance composed of shared genera (Supplementary Table 4; Data 2).

Taxonomic profile of 40 most abundant bacterial genera in bovine clinical mastitis (CM) and healthy (H) milk samples. (a) Relative abundance through PathoScope (PS) and (b) relative abundance through MG-RAST (MR) analyses. The 39 most abundant bacterial genera are sorted by descending order of the relative abundance in 21samples, with the remaining genera grouped into the ‘Other genera’. Each stacked bar plot represents the abundance of bacteria in each sample of the corresponding category, where the last two bar plots depict overall relative abundance of bacterial genera between CM and H samples, respectively. (c) The circular plot illustrates the relative abundance of the top 40 bacterial genera in CM and H milk samples analysed through PS and MR. Taxa in both metagenomes are represented by different colored ribbons in both tools. The relative abundancies are illustrated by the sizes of each color segment in the outer circle and the inner blue colored bars. Part of the microbiome is shared by both sample categories (CM-H milk) and part is analytic tool specific (PS-MR). Notable differences between the bacterial populations are those where the taxon is abundant in CM samples and effectively undetected in the H milk. Sample names: suffix ending in C refers to clinical (CM) and that ending with H refers to healthy (H) milk samples.
CM-associated bacteria changes at the strain level
We further investigated whether strain level relative abundances of the bacteria differed between CM and H samples (Figs 3 and 4). The CM milk metagenome had significantly (p = 0.001) higher number of bacterial strains than the H milk, and among the detected strains, 62.85% had unique association with bovine CM, and 7.63% were solely found in H milk (Fig. 1a). The presence of few predominating bacterial species in both categories of samples suggests that the crucial differences might be occurring at the strain level, and most of the species identified in each sample were represented by a single strain. The CM milk metagenome was dominated by 26 strains (7.16%) of Acinetobacter species while Pseudomonas, Streptococcus, Corynebacterium, Staphylococcus, Enterococcus, Bacillus and Escherichia species were represented by 22, 16, 12, 11, 8, 7 and 6 different strains, respectively. However, in both metagenomes, Acinetobacter johnsonii XBB1 remained as the most abundant strain with a relative abundance of 39.03% and 31.23% in CM and H samples, respectively. The other predominant strains in CM metagenome were Campylobacter mucosalis, Bacillus mycoides, Klebsiella pneumoniae subsp. pneumoniae HS11286, Leclercia adecarboxylata, Escherichia coli str. K-12 substr. MG1655, Escherichia coli O157:H7 str. Sakai, Escherichia coli UMN026, Escherichia coli IAI39, Staphylococ cusaureus subsp. aureus NCTC 8325, Staphylococcus xylosus, Bacillus subtilis subsp. subtilis str. 168, Mycobacterium sp. Root 265 and Macrococcus caseolyticus. Importantly, this study demonstrated that 68.04% of the detected bacterial strains were exclusively found in CM milk metagenome, and among them Pantoea dispersa EGD-AAK13, Klebsiella oxytoca, Kluyvera intermedia, Shewanella oneidensis MR-1, Kluyvera ascorbata ATCC 33433, Klebsiella aerogenes KCTC 2190, Kluyvera cryocrescens NBRC 102467, Acinetobacter pittii PHEA-2, Pseudomonas mendocina ymp and Acinetobacter gyllenbergii NIPH 230 were the most predominant strains. Furthermore, most of these strains were previously unreported and possibly played an opportunistic role in the mammary gland pathogenesis (Supplementary Data 2; Table 5).

The species and/or strain level taxonomic profile microbiota associated with bovine clinical mastitis (CM). Sequences are assigned to different taxonomic index in PathoScope (PS) analysis using minimum identity of 95% and minimum alignment length 20 as cutoff parameters and the circular phylogenetic tree is constructed based on the neighbor-joining algorithm using FigTree. The round tree illustrates 363 unique strains of bacteria in CM milk metagenomes. The inner circle represents the root of the microbiome defined as bacteria present in all samples. The outer circles represent different strains of bacteria is defined as species (with different strains) present in >50% of samples of the corresponding groups. For the outer circles, the width of a segment is proportional to the observed incidence for that species. Different colors are assigned according to the taxonomic ranks of the bacteria. The species and/or strains in the phylogenetic tree are also available in Supplementary Data 2.

The species and/or strain level taxonomic representation of microbiome in bovine healthy (H) milk samples. Sequences are assigned to different taxonomic index in PathoScope (PS) analysis using minimum identity of 95% and minimum alignment length 20 as cutoff parameters and the circular phylogenetic tree is constructed based on the neighbor-joining algorithm using FigTree. The round tree illustrates 146 unique strains of bacteria in H milk metagenomes. The inner circle represents the root of the microbiome defined as bacteria present in all samples. The outer circles represent different strains of bacteria is defined as species (with different strains) present in >50% of samples of the corresponding groups. For the outer circles, the width of a segment is proportional to the observed incidence for that species. Different colors are assigned according to the taxonomic ranks of the bacteria. The species and/or strains in the phylogenetic tree are also available in Supplementary Data 2.
CM-associated changes of archaea and viruses at the genus level
Another noteworthy finding of this study is the detection of archaeal (relative abundance 0.13%, p = 0.025) and viral (relative abundance 0.38%, p = 0.019) components of the microbiome both in CM and H milk samples (Fig. 5a,b; Supplementary Data 2). The CM metagenome was dominated by Methanosarcina (41.94%), Methanococcoides (19.58%), Methanococcus (12.30%), Methanocaldococcus (2.59%), Methanobrevibacter (1.85%), Thermococcus (1.79%) and Methanosphaera (1.53%) archaeal genera with a lower relative abundance (<0.05%) of the rest of the genera (Fig. 5a). Interestingly, none of the archaeal genera were detected in one CM sample (Ctg3C2). In contrast, Methanoplanus (14.69%), Methanoculleus (12.85%), Euryarchaeota (4.67%) and Haloarcula (1.50%) were the most abundant archaeal genera in H samples. The viral fraction of the current bovine milk microbiome was largely dominated by the members of the Caudovirales order, represented by the Podoviridae, Siphoviridae and Myoviridae families. The predominating viral genera found in CM were Epsilon15-like viruses (15.78%), P2-like viruses (10.12%), Myovirus (8.18%), Lambda-like viruses (8.06%), Bpp-1-like viruses (7.12%), phiKZ-like viruses (4.35%), Betaretrovirus (2.01%), P1-like viruses (1.79%) and T4-like viruses (1.79%). The H milk, however, had a relatively higher abundance of Siphovirus (55.85%), Podovirus (12.49%), T1-like viruses (3.44%) and P22-like viruses (1.71%) (Fig. 5b).

Taxonomic abundance of top 40 archaeal and viral genera in clinical mastitis (CM) and healthy (H) milk through MG-RAST (MR). (a) The relative abundance of 39 most abundant archaeal genera are sorted by descending order, with the remaining genera keeping into the ‘Other genera’. Archaeal genera are found in 20 samples, and absent in one clinical sample (Ctg3C2). (b) Taxonomic distribution of 35 viral genera detected in all of the 21 samples of clinical (CM) and healthy (H) milk metagenomes. The most abundant viral genera are sorted by descending order of the relative abundance. Each stacked bar plot represents the abundance of archaea and viruses in each sample of the corresponding category, where the last two bar plots depict overall relative abundance of archaeal and viral genera in both metagenome groups. Notable differences between the archaeal and viral populations are those where the taxon is abundant in clinical samples and effectively undetected in the healthy milk. Sample names: suffix ends with C refers to clinical (CM) and that ends with H refers to healthy (H) milk samples.
Microbial metabolic functions associated with CM
MR simultaneously analyzed and compared the taxonomic composition and functional profiles of our metagenomic sequences in several ways. On average, the putative genes with known and unknown protein functions were 3.94% and 5.51%, respectively, suggesting that a large proportion of the genes encoding for different functional properties are yet unknown (Supplementary Data 1). By comparing the number of genes assigned to each KEGG pathway between the groups, we found a series of significant differences (p = 0.001) that lead to the functional divergence between the CM and H milk microbiome groups. The PCoA analysis of functional components showed significant differences between the CM and H samples indicating significant functional differences (p = 0.035) (Supplementary Fig. 4). In the comparative analysis, we found that genes associated with metabolism (central carbohydrate, amino acids, cofactors, vitamins, prosthetic groups and pigment), substrate dependence, clustering-based subsystems, cell motility (bacterial chemotaxis, flagellar assembly, invasion of epithelial cells), phases, prophages, transposable elements and plasmids, regulation and cell signaling, stress response, virulence, disease and defense, and immune and infectious diseases were significantly (p < 0.05) overrepresented and positively correlated with bovine CM (Figs 6 and 7; Supplementary Data 3).

Projection of the clinical mastitis (CM) and healthy (H) milk metagenome onto KEGG pathways. The whole metagenome sequencing (WMS) reveals significant differences (p = 0.001) in functional microbial pathways. Heatmaps show the average relative abundance hierarchical clustering of the predicted KEGG Orthologs (KOs) functional pathways of the microbiome across all samples. The color bar at the bottom represents the relative abundance of putative genes. The color codes indicate the presence and completeness of each KEGG module, expressed as a value between -1 (low abundance) and 1 (high abundance). The yellow color indicates the more abundant patterns, whilst blue cells accounts for less abundant KOs in that particular sample. The color bar at the bottom represents the higher relative abundance of putative genes. Sample name: suffix ends with C refers to clinical mastitis (CM) and that ends with H refers to healthy (H) milk samples.

Functional annotation of the clinical mastitis (CM) and healthy (H) milk metagenome using different levels of SEED subsystem. Comparison of metagenomic profiles of CM and H milk microbiome at different levels of SEED subsystems (level 1–3). The selected subsystems showing significant (p < 0.05) differences between the two sample groups is shown. The less abundant subsystems in a given metagenome are shown in blue and subsystems that are more abundant are represented in yellow colors. The color codes indicated the presence and completeness of each subsystem module, expressed as a value between -1 (low abundance) and 1 (high abundance). The color bar at the bottom represents the higher relative abundance of putative genes. Sample name: suffix ends with C refers to clinical mastitis (CM) and that ends with H refers to healthy (H) milk samples.
Genes associated with citrate synthase (CS, gltA), fumarate hydratase class I (fumA, fumB), oxidative phosphorylation, bacterial translation, ribosome biogenesis and tRNA amino-acylation were significantly enriched in the metabolic pathways of CM associated microbiomes. The CM associated microbiome had significantly (p < 0.001) higher relative abundance (50.51%) of genes coding for benzoate degradation than the H milk biomes (36.41%). The CM samples had upregulation of genes for energy metabolism including carbon, sulfur, and methane metabolism compared to the H samples. The relative abundance of genes encoding ABC transporter (38.97%) and bacterial chemotaxis (68.61%) were significantly higher in CM microbes than those detected in H milk microbiomes (p < 0.005). Among the pathways in infectious diseases, genes coding for epithelial cell signaling, epithelial cells invasion, Legionellosis, Vibrio cholerae pathogenic cycle, Staphylococcus aureus, Salmonella and pathogenic Escherichia coli infection were most abundant in the CM metagenomes. Likewise, there was a predominant abundance of genes responsible for glutathione S-transferase (GST), breakpoint cluster region protein (BCR1), fumarate hydratase class II (fumC) and pyruvate kinase (pk) in different pathways causing mammary gland inflammation. The CM milk microbiomes had a significantly (p < 0.001) higher number of reads (64.29%) coding for severely combined immune deficient gene adenosine deaminase (ADA) compared to H milk microbes (28.58%) (Supplementary Fig. 5). Furthermore, sporulation related hypotheticals and CRISPR-associated proteins (Cas1, Cas2 and Cas3) remained higher in CM metagenomes compared to H milk microbes (Supplementary Data 3).
We found that the CM microbiome had significantly higher abundance of genes encoding for oxidative stress (36.46%), pathogenicity islands (10.13%), phage related transposable elements (19.48%), phage packaging machinery (6.37%), phage replication (6.70%) and phage regulatory gene expression (7.10%) compared to those of H milk biomes (p < 0.003). However, the phage lysogenic conversion related genes remained higher in abundance among the healthy milk microbes. A deeper look at microbial genes associated with regulation and cell signaling revealed that CM microbes had significantly higher expression of this gene compared to healthy milk microbiome (p = 0.001). Within this subsystem, genes coding for two-component regulatory system BarA-UvrY (SirA; CM = 85.78% vs H = 67.41%), pericellular trafficking and cell invasion- the membrane type-1 matrix metalloproteinase (MT1-MMP; CM = 86.59% vs H = 73.80%), programmed cell death (CM = 55.00% vs H = 28.57%) and intra-membrane regulatory proteolytic pathway- endoplasmic reticulum chaperon grp78 (BiP; CM = 92.85% vs H = 71.42%) were predominantly found to be associated with the onset of bovine CM. We also identified novel associations of biofilm formation (BF) properties among the microbes identified in both metagenomes. The relative abundance of genes coding for protein YjgK cluster linked to biofilm formation, biofilm PGA synthesis, deacetylase PgaB, N-glycosyltransferase PgaC and auxiliary protein PgaD were over-expressed in mastitis-causing pathogens (p = 0.035). In contrast, the genes coding for quorum sensing (QS) in particular to QS in Yersinia, Pseudomonas and Vibrio remained overexpressed in H milk metagenomes. Moreover, of the reads assigned to different levels of SEED subsystems (6.45 million), 2.63% mapped against 30 and 28 different resistance to antibiotic and toxic compounds (RATC) genes in CM and H milk metagenomes, respectively (Fig. 8; Supplementary Data 3). Among them, genes encoding multidrug resistance (efflux pumps, mdtABCD cluster, CmeABC operon), methicillin, vancomycin, and compounds (arsenic and chromium) resistance had two-fold higher relative abundances in CM microbiomes than H milk microbiomes. There was 5 to 7-fold overexpression of multidrug resistance to MAR locus and mercury resistance genes in CM microbes than in H milk organisms. In addition, CM-causing microorganisms harbored two additional resistance genes; multidrug resistance to operon (mdtRP) and aminoglycoside adenyltransferase (Supplementary Data 3).

Networks showing distribution of the resistance to antibiotic and toxic compounds (RATC) genes in bovine milk metagenomes. A total of 6.45 million reads mapped to different levels of SEED subsystems in MR pipeline, of which 2.63% reads mapped against 30 and 28 different RATC genes in CM and H milk metagenomes, respectively. Black lines with yellow circles demarcate the distribution of the resistant genes according to their class across the both metagenomes. The diameter of the circles indicates the relative abundance of the respective genes in both clinical mastitis and healthy milk samples. The two differentially expressed genes (multidrug resistance to operon, mdtRP and aminoglycoside adenyltransferase) in CM milk metagenome are highlighted in deep yellow circles.
Discussion
Over the past decade, metagenomics has helped shed light on the ‘known unknown’ component of the milk microbiome and enabled insights into its taxonomic composition, dynamics, and importance to cows udder health homeostasis. Metagenomic deep sequencing (WMS) of bovine milk has uncovered previously overlooked microbial populations of high complexity with potential roles in regulation of overall microbiome composition and function, and in the onset, progression, and treatment strategies of bovine CM. Yet today, 16Sr RNA partial gene sequencing remains the dominant technique for characterizing milk microbiomes, and findings are limited to bacterial identification at the genus level5,9,18, though this method has serious inherent limitations19. However, little is known about the association of other microbes (archaea and viruses), microbiome shift and particularly functional changes. The noteworthy findings of the present WMS study are the taxonomic profiling of bacteria at both the species and/or strain-level, the possible association of the archaeal and viral fractions with bacterial mastitis, and the crosstalk between the identified microbiomes and their functional genomics in the association of bovine CM.
The findings generated from shotgun metagenomic data are much higher in taxonomic resolution and predicted protein functions and are consistent with previous 16S rRNA partial gene based studies1,9,18. The core bacteria associated with bovine CM such as Acinetobacter, Pseudomonas, Klebsiella, Escherichia, Enterobacter, Staphylococcus, Streptococcus, Bacillus, Pantoea, Shewanella, Ralstonia etc. remained consistent with the metagenomic data regardless of the analytic tool. Though CM milk samples had relatively higher taxonomic abundance, their abundance remained more inconsistent corroborating several recent findings5,18,20. To date, around 50 bacterial genera have been reported in bovine milk through 16Sr RNA-based targeted amplicon sequencing1,9,18,21, while our current WMS study detected 356 and 251 bacterial genera in CM and H milk, respectively indicating the increased discriminatory power of this cutting-edge technology in identifying taxa in the milk microbiome10,16. The observed increase in phylum-level signature of Proteobacteria, Bacteroidetes, Firmicutes and Actinobacteria in CM milk independent of quarter, parity, and breeds of the cows is mostly consistent with many of the previous studies5,9,21. Furthermore, the CM milk metagenome had an inclusion of 68.04% previously unreported bacterial species and/or strains, most of which are opportunistic in nature. Until now, no substantial information was available regarding the association of different strains of Acinetobacter with bovine mastitis22. In a recent study, association of Acinetobacter causing bubaline CM7 has been reported, supporting our present findings. The H milk metagenome had higher relative abundance of soil or environmental microbes (Micromonospora)23 and animal skin microbes (Pseudomonas)24, which can potentially act as opportunistic infection leading to disease. Klebsiella pneumoniae is an opportunistic environmental pathogen, and transmission of this bacterium might occur from contaminated feces and bedding materials25. The gut microbiome plays a key role in maintenance of nutrition, host defense, and immune development26, and we revealed a close association between the gut microbiome and milk microbes in the pathogenesis of bovine CM as also reported previously27. Additional support for this finding includes the potential existence of an endogenous entero-mammary pathway, through which gut bacteria migrate to the mammary gland, and this could explain the predominating presence of gut bacteria such as the phyla Proteobacteria, Bacteroidetes, Firmicutes, Actinobacteria, Fusobacteria and Tenericutes, with Acinetobacter, Campylobacter, Bacillus, Enterobacter, Staphylococcus, Streptococcus and Kocuria genera in CM milk26,27. These pathogens use very efficient strategies to evade host defenses in order to colonize and invade mammary tissues through adhesion28, thereby damaging host cells and fighting with cow immune systems to produce clinical and/or chronic mastitis28,29,30.
Our study marks an additional step towards identifying the significant co-occurrence of archaea and viruses with bacterial population in bovine milk. In comparison to bacteria, the relative abundance and diversity of archaea31 and viruses32 remain substantially lower. Currently, there is no extensive evidence supporting the role of archaea and viruses in the pathogenesis of bovine mastitis. However, these microbes mostly seize the opportunity during the pathophysiological changes in the mammary glands created by bacteria33. Thus, it is hypothesized that archaea might follow the exact mechanisms of bacterial pathogens producing bovine CM31. Most of the detected viral genera belonged to the order Caudovirales which consists of the three families of tailed bacterial viruses (bacteriophages) infecting bacteria and archaea. The host range of Caudovirales is very broad and includes all major bacterial phyla found in our samples: Firmicutes, Bacteroidetes, Proteobacteria and Actinobacteria. This corresponded with an increased relative abundance of these bacterial taxa in CM milk samples together with an overrepresentation of Caudovirales taxa compared with H milk samples34. In addition, we found a significant association of Herpesvirales (Macavirus and Rhadinovirus genera) with bovine CM34,35. Our current findings demonstrated that viruses neither cause bovine mastitis directly nor play a role in the initiation of the disease process, but later, when bacterial infection of the udder occurs, they replicate in the immune and epithelial cells of the udder and/or milk ducts and may act as a predisposing factor as well as a primary etiological agent for more severe and prolonged mastitis36.
The KEGG pathways and SEED subsystems of the MR pipeline uncovered significant differences in microbial metabolic functions in both CM and healthy samples5,37 as supported by several previous reports on mastitis in lactating cows9 and women5. The CM microbiome had significantly higher abundance of Proteobacteria and Bacteroidetes, which are well-known utilizers of milk oligosaccharides through one carbon metabolism38. Genes associated with the TCA cycle (gltA, fumA) and energy metabolism (oxidative phosphorylation) remained overexpressed in CM microbiomes, which might be associated with host-pathogen interactions during the progression of bovine mastitis39. Increased benzoate degradation by different strains of Acinetobacter and Klebsiella in CM metagenome through TCA cycle is thought to promote bacterial growth and virulence factors expressed during pathogenesis40. To elucidate the role of bacterial cell to cell communication in bovine mastitis, we found that genes coding for bacterial chemotaxis (cheBR, motB, rbsB and tsr) remained predominantly abundant in CM milk microbiomes suggesting their role in the early phase of mastitis for attachment to or entry into the udder tissues and virulence regulation41. The p38 signaling pathway exerts its biological effects in the pathophysiology of bovine CM through several complex biologic processes including expression of many cytokines, transcription factors, cell surface receptors, enzymes and oxidative stress mediators42,43. The up-regulation of genes coding for programmed-cell death during host–pathogen interactions in CM is associated with increased secretion of bacterial toxins or pro-inflammatory mediators44. Biofilm formation can be a strain specific or genetically linked trait, representing a selective advantage in pathogenesis of mastitis. The relative overexpression of genes encoding the protein YjgK cluster linked to biofilm formation and biofilm PGA synthesis in CM microbiomes is in accordance with several earlier reports45. Moreover, biofilm formation can also be harmful to host tissues since they can promote the phagocyte release of lysosomal enzymes, proliferation of reactive oxygen and nitrogen species, and transfer of antibiotic resistance45. The observed increased abundance of genes for primary immune diseases (e.g., adenosine deaminase) in CM pathogens is responsible for inhibition of T cell maturation and lymphocytic proliferation46, very low CD4 count47, cell-to-cell communication47 and therefore could be used as a selective marker for bovine CM diagnosis. CRISPR/Cas systems are present in both pathogenic and commensal organisms found in bovine milk and play critical roles during the pathogenesis of mastitis by evading the hosts defense system particularly under stress conditions48. The type III and IV secretion systems found on the pathogenicity islands of CM associated microbes are capable of producing immunosuppression in cows by delivering effector proteins9,49. Phages, which are the regulators of bacterial population, play important and diverse roles in all bacterial ecosystems36, but their precise impact on the milk microbiome is far from being understood. The relative overrepresentation of genes coding for phage-related transposable elements, phage packaging machinery, phage replication and phage regulatory gene expression in CM microbes may suggest that bacteriophages participate in the horizontal gene transfer among the members of bovine milk microbiomes and ultimately to mammary gland pathogens34.
Bovine milk microbiomes are a wide source of resistance to antibiotic and toxic compounds (RATC) genes and the pathogenic bacteria within this potential reservoir are becoming more resistant. The current metagenomic deep sequencing provides a wealth of information not only on RATC genes, but on the entire gene content thereby enabling the identification of the community composition and metabolic profile. We found that all of the samples in both metagenomes harbored RATC genes (2.63%) indicating their wide and indiscriminate use in Bangladeshi dairy farms. However, most of the resistant genes in RATC functional groups remained predominantly higher in CM milk microbes. While our knowledge of the uncontrolled spread of antibiotics resistant genes in bovine mastitis pathogens50 is increasing, information on heavy metal resistance is not yet available. This worrisome trend in increasing RATC against mastitis pathogens has become a major concern for the dairy farmers of Bangladesh, given the seriousness of such problems; effective therapies using alternative medicines are needed for successful prevention and control of bovine mastitis.
Conclusions
In this study, the metagenomics of milk samples from bovine with clinical mastitis (CM) and healthy (H) controls clearly show that the microbiome composition in CM milk samples are significantly different from H milk. Furthermore, some of the detected microbes (bacteria, 68.04%, archaea, 31.82% and viruses, 40.00%) are solely found only in CM samples. The co-relations of the microbiome composition and functional metagenomics in the progression of clinical mastitis are also evidenced by abundance differences in metabolic pathways related to bacterial colonization, proliferation, chemotaxis and invasion, immune-diseases, oxidative stress, regulation and cell signaling, antimicrobial resistant genes, biofilm formation, phage and prophases etc. between CM and H samples. The presence of human pathogens including Escherichia coli O157:H7 str. Sakai, Salmonella enterica subsp. enterica serovar Typhi str. CT18, Salmonella enterica subsp. enterica serovar Typhimurium str. LT2, Bacillus cereus ATCC 14579 etc. in bovine milk and RATC genes in milk microbiome are serious concerns for public and animal health since diseased animals are improperly handled in Bangladesh. Because of the limitations we faced with fewer samples, it would be interesting to conduct similar trials using a larger sample size with a different animal population (breed, parity, body condition, lactation) and matrices prior to undertaking a metagenomics sequencing venture to elucidate the progression of the disease. Furthermore, such studies would also be enhanced by the inclusion of gut microbiome sampling in addition to the milk samples for direct testing of microbial transfer across this axis.
Methods
Study population and sampling
Details of study population and collected samples are presented in Supplementary Table 1. A total of 21 milk samples (14, CM and 7, H) from 21 lactating crossbred cows at their early stage of lactation (within 10–40 days of calving) were collected from three districts of Bangladesh (Chattogram = 12, Dhaka = 3, Gazipur = 6). Cows were diagnosed with CM using the California mastitis test51. Two CM and one H milk samples were collected from the same farm. Approximately 15–20 ml of milk from each cow was collected in a sterile falcon tube during the morning milking (8.0–10.0 am) with an emphasis on pre-sampling disinfection of teat-ends and hygiene during sampling1,51. The samples were kept in an ice box (at 4 °C temp) immediately after collection, transported to the laboratory following similar transport protocols, and stored at −20 °C until DNA extraction.
DNA extraction and sequencing
Genomic DNA (gDNA) was extracted by an automated DNA extraction platform (Promega, UK) following previously described protocols5,17. DNA quantity and purity was determined with NanoDrop (ThermoFisher, USA) by measuring 260/280 absorbance ratios. Sequencing libraries were prepared with Nextera XT DNA Library Preparation Kit52 according to the manufacturer’s instructions and paired-end (2 × 150 bp) sequencing was performed on a NextSeq 500 machine (Illumina Inc., USA) at the George Washington University Genomics Core facility. Our metagenomic DNA yielded 483.38 million reads with an average of 23.01 million (maximum = 35.10 million, minimum = 6.77 million) reads per sample (Supplementary Data 1).
Sequence reads preprocessing
The resulting FASTQ files were concatenated and filtered through BBDuk14 (with options k = 21, mink = 6, ktrim = r, ftm = 5, qtrim = rl, trimq = 20, minlen = 30, overwrite = true) to remove Illumina adapters, known Illumina artifacts, and phiX. Any sequence below these thresholds or reads containing more than one ‘N’ were discarded. On average, 20.16 million reads per sample (maximum = 32.33 million, minimum = 4.71 million) passed the quality control step (Supplementary Data 1).
Microbiome community analysis
We analyzed the WMS data using mapping-based and assembly-based hybrid methods of PathoScope 2.0 (PS)53 and MG-RAST 4.0 (MR)8, respectively. In PS analysis, a ‘target’ genome library was constructed containing all bacterial and archaeal sequences from the NCBI Database (https://en.wikipedia.org/wiki/National_Center_for_Biotechnology_Information) using the PathoLib module. The reads were then aligned against the target libraries using the very sensitive Bowtie2 algorithm16,17 and filtered to remove the reads aligned with the cattle genome (bosTau8) and human genome (hg38) as implemented in PathoMap (-very-sensitive-local -k 100–score-min L, 20, 1.0). Finally, the PathoID54 module was applied to obtain accurate read counts for downstream analysis. In these samples, an average of 12.90 million aligned reads per sample mapped to the target reference genome libraries (96.24%) after filtering the cow and human genome (Supplementary Data 1). The raw sequences were simultaneously uploaded in MR server (release 4.0) with proper embedded metadata and were subjected to the quality filter containing dereplication and removal of host DNA by screening55 for taxonomic and functional assignment.
Diversity analysis
Alpha diversity (diversity within samples) was estimated using the Shannon index for both PS and MR reads. To test beta diversity (differences in the organismal structure) of the milk microbiome, a principal coordinate analysis (PCoA) was performed based on weighted-UniFrac distances (for PS data) through Phyloseq R56 and Bray-Curtis dissimilarity matrix57 for MR data. In addition, non-metric multidimensional scaling (NMDS) on PS data was also used for beta diversity58 analysis between the sample groups59. Taxonomic abundance was determined by applying the “Best Hit Classification” option using the NCBI database as a reference with the following settings: maximum e-value of 1 × 10−30, minimum identity of 95% for bacteria, 60% for archaea and viruses and a minimum alignment length of 20 as the set parameters. The phylogenetic origin of the metagenomic sequences was projected against the NCBI taxonomic tree and determined by the lowest common ancestor (LCA) with the same cutoff mentioned above. Two phylogenetic trees consisting of 363 and 146 bacterial strains in CM and H metagenomes, respectively, with >80% taxonomic identity were constructed using the neighbor-joining method in Clustal W (version 2.1)60 and visualized with FigTree (version 1.5.1)14.
Statistical analysis
The characteristics of cows with and without CM were compared using Fisher’s exact test for categorical variables and Mann-Whitney U test for quantitative variables5,7,8,9. The Shapiro-Wilk test was used to check normality of the data and the non-parametric test Kruskal-Wallis rank sum test was used to evaluate differences in the relative percent abundance of taxa in CM and H groups. The statistical analyses for the MR data were initially performed by embedded calls to statistical tests in the pipeline and validated further using SPSS (SPSS, Version 23.0, IBM Corp., NY USA) using above mentioned tests. For the functional abundance profiling, the statistical tests were applied at different KEGG and SEED subsystem levels in the MR pipeline. Differences between the pipelines were evaluated using ANOVA and the Friedman rank sum test. A significance level of alpha = 0.05 was used for all tests8.
Data Availability
The sequence data reported in this paper have been deposited in the NCBI database (BioProject PRJNA529353) and are available from the corresponding author upon reasonable request.
References
Falentin, H. et al. Bovine teat microbiome analysis revealed reduced alpha diversity and significant changes in taxonomic profiles in quarters with a history of mastitis. Front. Microbiol. 7, 480 (2016).
Aitken, S. L., Christine, M. C. & Lorraine, M. S. Immunopathology of mastitis: insights into disease recognition and resolution. J. Mammary Gland Biol. Neoplasia 16(4), 291–304 (2011).
Lin, S. et al. Mammary inflammatory gene expression was associated with reproductive stage and regulated by docosahexenoic acid: in vitro and in vivo studies. Lipids Health Dis. 15(1), 215 (2016).
Rinaldi, M., Li, R. W. & Capuco, A. V. Mastitis associated transcriptomic disruptions in cattle. Vet. Immun. Immunopath. 138(4), 267–279 (2010).
Patel, S. H. et al. Culture independent assessment of human milk microbial community in lactational mastitis. Sci. Rep. 7(1), 7804 (2017).
Kateete, D. P. et al. Prevalence and antimicrobial susceptibility patterns of bacteria from milkmen and cows with clinical mastitis in and around Kampala, Uganda. PLoS One 8(5), p.e63413 (2013).
Catozzi, C. et al. The microbiota of water buffalo milk during mastitis. PLoS One 12(9), e0184710 (2017).
D’Argenio, V., Giorgio, C., Vincenza, P. & Francesco, S. Comparative metagenomic analysis of human gut microbiome composition using two different bioinformatic pipelines. BioMed Res. Int. 325340 (2014).
Cremonesi, P. et al. Milk microbiome diversity and bacterial group prevalence in a comparison between healthy Holstein Friesian and Rendena cows. PLoS One 13(10), e0205054 (2018).
Jovel, J. et al. Characterization of the gut microbiome using 16S or shotgun metagenomics. Front. Microbiol. 7, 459 (2016).
Salvetti, E. et al. Whole-metagenome-sequencing-based community profiles of Vitisvinifera L. cv. Corvina berries withered in two post-harvest conditions. Front. Microbiol. 7, 937 (2016).
Bicalho, M. L. S., Machado, V. S., Higgins, C. H., Lima, F. S. & Bicalho, R. C. Genetic and functional analysis of the bovine uterine microbiota. Part I: metritis versus healthy cows. J. Dairy Sci. 100, 3850–3862 (2017).
Blackburn, M. R. & Linda, F. T. Adenosine deaminase deficiency: unanticipated benefits from the study of a rare immunodeficiency. The J. Immunol. 188(3), 933–935 (2012).
Stewart, R. D. et al. Assembly of 913 microbial genomes from metagenomic sequencing of the cow rumen. Nat. Commun. 9(1), 870 (2018).
Vaibhav, D. B., Anju, P. K., Keyur, D. B., Navin, R. S. & Chaitanya, G. J. Analysis of virulence associated and antibiotic resistance genes of microbes in subclinical mastitis affected cattle milk by pyrosequencing approach. J. Vet. Sci. Med. Diag. 2(2), 3 (2014).
Oniciuc, E. et al. The Present and Future of Whole Genome Sequencing (WGS) and Whole Metagenome Sequencing (WMS) for Surveillance of Antimicrobial Resistant Microorganisms and Antimicrobial Resistance Genes across the Food Chain. Genes 9(5), 268 (2018).
Pärnänen, K. et al. Maternal gut and breast milk microbiota affect infant gut antibiotic resistome and mobile genetic elements. Nat. Commun. 9(1), 3891 (2018).
Oikonomou, G. et al. Microbiota of cow’s milk; distinguishing healthy, sub-clinically and clinically diseased quarters. PLoS One 9(1), e85904 (2014).
Ranjan, R., Rani, A., Metwally, A., McGee, H. S. & Perkins, D. L. Analysis of the microbiome: Advantages of whole genome shotgun versus 16S amplicon sequencing. Biochem. Biophys. Res. Commun. 469(4), 967–77 (2015).
Ganda, E. K. et al. Longitudinal metagenomic profling of bovine milk to assess the impact of intramammary treatment using a third-generation cephalosporin. Sci. Rep. 6, 37565, https://doi.org/10.1038/srep3756512 (2016).
Bhatt, V. D. et al. Milk microbiome signatures of subclinical mastitis‐affected cattle analysed by shotgun sequencing. J. Appl. Microbiol. 112(4), 639–650 (2012).
Montaña, S. et al. The genetic analysis of an Acinetobacter johnsonii clinical strain evidenced the presence of horizontal genetic transfer. PLoS One 11(8), e0161528 (2016).
Carro, L. et al. Genome-based classification of micromonosporae with a focus on their biotechnological and ecological potential. Sci. Rep. 8(1), 525 (2018).
Sela, S. et al. Phenotypic and genotypic characterization of Pseudomonas aeruginosa strains isolated from mastitis outbreaks in dairy herds. J. Dairy Res. 74, 425–429 (2007).
Podder, M. P. et al. Klebsiella species associated with bovine mastitis in Newfoundland. PLoS One 9(9), e106518 (2014).
Maga, E. A., Weimer, B. C. & Murray, J. D. Dissecting the role of milk components on gut microbiota composition. Gut Microbes 4(2), 136–9 (2013).
Ma, C. et al. Cow-to-mouse fecal transplantations suggest intestinal microbiome as one cause of mastitis. Microbiome 6(1), 200 (2018).
Gomes, F., Maria, J. S. & Mariana, H. Bovine mastitis disease/pathogenicity: evidence of the potential role of microbial biofilms. Pathogens Dis. 74(3) (2016).
Hoque, M. N. et al. Molecular characterization of Staphylococcus aureus strains in bovine mastitis milk in Bangladesh. Int. J. Vet. Sci. Med. 6, 53–60 (2018).
Leimbach, A. et al. Whole-genome draft sequences of six commensal fecal and six mastitis-associated Escherichia coli strains of bovine origin. Genome Announc. 4(4), e00753–16 (2016).
Lurie-Weinberger & Gophna, M. N. Archaea in and on the human body: health implications and future directions. PLoS Pathog. 11(6), e1004833 (2015).
Marcó, M. B., Moineau, S. & Quiberoni, A. Bacteriophages and dairy fermentations. Bacteriophage 2, 149–158 (2012).
Lagier, J. C. et al. Many more microbes in humans: enlarging the microbiome repertoire. Clinic. Infect. Dis. 65(1), S20–S29 (2017).
Shkoporov, A. N. & Hill, C. Bacteriophages of the Human Gut: The “Known Unknown” of the Microbiome. Cell Host Microbe 25(2), 195–209 (2019).
Riaz, A., Kifayatullah, M. H. & Naeem, A. Recent Understanding of the Classification and Life Cycle of Herpesviruses: A Review. Sci. Letters 5(2), 195–207 (2017).
Abeles, S. R. & Pride, D. T. Molecular bases and role of viruses in the human microbiome. J. Molecular Biol. 426(23), 3892–3906 (2014).
Kanehisa, M. et al. New approach for understanding genome variations in KEGG. Nucleic Acids Res. 47, D590–D595 (2019).
Li, N. et al. Variation in raw milk microbiota throughout 12 months and the impact of weather conditions. Sci. Rep. 8(1), 2371 (2018).
Frese, S. A., Parker, K., Calvert, C. C. & Mills, D. A. Diet shapes the gut microbiome of pigs during nursing and weaning. Microbiome 3, 2 (2015).
Li, X., Ding, X. Z., Wan, Y. L., Liu, Y. M. & Du, G. Z. Comparative proteomic changes of differentially expressed whey proteins in clinical mastitis and healthy yak cows. Genet. Mol. Res. 13(3), 6593–6601 (2014).
Eloe-Fadrosh, E. A. & Rasko, D. A. The human microbiome: from symbiosis to pathogenesis. Annu. Rev. Med. 64, 145–163 (2013).
Matilla, M. A. & Krell, T. The effect of bacterial chemotaxis on host infection and pathogenicity. FEMS Microbiol. Reviews 42(1), 24 (2017).
Koul, H. K., Pal, M. & Koul, S. Role of p38 MAP kinase signal transduction in solid tumors. Genes Cancer 4(9–10), 342–59 (2013).
Song, H., Ki, S. H., Kim, S. G. & Moon, A. Activating transcription factor 2 mediates matrix metalloproteinase-2 transcriptional activation induced by p38 in breast epithelial cells. Cancer Res. 66(21), 10487–96 (2006).
Long, E. et al. Escherichia coli induces apoptosis and proliferation of mammary cells. Cell Death Differ. 8, 808–816 9 (2001).
Gomes, F., Saavedra, M. J. & Henriques, M. Bovine mastitis disease/pathogenicity: evidence of the potential role of microbial biofilms. Pathogens Dis. 74(3), 1–7 (2016).
Farzaneh, M., Haghkhah, M., Nazifi, S., Lari, M.A. & Fani, M.M. Evaluation of milk adenosine deaminase activities in dairy cattle with subclinical mastitis and their correlation with milk quality. Iranian J. Vet. Clin. Sci. 12(1) (2018).
Baba, K. et al. Adenosine deaminase activity is a sensitive marker for the diagnosis of tuberculous pleuritis in patients with very low CD4 counts. PLoS One 3(7), e2788 (2008).
Sampson, T. R. & Weiss, D. S. Alternative Roles for CRISPR/Cas Systems in Bacterial Pathogenesis. PLoS Pathog. 9(10), e1003621 (2013).
Cheng, D. et al. Prevalence and Isoforms of the Pathogenicity Island ETT2 Among Escherichia coli Isolates from Colibacillosis in Pigs and Mastitis in Cows. Current Microbiol. 64(1), 43–49 (2011).
Cheng, J. et al. Antimicrobial resistance profiles of 5 common bovine mastitis pathogens in large Chinese dairy herds. J. Dairy Sci. 102, 1–11 (2018).
Hoque, M. N. et al. Different screening tests and milk somatic cell count for the prevalence of subclinical bovine mastitis in Bangladesh. Trop. Anim. Health Prod. 47(1), 79–86 (2015).
Head, S. R. et al. Library construction for next-generation sequencing: overviews and challenges. Biotechniques 56(2), 61–77 (2014).
Hong, C. et al. PathoScope 2.0: a complete computational framework for strain identification in environmental or clinical sequencing samples. Microbiome 2(1), 33 (2014).
Francis, O. E. et al. Pathoscope: species identification and strain attribution with unassembled sequencing data. Genome Res. 23(10), 1721–1729 (2013).
Zheng, W. et al. Metagenomic sequencing reveals altered metabolic pathways in the oral microbiota of sailors during a long sea voyage. Sci. Rep. 5, 9131 (2015).
McMurdie, P. J. & Susan, H. Phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS One 8(4), e61217 (2013).
Janssens, P. L. H. R. et al. Long-term green tea supplementation does not change the human gut microbiota. PLoS One 11(4), e0153134 (2016).
Guan, Y. et al. Comparison of the gut microbiota composition between wild and captive sika deer (Cervus nippon hortulorum) from feces by high-throughput sequencing. AMB Express 7(1), 212 (2017).
Larkin, M. A. et al. Clustal W and Clustal X version 2.0. Bioinformatics 23, 2947–8 (2007).
Acknowledgements
The Bangladesh Bureau of Educational Information and Statistics (BANBEIS), Ministry of Education, Government of the People’s Republic of Bangladesh (Grant No. LS2017313) supported this work. The authors also thank Keylie Gibson and Stephanie Warnken, PhD students at the Computational Biology Institute, Milken Institute School of Public Health, the George Washington University, USA and Ruhshan Ahmed Abir, Bio-Bio-1, Bangladesh for their technical support in learning basic bioinformatics operations.
Author information
Authors and Affiliations
Contributions
M.N.H., M.S., A.Z.S. and M.A.H. conceived and designed the overall study and M.N.H. and R.A.C. carried out laboratory works including DNA extractions, quality control and preparation for sequencing. M.A.H., R.A.C. and K.A.C. contributed reagents/materials/analysis tools and sequencing. M.N.H. and A.I. conceived, developed methods and executed data analysis and M.N.H. interpreted the results and prepared the manuscript. M.S., K.A.C. and M.A.H. contributed intellectually to the interpretation and presentation of the results. Finally, all authors have approved the manuscript for submission.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Hoque, M.N., Istiaq, A., Clement, R.A. et al. Metagenomic deep sequencing reveals association of microbiome signature with functional biases in bovine mastitis. Sci Rep 9, 13536 (2019). https://doi.org/10.1038/s41598-019-49468-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-49468-4
- Springer Nature Limited