
Abstract
Inherited hearing loss is associated with gene mutations that result in sensory hair cell (HC) malfunction. HC structure is defined by the cytoskeleton, which is mainly composed of actin filaments and actin-binding partners. LIM motif-containing protein kinases (LIMKs) are the primary regulators of actin dynamics and consist of two members: LIMK1 and LIMK2. Actin arrangement is directly involved in the regulation of cytoskeletal structure and the maturation of synapses in the central nervous system, and LIMKs are involved in structural plasticity by controlling the activation of the actin depolymerization protein cofilin in the olfactory system and in the hippocampus. However, the expression pattern and the role of LIMKs in mouse cochlear development and synapse function also need to be further studied. We show here that the Limk genes are expressed in the mouse cochlea. We examined the morphology and the afferent synapse densities of HCs and measured the auditory function in Limk1 and Limk2 double knockout (DKO) mice. We found that the loss of Limk1 and Limk2 did not appear to affect the overall development of the cochlea, including the number of HCs and the structure of hair bundles. There were no significant differences in auditory thresholds between DKO mice and wild-type littermates. However, the expression of p-cofilin in the DKO mice was significantly decreased. Additionally, no significant differences were found in the number or distribution of ribbon synapses between the DKO and wild-type mice. In summary, our data suggest that the Limk genes play a different role in the development of the cochlea compared to their role in the central nervous system.
Similar content being viewed by others


Introduction
The cochlea is the primary sensory organ in the inner ear for hearing. There are two types of sensory hair cells (HCs) – inner hair cells (IHCs) and outer hair cells (OHCs) – and different supporting cells (SCs), including Deiters’ cells, pillar cells, Hensen’s cells, inner border cells, and inner phalangeal cells1,2,3 (Supplementary Fig. 1). The HCs serve as mechanosensory receptors and convert mechanical sound stimuli into electric signals4,5,6,7. When sound stimulation occurs, the hair bundles of HCs are deflected due to the shearing of the sensory epithelium and the tectorial membrane in the regions of the stimulus8,9,10. Deflection of the hair bundles opens mechanosensitive channels, which induces a depolarizing current that in turn induces persistent and graded receptor potentials in the HCs11,12. The electromotility of OHCs, which form the foundation for sound amplification in the cochlea, is largely dependent on the properties of prestin, which is a motor protein unique to mammals13,14,15. Conclusive evidence for the requirement of prestin for cochlear amplification was obtained from its genetic deletion in mice, which led to the loss of electromotility in isolated OHCs and to a 50-dB decrease in cochlear sensitivity in vivo16,17. The receptor potential of IHCs is responsible for regulating the structure of synaptic vesicles and for the release of glutamate from the ribbon synapse18,19. Abnormalities in the structure or function of HCs usually lead to hearing loss.
LIM motif-containing protein kinases (LIMKs) modulate actin arrangements20,21,22,23, and they consist of two N-terminal LIM domains, a PDZ domain, and a C-terminal kinase domain. LIMK1 and LIMK2 are the two isoforms of LIMK24. In the central nervous system, inhibition of LIMKs reduces actin assembly of the growth cone peripheral region of chick dorsal root ganglion neurons25. Transient overexpression of Limk1 promotes axon formation, but permanent overexpression of Limk1 damages the growth cone and leads to axon retraction26. Cofilin is an actin depolymerizing factor that is inactivated by phosphorylation by LIMKs, and loss of Limk1 shows a significant decrease in the phosphorylation of cofilin in the central nervous system with clustered accumulations of actin filaments along the dendrites, which are smaller than normal27. Additionally, Limk1 knockout mice show increased miniature excitatory postsynaptic currents and enhanced synaptic depression, thus highlighting the role of Limk1 in synaptic function28. Previous reports show that the LIMK-mediated pathway has a profound influence on the motility of OHCs, and LIMK-mediated phosphorylation of cofilin increases both the electromotility and OHC length in guinea pigs29,30. Limk1 is expressed in the mouse brain during embryogenesis31, and Limk2 is expressed in the placenta, brain, and kidney32. However, the detailed expression pattern and the roles of Limk1 and Limk2 during the development of the mouse cochlea need to be further investigated. Here we report the expression pattern of LIMK1/2 in the mouse cochlea and the role of these proteins in auditory function and HC morphology in the mouse cochlea using a Limk1 and Limk2 double knock out (DKO) mouse model33,34.
Results
The expression of LIMKs in the mouse cochlea
To determine the relative expression of LIMKs in the cochlea, we first immunolabeled LIMK1 and LIMK2 in the cochlear epithelia of postnatal day 21 (P21) wild-type (WT) mice with myosin7a, which is a specific HC marker with a cytoplasmic expression pattern, and sox2, which has a nuclear expression pattern and labels the SCs, including Hensen’s cells, Deiters’ cells, pillar cells, inner phalangeal cells, and inner border cells. Confocal imaging of the whole-mount organ of Corti showed that LIMK1 was mainly expressed in the cytoplasm of HCs and SCs at P21, while almost all of the LIMK2 was in the nuclei of HCs and SCs (Fig. 1a, Supplementary Fig. 2). The same expression pattern of LIMKs at P21 was seen at P30 (Fig. 1a. Supplementary Fig. 2). When looking at earlier time points, we found that the expression of LIMK2 was mainly in the cytoplasm at P3 but transferred into the nucleus during postnatal development (Fig. 2a). There was no difference in the immunolabeling of either LIMK1 or LIMK2 from the apical to basal turns, and together these results confirm that the expression pattern of LIMKs is maintained after the auditory system is fully matured. In order to confirm the changes in the expression of the Limk genes, q-PCR was performed in the WT cochlea. We found that Limk1 expression was significantly increased at P3 compared with E16 and then decreased significantly during postnatal development. The expression of Limk2 was not significantly different at P3 compared with E16, but the subsequent reduction in Limk2 expression was similar to that of Limk1 (p < 0.01, n = 5) (Fig. 1b).

Expression of LIMKs in WT mouse cochlea. (a) Immunofluorescence staining showed that LIMK1 and LIMK2 were expressed in the cochlear epithelium in the P21 and P30 mice. Myosin7a was used as a marker for HCs. Images were taken from the basal turn of the sensory epithelium. There was no difference in the immunolabeling of LIMK1 and LIMK2 from the apical to basal turns. Scale bar = 10 µm. (b) RT-qPCR results show the changes in expression of Limks in the mouse cochlea from embryonic development to adult. β-actin was used as the internal control. Data are presented as mean ± SD. (p < 0.01, n = 5).

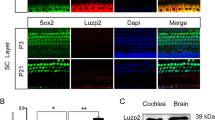
Analysis of LIMK expression in the DKO mice at the mRNA and protein level. (a) The cochleae of P3 DKO mice were immunolabeled with LIMK1 and LIMK2 antibodies. Scale bar = 10 µm. Images were taken from the basal turn of the sensory epithelium. There was no difference in the immunolabeling of LIMK1 and LIMK2 from the apical to basal turns. (b) RT-PCR was performed to analyze the DKO mice. Total cochlear RNA was extracted from P3 DKO and WT mice. Brn3.1 was used as the positive control, and β-actin was used as the internal control. (c) Western blot was performed to analyze DKO mice using antibodies against LIMK1 and LIMK2. Proteins from the brain and cochlea were extracted from P3 DKO and WT mice, and GAPDH was used as the internal control.
The Limk1 and Limk2 genes are knocked out in the DKO mouse cochlea
The DKO mice were genotyped as described previously28,34,35 (Table 1). The coding exon of the LIM and PDZ domains were replaced by the neomycin cassette to create the Limk1 KO mice, and deleting exons 3–5 generated the Limk2 KO mice. To confirm that the Limk1 and Limk2 genes were knocked out in the mouse cochlea, we dissected the cochleae and brains from P3 WT and DKO mice. The immunolabeling of LIMK1 and LIMK2 was seen in HCs of the WT mouse cochlea, but almost none was detected in the DKO mouse cochlea (Fig. 2a). The RT-PCR results confirmed that the Limk1 and Limk2 genes were knocked out in the DKO mice (Fig. 2b, Table 2). Western blots showed that specific bands with a molecular mass around 72 kDa, consistent with the expected sizes of LIMK1 and LIMK2, were detected in the cochlear and brain tissues from WT mice, while only very faint bands were seen in DKO mice (Fig. 2c). The faint signal in the DKO mice might be due to the non-specific binding of the antibody. These results confirmed that the Limk1 and Limk2 genes had been successfully knocked out in the DKO mouse cochleae.
No alteration of general cochlear development in the DKO mice
LIMK1 and LIMK2 have been suggested to play fundamental roles in cell cytoskeleton regulation, and thus we hypothesized that the DKO mice would show cochlear developmental deficits. The cochleae of P30 DKO mice were dissected, and we performed immunofluorescence reactions to examine the structure of the cochlea. Unexpectedly, the cochleae from DKO mice were morphologically indistinguishable from the cochleae of P7 and P30 WT mice, and we did not observe any significant HC loss in P7 and P30 DKO mice. Even at P120 we did not observe any significant differences in morphology or HC populations between WT and DKO mice (Fig. 3a,b, Supplementary Fig. 3). Prestin is the motor protein that is related to OHC motile activity, and in order to confirm the expression and structure of prestin, immunolabeling was performed in P30 DKO and WT mice. The expression of prestin in DKO mice was not obviously different compared to WT mice (Fig. 3c). FM1-43, as a marker of functional mechanotransduction channels in HCs, was used to investigate the HC function in DKO mice. No obvious signals were seen in either WT mice or DKO mice at P0, which indicates that the mechanotransduction channels in HCs are immature at this time. At P14, there was no difference in the signal between DKO mice and WT mice (Supplementary Fig. 5a).

The auditory HCs are morphologically normal in the DKO mice. (a) Auditory HCs of P7, P30, and P120 mice were stained with antibodies against myosin7a and imaged using a confocal microscope. Scale bar = 10 µm. Images were taken from the basal turn of the cochlea. There was no difference in the staining from the apical to basal turns. (b) The HCs were counted and compared with age-matched WT mice (p > 0.05, n = 4). Data are presented as mean ± SD. (c) Auditory OHCs of P30 mice were stained with antibodies against prestin and imaged using a confocal microscope. Images were taken from the basal turn of the cochlea. Scale bar = 10 µm.
The stereocilia structures of HCs in DKO mice are intact
Stereocilia are actin-based protrusions on sensory epithelium HCs that are deflected by sound stimuli to initiate the transformation of mechanical energy into electrical signals, and proper stereocilia structure is essential for maintaining the auditory function of HCs. Immunofluorescence staining of phalloidin showed that the stereocilia polarity of P30 cochlear HCs in DKO mice was normal (Fig. 4a). We then performed scanning electron microscopy to examine the structure of the cochlear HC stereocilia at higher resolution, but still no differences were seen between DKO and WT mice along the tonotopic regions (Fig. 4b). These results suggest that the knockout of LIMKs does not affect the morphology of cochlear HC stereocilia in mice.

The auditory HC stereocilia are morphologically normal in DKO mice. (a) Auditory HC stereocilia of DKO and WT mice were stained with FITC-conjugated phalloidin and imaged using a confocal microscope. Images were taken from the basal turn of the cochlea, and there was no difference from the apical to basal turns. Scale bar = 10 µm. (b) Low magnification and high magnification scanning electron microscope images of OHC stereocilia bundles of DKO and WT mice. Images were taken from the middle turn of P30 mice. Scale bar = 5 µm.
Knockout of Limk1 and Limk2 does not change IHC ribbon synapses
Because p-cofilin regulates the length of the postsynaptic density of asymmetric synapses in the hippocampus of the mouse36, we assessed whether there was any potential change in the molecular composition and/or amount of cochlear ribbon synapses in the HCs of the P14 and P30 DKO mice by immunolabeling the pre-synaptic ribbons and post-synaptic ribbon terminals with antibodies against CtBP2 and PSD95, respectively (Fig. 5a, Supplementary Fig. 4). As specialized components of the pre-synaptic ribbons, the ribbon synapses contain three members of transcriptional repressors: CtBP1, CtBP2, and an alternative splice form called RIBEYE. The signals of CtBP2 were seen in the lateral aspects of the IHCs37. Consistent with a previous study, the PSD95 signal was concentrated at the terminus and the plasma membrane of IHCs38, and this staining did not interfere with the detection of the target signal. The red dot-like staining of CtBP2 at the base of the IHCs overlapped with the PSD95 signal. The synapse densities of the IHCs were comparable between DKO mice and WT controls in all three turns of the cochlea, and no significant differences were seen in the amount of CtBP2 and PSD95-positive puncta in DKO mice compared to WT mice (Fig. 5b,c,e,f). Because nerve fibers will not trigger an action potential when pre-synapses are disconnected from post-synapses, we analyzed the co-localization of CtBP2 and PSD95. We found that almost all of the CtBP2 puncta were overlapping with PSD95 in both WT and DKO mice (Fig. 5d,g). Together, our results indicate that knockout of LIMKs did not change cochlear IHC synapses during postnatal development.

The ribbon synapses were normal in DKO mice. (a) Ribbon synapses of P14 and P30 DKO and WT mice were stained with the ribbon synapse-specific markers CtBP2 and PSD95 and imaged under a confocal microscope. Images were taken from the basal turn of the cochlea, and there was no difference from the apical to basal turns (b,c,e,f). The total numbers of synapses from the 8-kHz to 32-kHz region were counted and compared between WT and DKO mice (d,g). The numbers of functional synapses in DKO mice were compared with WT mice. No significant differences were seen for any measurements (p > 0.05, n = 4). Scale bar = 10 µm. Data are presented as mean ± SD.
Phosphorylated cofilin is significantly decreased in P30 DKO mice
Because cofilin, which is regulated by LIMKs, has a strong influence in the formation of synapses in mature neurons in the hippocampus39,40, we analyzed the expression of cofilin and p-cofilin by Western blot and immunofluorescence in P30 DKO and WT mice. The Western blot data suggested that the levels of cofilin were similar between the DKO and WT mice (Fig. 6a,b), but the expression of p-cofilin was significantly decreased in the DKO mice (p < 0.05, n = 4) (Fig. 6a,b). The immunostaining data also showed that the immunofluorescence intensity of cofilin in HCs was similar between the DKO and WT mice (Fig. 6c,d), but the immunofluorescence intensity of p-cofilin was significantly decreased in the DKO mice (p < 0.05 n = 3) (Fig. 6c,d). In order to determine if knockout of LIMKs altered other synapse-related or actin-related genes, we measured the mRNA levels of synapse-related genes and actin-related genes using RT-qPCR in the cochleae of P30 WT and DKO mice. Cofilin, formin, profilin, myosin, tropomyosin, CtBP2, and PSD95 showed no significant differences compared with WT mice (Supplementary Fig. 5b), while the expression of Arp2, Arp3, and TESK1 were significantly increased and Hsp25 was significantly decreased in DKO mice compared with WT controls (p < 0.05 n = 5) (Fig. 6e).

The effect of loss of LIMKs on cofilin. (a) Western blot was performed to detect the expression level of cofilin and p-cofilin in both P30 WT and DKO mice, n = 3. (b) Quantification of the Western blot (p < 0.01). (c) Auditory HCs of P30 DKO and WT mice were stained with cofilin and p-cofilin antibodies. Images were taken from the basal turn of the cochlea, and there was no difference from the apical to basal turns. Scale bar = 10 µm. (d) Quantification of cofilin and p-cofilin immunolabeling in the basal turns of OHCs (p < 0.05) n = 3. (e) RT-qPCR was performed with P30 WT and DKO mice cochleae (p < 0.05, n = 5). Data are presented as mean ± SD.
DKO mice show no changes in auditory function
Although DKO mice have been reported to be generally healthy35, their auditory function has remained unknown. We therefore first measured their auditory brainstem response (ABR). Auditory thresholds showed no difference between the DKO mice and WT controls at P30 (Fig. 7a) or P120 (Fig. 7b). The ABR reflects the overall activity of the neural output of the cochlea, while the distortion-product otoacoustic emissions (DPOAEs) depend only on the normal function of the OHCs41. We measured the DPOAE of P30 DKO mice, and no obvious differences were found in DPOAE thresholds compared with WT mice (Fig. 7c). Finally, we measured the compound action potential (CAP) of different frequencies at 90 dB in response to tone bursts, in which the N1 amplitude provides a physiological representation of the mouse audiogram42. We found no difference in cochlear sensitivity at frequencies between 4 kHz and 32 kHz between DKO and WT mice (Fig. 7d). Together these results show that the DKO mice have full auditory function.

Auditory measurements show normal auditory function in DKO mice. (a) ABR thresholds of P30 DKO and WT mice were measured at 4, 8, 12, 16, 24, and 32 kHz. (b) ABR thresholds of P120 DKO and WT mice were measured at 4, 8, 12, 16, 24, and 32 kHz. (c) DPOAE thresholds were measured in response to tone bursts of 4, 8, 16, 20, 24, 28, 32, 36, and 40 kHz in P30 DKO and WT mice. (d) CAP amplitudes of P30 DKO and WT mice were measured in response to tone bursts of 4, 8, 16, and 32 kHz at 90 dB SPL. No significant differences were observed between DKO and WT mice (p > 0.05). Data are presented as mean ± SD.
Discussion
LIMKs modulate the architecture of the cytoskeleton through the phosphorylation of the cofilin family of proteins43, which consists of non-muscle type cofilin-1, muscle type cofilin-2, and actin depolymerization factor (ADF). This inactivation of cofilin is modulated by a number of extracellular signals, for example, LIMKs, TES kinases, and Nck-interacting kinase-related kinase44. Cofilin is phosphorylated at Ser-3 by several kinases45, and the major kinases of cofilin are LIMKs, testicular protein kinase 1/246,47, and the Nck-interacting kinase-related kinase48 in mammals. P-cofilin is dephosphorylated in mammals by the three members of the slingshot protein phosphatase family, SSH1, SSH2, and SSH349,50,51.
LIMK1 is known to play an indispensable role in dendritic spine function and synapse function, and previous studies suggested that Limk1 KO mice have severely impaired long-term hippocampal potentiation and severely impaired dendritic spine morphology28,52,53. LIMKs might contribute to synaptic regulation through the modulation of plasticity and cellular processes, and actin is actively involved in the regulation of synaptic plasticity and signaling through presynaptic neurotransmitter release. Previous studies also showed that 15–20% of the p-cofilin was still present in DKO mice, which indicates that other compensatory signaling pathways are capable of regulating cofilin phosphorylation in the central nervous system35. Because LIMKs have an indispensable role in the development of the brain, we hypothesized that LIMKs would have a significant effect on the development and function of the mouse cochlea. A previous study used a patch clamp to isolate guinea pig OHCs and performed immunostaining of LIMKs in guinea pig OHCs in vitro, and they found that both LIMKs were abundant in both the cytoplasm and nuclei of OHCs30. In this study, we found that LIMKs were expressed in the WT mouse cochlea by whole-mount immunostaining. However, most of the antibody signal was seen in the cytoplasm of HCs and SCs, while the signal for LIMK2 antibody was mainly seen in the nuclei of HCs and SCs in the adult mice (Fig. 1, Supplementary Fig. 2). The difference in expression pattern might due to the different animal species or due to the different immunostaining methods.
The different subcellular locations of LIMK1 and LIMK2 might indicate distinct roles of LIMKs in HCs. Although knockout of both LIMK1 and LIMK2 proteins in the DKO mice decreased the levels of p-cofilin in the mouse cochlea, the auditory function and structural arrangements of hair bundles and ribbon synapses showed no alterations compared to age-matched WT mice up to P120. However, it is possible that older DKO mice might have more severe hearing loss compared to WT controls. As for aging DKO mice, whether the loss of LIMK1 and LIMK2 will cause hearing loss is still unknown and needs to be further studied. Moreover, the effect of LIMKs in the development of dendritic spines on auditory nerve dendrites was not investigated in the current study and should be investigated in the future.
The actin dynamics are not only indispensable for maintaining the cytoskeleton and for extending cellular protrusions, but are also indispensable for internal cell rearrangements. The mechanical forces that drive intracellular processes are provided by actin filament formation at intracellular membranes. The dynamics of the actin cytoskeleton require the formation of rod-like actin polymers, known as filaments, from actin monomers. The process by which G-actin is removed from the trailing end of an actin filament and is added onto the opposing fast-growing end of F-actin is referred as actin treadmilling54, and the cofilin/ADF proteins are critical intermediates that rely on the upstream proteins to modulate actin filament assembly.
Prestin is a tetrameric protein expressed in the lateral wall plasma membrane of OHCs and is necessary for the electromotility of OHCs. Prestin has been under intense investigation due to its crucial role in cochlear amplification, which enhances sound-induced vibration in the cochlea to activate the mechanical processes behind hearing sensitivity. Prestin is located in the lateral wall membrane of OHCs, along the region of a well-organized actin-based cortical cytoskeleton15. Prestin KO mice lack an organized cytoskeleton, suggesting that prestin is essential for maintaining such structures55. However, in our study we did not find an auditory phenotype in the DKO mice, and this indicates that the motility of OHCs in DKO mice was not significantly different from WT. Also, there were no obvious differences in the amount of functional mechanotransduction channels between DKO mice compared to WT. The depolymerization of actin plays an important role in the modulation of the motility of OHCs through the ROCK-LIMK1 signaling pathway. Furthermore, the active ROCK-PKCα signaling cascade with lysophosphatidic acid leads to phosphorylation of Ser-726, which is well known as an actin-spectrin dissociation promoter that regulates the length of OHCs and enhances the OHCs’ electromotile amplitude. Thus, we also speculate that the ROCK-PKCα pathway and the ROCK-LIMK1 pathway in OHCs engage in active cross talk and work in parallel with each other29,30. ROCK-PKCα might also be a compensatory signaling pathway for OHC motility in the DKO mice.
Cofilin contributes to the dynamics of actin filaments. Cofilin promotes depolymerization at low concentrations but promotes nucleation at high concentrations56. Previous studies in the central nervous system showed that diminished expression of p-cofilin induces synaptic loss in the hippocampus36,57. However, although the expression of p-cofilin protein is significantly decreased in the DKO mice, this decrease does not change cochlear development or auditory function in the mice, which is different from the previous reports of the effect of cofilin in the central nervous system36,57. Several possible reasons might explain the difference between the effect of cofilin in the brain and auditory system. First, there are several important regulators of the cytoskeleton, including the actin nucleator formin, the actin-sequestering protein profilin, the actin-related protein 2/3 complex58, ADF/cofilin46,58, the actin motor protein myosin59, and the actin-stabilizing protein tropomyosin60. Thus, cofilin might not be the key factor regulating the actin cytoskeleton in the inner ear. This hypothesis is supported by our qPCR data. We did not find any statistical difference in synapse-related genes (CtBP2 and PSD95) or some actin-related genes (cofilin, formin, profilin, myosin, and tropomyosin) between DKO and WT mice, indicating that the homeostasis between actin polymerization and depolymerization is unchanged in the DKO mice. Second, the p38/MK2/Hsp25 and Cdc42/PAK/LIMK1 signaling pathways work in parallel to modulate specific actin-related proteins in mesenchymal cells61,62. RhoA/ROCK/PKCα might be a compensatory signaling pathway that regulates the motility of OHCs. Thus, these alternative signaling pathways might modulate the cytoskeleton in the inner ear and compensate for the lack of the LIMK proteins. The Arp2/3 complex, which can be phosphorylated by p21-activated kinase 1 (PAK1) to promote actin polymerization, was significantly increased in DKO mice63. TESK1 is another signaling molecule working in parallel to LIMKs and can also phosphorylate cofilin to inhibit the depolymerization of actin64. The significant elevation of TESK1 expression seen here indicates that this compensatory signaling plays a role in actin dynamics. The phosphorylation of Hsp25 has been associated with modification of the actin cytoskeleton61, and the reduction of Hsp25 might also be a compensatory pathway for the regulation of actin dynamics (Fig. 6e). Third, cofilin and ADF play redundant roles during the development of the kidney, and conditional knockout of cofilin-1 in the ureteric buds of mice and the Wolffian ducts did not cause an obvious effect during the development of renal morphology, which is in agreement with our findings44. Our study shows that the ratios of cofilin and ADF differed in different cell types and at various developmental periods. Thus, the decrease in p-cofilin levels seen in our DKO mice might be compensated for by other mechanisms that depend on the concentration of cofilin. This might also explain why no significant defects in cochlear development or auditory function were seen in DKO mice in spite of the fact that the p-cofilin level in DKO mice decreased significantly. Rac and Cdc42 are upstream regulators of LIMKs, and the lack of Rac or Cdc42 causes disorganization of hair bundles. These processes can also be controlled by Lis1, which is a potential upstream regulator of both Cdc42 and Rac165,66; however, as one of their downstream effectors, LIMKs should also have negative effects on actin dynamics. There are around 40 known eukaryotic LIM proteins in different species based on the presence of LIM domains. The LIM superclass of genes is divided into 14 classes – LMO7, ENIGMA, LIMK, LASP, LHX, MICAL, PXN, EPLIN, ABLIM, CRP, LMO, PINCH, TES, and ZYX – any of which might compensate for the knockout of Limk1 and Limk2 in DKO mice.
In summary, our results demonstrate the expression patterns of LIMK1 and LIMK2 in the mouse cochlea. Knockout of Limk1 and Limk2 in DKO mice significantly decreased the level of p-cofilin in the cochlear tissue, but because of the compensatory role of Arp2/3, TESK1, and Hsp25, the loss of the LIMK proteins did not significantly change the number and function of ribbon synapses or the overall cochlear structure or auditory function.
Material and Methods
Animals and genotyping
The Limk1 and Limk2 DKO mice were the kind gift of Dr. Zikai Zhou of Southeast University, China. The genotyping of the transgenic mice was performed as described previously35. Primers used for genotyping are shown in Table 1. All mouse experiments were conducted following protocols approved by the Animal Care and Use Committee of Southeast University and were agreement with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. All efforts were performed to minimize the number of animals used in these experiments and to prevent their suffering.
Immunohistochemistry
The cochleae were placed in the medium containing 3 mM FM1-43 (Thermo Fisher, F35355) for 90 s and washed three times in PBS (pH 7.2). The cochleae then were dissected and fixed with 4% polyoxymethylene for 1 h and permeabilized with 0.5% Triton X-100 for 1 hour. The sensory epithelia were then incubated with the following primary antibodies overnight at 4 °C: anti-LIMK1 (Santa Cruz, sc-8387); anti-LIMK2 (Santa Cruz, sc-5577); anti-MYO7A (Proteus Bioscience, 25-6790); anti-SOX2 (Santa Cruz, sc-17320, sc-365823); anti-CtBP2 (C-terminal-binding protein 2), anti-IgG1 (BD Biosciences, 612044), anti-PSD95 IgG2a (Millipore, MAB1596), anti-cofilin (Abcam, ab42824), anti p-cofilin (Santa Cruz, sc-12912), and anti-prestin (Santa Cruz, sc-22692). Phalloidin (Invitrogen, A34055) was used to stain the actin cytoskeleton, and DAPI was used to label the nuclei. The tissues were washed three times with PBST (0.1 M phosphate buffer, pH 7.2, with 0.1% Triton X-100) and incubated for 1 h (37 °C) with DAPI (Sigma-Aldrich, D9542) and suitable secondary antibody (Abcam: ab150075, ab150105, ab150074, ab150073, ab150107; Invitrogen: A21131, A21124). Finally, the sensory epithelia were mounted on glass slides with Fluoromount-G mounting medium. Images were taken using a Zeiss LSM700 confocal microscope.
Auditory measurement
A TDT System III workstation running SigGen32 software (Tucker-Davis Technologies, USA) was used to test mice for ABR and CAP recordings as previously described3,14,67. Mice were anesthetized using 10 mg/kg xylazine and 80 mg/kg ketamine, and the body temperature was kept at 38 °C with a thermostatic heating pad. Three subdermal electrodes were used to measure the ABRs. The reference and grounding electrodes were inserted subdermally behind the ears, and the recording electrode was inserted at the vertex of the skull. In order to confirm that the equipment worked well, the ABR threshold of mice after 120 dB noise exposure for 2 hours was used as the positive control. In order to record CAPs, the reference and grounding electrodes were inserted behind the ears and a silver electrode was placed on the round window membrane. The TDT hardware and software (BioSig and SigGen) were used to generate stimuli and record the signal. The acoustic stimuli parameters were as follows: 10 ms tone bursts for ABR and CAP with a 0.5 ms rise/fall time and cos2 gating. The stimuli were played using a broadband speaker (MF1; TDT) that was kept 10 cm in front of the head of the mouse. ABR thresholds were tested at 4, 8, 12, 16, 24, and 32 kHz. For each frequency, the test was measured in a downward sequence beginning with 90 dB SPL and decreasing in 5 dB steps until the ABR response disappeared. The threshold was judged as the lowest level at which a stable wave III response could be seen and repeated in the ABR test. We usually focused on the peak-to-peak amplitude, which was performed at 90 dB SPL to generate I/O functions at 4, 8, 16, and 32 kHz in the CAP test. For DPOAE, the primary tones f1 and f2 were generated and shaped using the BioSig software and TDT hardware. DPOAEs were recorded in the form of level/frequency functions at 4, 8, 12, 16, 20, 24, 28, 32, 36, and 40 kHz, and the surrounding noise floor and 2f1-f2 DPOAE amplitudes were subtracted. The DPOAE threshold was calculated offline as a way of identifying and interpolating the data when the signal was at least 5 dB SPL and greater than two standard deviations above the noise floor. If no DPOAE response was recorded even at our equipment’s limitation of 90 dB SPL, we defined the threshold as 90 dB.
Scanning electron microscopy (SEM)
The cochlear specimens from P30 mice were fixed in 2.5% glutaraldehyde (Sigma-Aldrich, G5882) diluted in 0.1 M phosphate buffer (pH 7.2). A hole was poked in the apical region of the cochlea, and the fixative was flushed through the round window and oval window. The cochleae were kept for 24 hours at 4 °C, washed three times in PBS, and then decalcified for 3 hours in 0.5 M EDTA. The tissues were post-fixed for 2 hours at 4 °C in 1% osmium tetroxide and dehydrated in ethanol and critically point dried with liquid CO2 (CPD300, Leica). After applying an electrically conductive coating, the cochlear tissues were imaged on a Quanta 250 FEG scanning electron microscope.
RT-qPCR
Total cochlear RNA was extracted with ExTrizol Reagent (Protein Biotechnology, PR910), and reverse transcription of mRNA to cDNA was performed with a cDNA synthesis kit according to the manufacturer’s instructions (Thermo Fisher Scientific, K1622). The qPCR was performed using an Applied Biosystems CFX96 qPCR system (Bio-Rad, Hercules, CA, USA) and the SYBR Green (Rox) qPCR Master Mix (Roche Life Science, 04913850001). Specific primers were designed for target DNA or mRNA. The qPCR protocols were set as follows: 15-second denaturation at 95 °C followed by 40 cycles of 15-second denaturing at 95 °C, 60 seconds annealing at 60 °C, and 20-second extension at 72 °C. The expression of mRNA was normalized using the values of β-actin and Gapdh. The results were analyzed using the comparative cycle threshold (ΔΔCt) method.
Western blot
Cochleae from around 10 mice were dissected in cold PBS, and the cells were lysed with RIPA Lysis Buffer (Protein Biotechnology, PP109) and Cocktail-1 (Roche, 4906845001) and Cocktail-2 (Roche, 04693132001) at 4 °C. The protein concentration was measured with a BCA protein quantification kit (Protein Biotechnology, PP20). GAPDH was used as the reference protein. Donkey anti-goat immunoglobulin G (Abcam, ab6789, ab6721), peroxidase-conjugated goat anti-rabbit, and goat anti-mouse were used as the secondary antibodies. Polyvinylidene fluoride membranes were used to bind proteins, and the target proteins were detected using the SuperSignal West Dura chemiluminescent substrate kit (Thermo Scientific, 34075). Semi-quantification of the Western blot band was performed using Image J software. The background values were subtracted and the resulting signal was normalized to the GAPDH intensity and the relative optical density ratio was calculated. Each experiment was repeated at least three times.
Cell and synapse quantification
To quantify the immunopositive cells, the entire sensory epithelia were divided into three turns. For synapse quantification, immunolabeled puncta of CtBP2 and PSD95 in IHCs were quantified from the images. Each image was acquired at 63× magnification under identical z-stack conditions with 1-µm intervals. The binary file was converted to a 3D z-y projection to measure ribbon dispersion. The synapses were manually counted one by one from the 8 kHz to 32 kHz (with each image containing ~12 IHCs) region of sensory epithelia. The number of functional synapses were manually counted by visualizing the presence of the overlapping CtBP2 and PSD95 signals.
Statistical analysis
All of the data in this study are shown as the mean ± SD, and all experiments were repeated at least three times. Statistical analyses were performed using Microsoft Excel and GraphPad Prism 6 software. For the cell counting experiments, n represents the number of mice. Two-tailed, unpaired Student’s t-tests were performed to determine the statistical differences between two groups, and one-way ANOVA followed by a Dunnett’s multiple comparisons test was conducted when comparing more than two groups. A value of p < 0.05 was considered to be statistically significant.
References
Fekete, D. M., Muthukumar, S. & Karagogeos, D. Hair cells and supporting cells share a common progenitor in the avian inner ear. The Journal of neuroscience: the official journal of the Society for Neuroscience 18, 7811–7821 (1998).
Li, A. et al. Novel compounds protect auditory hair cells against gentamycin-induced apoptosis by maintaining the expression level of H3K4me2. Drug Delivery 25, 1033–1043, https://doi.org/10.1080/10717544.2018.1461277 (2018).
Liu, W. et al. Wnt Signaling Activates TP53-Induced Glycolysis and Apoptosis Regulator and Protects Against Cisplatin-Induced Spiral Ganglion Neuron Damage in the Mouse Cochlea. Antioxid Redox Signal. https://doi.org/10.1089/ars.2017.7288 (2018).
Savary, E. et al. Cochlear stem/progenitor cells from a postnatal cochlea respond to Jagged1 and demonstrate that notch signaling promotes sphere formation and sensory potential. Mechanisms of development 125, 674–686, https://doi.org/10.1016/j.mod.2008.05.001 (2008).
Li, H. et al. Meclofenamic Acid Reduces Reactive Oxygen Species Accumulation and Apoptosis, Inhibits ExcessiveAutophagy, and Protects Hair Cell-Like HEI-OC1 Cells From Cisplatin-Induced Damage. Frontiers in cellular neuroscience 12, 139, https://doi.org/10.3389/fncel.2018.00139 (2018).
Zhang, Y. et al. Characterization of Lgr6+ Cells as an Enriched Population of Hair Cell Progenitors Compared to Lgr5+ Cells for Hair Cell Generation in the Neonatal Mouse Cochlea. Front Mol Neurosci 11, 147, https://doi.org/10.3389/fnmol.2018.00147 (2018).
He, Z. et al. Autophagy protects auditory hair cells against neomycin-induced damage. Autophagy 13, 1884–1904, https://doi.org/10.1080/15548627.2017.1359449 (2017).
Petit, C. & Richardson, G. P. Linking genes underlying deafness to hair-bundle development and function. Nature neuroscience 12, 703–710, https://doi.org/10.1038/nn.2330 (2009).
Schwander, M., Kachar, B. & Muller, U. Review series: The cell biology of hearing. The Journal of cell biology 190, 9–20, https://doi.org/10.1083/jcb.201001138 (2010).
He, Z. et al. Role of Autophagy in Auditory System Development and Survival. Journal of Otorhinolaryngology, Hearing and Balance Medicine 1, 7, https://doi.org/10.3390/ohbm1010007 (2018).
Ciganovic, N., Wolde-Kidan, A. & Reichenbach, T. Hair bundles of cochlear outer hair cells are shaped to minimize their fluid-dynamic resistance. Scientific reports 7, 3609, https://doi.org/10.1038/s41598-017-03773-y (2017).
Yan, W. et al. A Three-Dimensional Culture System with Matrigel Promotes Purified Spiral Ganglion Neuron Survival and Function In Vitro. Mol Neurobiol 55, 2070–2084, https://doi.org/10.1007/s12035-017-0471-0 (2018).
Chen, Y. et al. Hedgehog Signaling Promotes the Proliferation and Subsequent Hair Cell Formation of Progenitor Cells in the Neonatal Mouse Cochlea. Front Mol Neurosci 10, 426, https://doi.org/10.3389/fnmol.2017.00426 (2017).
Wang, Y. et al. Loss of CIB2 Causes Profound Hearing Loss and Abolishes Mechanoelectrical Transduction in Mice. Front Mol Neurosci 10, 401, https://doi.org/10.3389/fnmol.2017.00401 (2017).
Spector, A. A., Deo, N., Grosh, K., Ratnanather, J. T. & Raphael, R. M. Electromechanical models of the outer hair cell composite membrane. J Membr Biol 209, 135–152, https://doi.org/10.1007/s00232-005-0843-7 (2006).
Belluscio, L., Lodovichi, C., Feinstein, P., Mombaerts, P. & Katz, L. C. Odorant receptors instruct functional circuitry in the mouse olfactory bulb. Nature 419, 296–300, https://doi.org/10.1038/nature01001 (2002).
Elgoyhen, A. B. & Franchini, L. F. Prestin and the cholinergic receptor of hair cells: positively-selected proteins in mammals. Hear Res 273, 100–108, https://doi.org/10.1016/j.heares.2009.12.028 (2011).
Fettiplace, R. Hair Cell Transduction, Tuning, and Synaptic Transmission in the Mammalian Cochlea. Comprehensive Physiology 7, 1197–1227, https://doi.org/10.1002/cphy.c160049 (2017).
Fettiplace, R. & Hackney, C. M. The sensory and motor roles of auditory hair cells. Nature reviews. Neuroscience 7, 19–29, https://doi.org/10.1038/nrn1828 (2006).
Gorovoy, M. et al. LIM kinase 1 coordinates microtubule stability and actin polymerization in human endothelial cells. The Journal of biological chemistry 280, 26533–26542, https://doi.org/10.1074/jbc.M502921200 (2005).
Mizuno, K. et al. Identification of a human cDNA encoding a novel protein kinase with two repeats of the LIM/double zinc finger motif. Oncogene 9, 1605–1612 (1994).
Nunoue, K., Ohashi, K., Okano, I. & Mizuno, K. LIMK-1 and LIMK-2, two members of a LIM motif-containing protein kinase family. Oncogene 11, 701–710 (1995).
Okano, I. et al. Identification and characterization of a novel family of serine/threonine kinases containing two N-terminal LIM motifs. The Journal of biological chemistry 270, 31321–31330 (1995).
Liu, W. et al. Electroacupuncture Regulates Hippocampal Synaptic Plasticity via miR-134-Mediated LIMK1 Function in Rats with Ischemic Stroke. Neural plasticity 2017, 9545646, https://doi.org/10.1155/2017/9545646 (2017).
Endo, M., Ohashi, K. & Mizuno, K. LIM kinase and slingshot are critical for neurite extension. The Journal of biological chemistry 282, 13692–13702, https://doi.org/10.1074/jbc.M610873200 (2007).
Rosso, S. et al. LIMK1 regulates Golgi dynamics, traffic of Golgi-derived vesicles, and process extension in primary cultured neurons. Molecular biology of the cell 15, 3433–3449, https://doi.org/10.1091/mbc.E03-05-0328 (2004).
Rust, M. B. ADF/cofilin: a crucial regulator of synapse physiology and behavior. Cellular and molecular life sciences: CMLS 72, 3521–3529, https://doi.org/10.1007/s00018-015-1941-z (2015).
Meng, Y. et al. Abnormal spine morphology and enhanced LTP in LIMK-1 knockout mice. Neuron 35, 121–133 (2002).
Park, C. & Kalinec, F. PKCalpha-Mediated Signals Regulate the Motile Responses of Cochlear Outer Hair Cells. Biophys J 108, 2171–2180, https://doi.org/10.1016/j.bpj.2015.03.044 (2015).
Matsumoto, N., Kitani, R., Maricle, A., Mueller, M. & Kalinec, F. Pivotal role of actin depolymerization in the regulation of cochlear outer hair cell motility. Biophys J 99, 2067–2076, https://doi.org/10.1016/j.bpj.2010.08.015 (2010).
Xie, J. et al. Aberrant expression of LIMK1 impairs neuronal migration during neocortex development. Histochem Cell Biol 147, 471–479, https://doi.org/10.1007/s00418-016-1514-8 (2017).
Nomoto, S., Tatematsu, Y., Takahashi, T. & Osada, H. Cloning and characterization of the alternative promoter regions of the human LIMK2 gene responsible for alternative transcripts with tissue-specific expression. Gene 236, 259–271 (1999).
Takahashi, H., Funakoshi, H. & Nakamura, T. LIM-kinase as a regulator of actin dynamics in spermatogenesis. Cytogenet Genome Res 103, 290–298, https://doi.org/10.1159/000076815 (2003).
Takahashi, H., Koshimizu, U., Miyazaki, J. & Nakamura, T. Impaired spermatogenic ability of testicular germ cells in mice deficient in the LIM-kinase 2 gene. Dev Biol 241, 259–272, https://doi.org/10.1006/dbio.2001.0512 (2002).
Meng, Y. et al. Regulation of ADF/cofilin phosphorylation and synaptic function by LIM-kinase. Neuropharmacology 47, 746–754, https://doi.org/10.1016/j.neuropharm.2004.06.030 (2004).
Yang, X. et al. Transient inhibition of LIMKs significantly attenuated central sensitization and delayed the development of chronic pain. Neuropharmacology 125, 284–294, https://doi.org/10.1016/j.neuropharm.2017.06.031 (2017).
Tong, M., Brugeaud, A. & Edge, A. S. Regenerated synapses between postnatal hair cells and auditory neurons. J Assoc Res Otolaryngol 14, 321–329, https://doi.org/10.1007/s10162-013-0374-3 (2013).
Shi, L. et al. Ribbon synapse plasticity in the cochleae of Guinea pigs after noise-induced silent damage. PloS one 8, e81566, https://doi.org/10.1371/journal.pone.0081566 (2013).
Yuen, G. S., McEwen, B. S. & Akama, K. T. LIM kinase mediates estrogen action on the actin depolymerization factor Cofilin. Brain Res 1379, 44–52, https://doi.org/10.1016/j.brainres.2010.07.067 (2011).
Sheng, M. & Hoogenraad, C. C. The postsynaptic architecture of excitatory synapses: a more quantitative view. Annu Rev Biochem 76, 823–847, https://doi.org/10.1146/annurev.biochem.76.060805.160029 (2007).
Gilels, F., Paquette, S. T., Beaulac, H. J., Bullen, A. & White, P. M. Severe hearing loss and outer hair cell death in homozygous Foxo3 knockout mice after moderate noise exposure. Scientific reports 7, 1054, https://doi.org/10.1038/s41598-017-01142-3 (2017).
Ryugo, D. K. Projections of low spontaneous rate, high threshold auditory nerve fibers to the small cell cap of the cochlear nucleus in cats. Neuroscience 154, 114–126, https://doi.org/10.1016/j.neuroscience.2007.10.052 (2008).
Endo, M. et al. Control of growth cone motility and morphology by LIM kinase and Slingshot via phosphorylation and dephosphorylation of cofilin. The Journal of neuroscience: the official journal of the Society for Neuroscience 23, 2527–2537 (2003).
Ohashi, K. Roles of cofilin in development and its mechanisms of regulation. Dev Growth Differ 57, 275–290, https://doi.org/10.1111/dgd.12213 (2015).
Yang, N. et al. Cofilin phosphorylation by LIM-kinase 1 and its role in Rac-mediated actin reorganization. Nature 393, 809–812, https://doi.org/10.1038/31735 (1998).
Sumi, T., Matsumoto, K., Takai, Y. & Nakamura, T. Cofilin phosphorylation and actin cytoskeletal dynamics regulated by rho- and Cdc42-activated LIM-kinase 2. The Journal of cell biology 147, 1519–1532 (1999).
Toshima, J., Toshima, J. Y., Takeuchi, K., Mori, R. & Mizuno, K. Cofilin phosphorylation and actin reorganization activities of testicular protein kinase 2 and its predominant expression in testicular Sertoli cells. The Journal of biological chemistry 276, 31449–31458, https://doi.org/10.1074/jbc.M102988200 (2001).
Nakano, K. et al. Cofilin phosphorylation and actin polymerization by NRK/NESK, a member of the germinal center kinase family. Experimental cell research 287, 219–227 (2003).
Niwa, R., Nagata-Ohashi, K., Takeichi, M., Mizuno, K. & Uemura, T. Control of actin reorganization by Slingshot, a family of phosphatases that dephosphorylate ADF/cofilin. Cell 108, 233–246 (2002).
Ohta, Y. et al. Differential activities, subcellular distribution and tissue expression patterns of three members of Slingshot family phosphatases that dephosphorylate cofilin. Genes to cells: devoted to molecular & cellular mechanisms 8, 811–824 (2003).
Mizuno, K. Signaling mechanisms and functional roles of cofilin phosphorylation and dephosphorylation. Cellular signalling 25, 457–469, https://doi.org/10.1016/j.cellsig.2012.11.001 (2013).
Jarrold, C., Baddeley, A. D. & Phillips, C. Long-term memory for verbal and visual information in Down syndrome and Williams syndrome: performance on the Doors and People test. Cortex; a journal devoted to the study of the nervous system and behavior 43, 233–247 (2007).
Todorovski, Z. et al. LIMK1 regulates long-term memory and synaptic plasticity via the transcriptional factor CREB. Molecular and cellular biology 35, 1316–1328, https://doi.org/10.1128/MCB.01263-14 (2015).
Rottner, K., Faix, J., Bogdan, S., Linder, S. & Kerkhoff, E. Actin assembly mechanisms at a glance. J Cell Sci 130, 3427–3435, https://doi.org/10.1242/jcs.206433 (2017).
He, D. Z. et al. Changes in plasma membrane structure and electromotile properties in prestin deficient outer hair cells. Cytoskeleton (Hoboken) 67, 43–55, https://doi.org/10.1002/cm.20423 (2010).
Andrianantoandro, E. & Pollard, T. D. Mechanism of actin filament turnover by severing and nucleation at different concentrations of ADF/cofilin. Molecular cell 24, 13–23, https://doi.org/10.1016/j.molcel.2006.08.006 (2006).
Cichon, J. et al. Cofilin aggregation blocks intracellular trafficking and induces synaptic loss in hippocampal neurons. The Journal of biological chemistry 287, 3919–3929, https://doi.org/10.1074/jbc.M111.301911 (2012).
Molinie, N. & Gautreau, A. The Arp2/3 Regulatory System and Its Deregulation in Cancer. Physiol Rev 98, 215–238, https://doi.org/10.1152/physrev.00006.2017 (2018).
Xiong, A. et al. The Shaker-1 Mouse Myosin VIIa Deafness Mutation Results in a Severely Reduced Rate of the ATP Hydrolysis Step. The Journal of biological chemistry, https://doi.org/10.1074/jbc.M117.810119 (2017).
Shchepkin, D. V. et al. Cooperativity of myosin interaction with thin filaments is enhanced by stabilizing substitutions in tropomyosin. J Muscle Res Cell Motil 38, 183–191, https://doi.org/10.1007/s10974-017-9472-x (2017).
Gamell, C., Susperregui, A. G., Bernard, O., Rosa, J. L. & Ventura, F. The p38/MK2/Hsp25 pathway is required for BMP-2-induced cell migration. PloS one 6, e16477, https://doi.org/10.1371/journal.pone.0016477 (2011).
Gong, J. et al. Insm1a Regulates Motor Neuron Development in Zebrafish. Front Mol Neurosci 10, 274, https://doi.org/10.3389/fnmol.2017.00274 (2017).
Dayel, M. J. & Mullins, R. D. Activation of Arp2/3 complex: addition of the first subunit of the new filament by a WASP protein triggers rapid ATP hydrolysis on Arp2. PLoS Biol 2, E91, https://doi.org/10.1371/journal.pbio.0020091 (2004).
Wang, L., Buckley, A. F. & Spurney, R. F. Regulation of cofilin phosphorylation in glomerular podocytes by testis specific kinase 1 (TESK1). Scientific reports 8, 12286, https://doi.org/10.1038/s41598-018-30115-3 (2018).
Kholmanskikh, S. S. et al. Calcium-dependent interaction of Lis1 with IQGAP1 and Cdc42 promotes neuronal motility. Nature neuroscience 9, 50–57, https://doi.org/10.1038/nn1619 (2006).
Kirjavainen, A., Laos, M., Anttonen, T. & Pirvola, U. The Rho GTPase Cdc42 regulates hair cell planar polarity and cellular patterning in the developing cochlea. Biol Open 4, 516–526, https://doi.org/10.1242/bio.20149753 (2015).
Pan, H. et al. Auditory Neuropathy after Damage to Cochlear Spiral Ganglion Neurons in Mice Resulting from Conditional Expression of Diphtheria Toxin Receptors. Scientific reports 7, 6409, https://doi.org/10.1038/s41598-017-06600-6 (2017).
Acknowledgements
This work was supported by grants obtained from the Strategic Priority Research Program of the Chinese Academy of Science (XDA16010302). The National Key R&D Program of China (2015CB965000, 2017YFA0104303, and 2017YFA0103903), the National Natural Science Foundation of China (Nos 81622013, 81470692, 31500852, 81470701, 81670938, 31571530, and 81570910), the Natural Science Foundation of Jiangsu Province (BK20150022), the Fundamental Research Funds for the Central Universities (2242018k1G011), Boehringer Ingelheim Pharma GmbH, the Yingdong Huo Education Foundation, Excellence Project of Southeast University.
Author information
Authors and Affiliations
Contributions
K.S., R.C. and F.C. conceived and designed the experiments. Q.F., Y.Z., P.D., H.P., Z.H., C.C., D.L., B.S., J.G., M.L. and X.W. performed the experiments. Q.F., M.G., S.S., Z.Z., Y.Z. and J.W. analyzed the data. Q.F., S.S., R.C., T.W., and F.C. wrote the paper.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Fang, Q., Zhang, Y., Da, P. et al. Deletion of Limk1 and Limk2 in mice does not alter cochlear development or auditory function. Sci Rep 9, 3357 (2019). https://doi.org/10.1038/s41598-019-39769-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-39769-z
- Springer Nature Limited