
Abstract
In this study we show Yersinia pseudotuberculosis secretes membrane vesicles (MVs) that contain different proteins and virulence factors depending on the strain. Although MVs from Y. pseudotuberculosis YPIII and ATCC 29833 had many proteins in common (68.8% of all the proteins identified), those located in the outer membrane fraction differed significantly. For instance, the MVs from Y. pseudotuberculosis YPIII harbored numerous Yersinia outer proteins (Yops) while they were absent in the ATCC 29833 MVs. Another virulence factor found solely in the YPIII MVs was the cytotoxic necrotizing factor (CNFy), a toxin that leads to multinucleation of host cells. The ability of YPIII MVs to transport this toxin and its activity to host cells was verified using HeLa cells, which responded in a dose-dependent manner; nearly 70% of the culture was multinucleated after addition of 5 µg/ml of the purified YPIII MVs. In contrast, less than 10% were multinucleated when the ATCC 29833 MVs were added. Semi-quantification of CNFy within the YPIII MVs found this toxin is present at concentrations of 5 ~ 10 ng per µg of total MV protein, a concentration that accounts for the cellular responses seen.
Similar content being viewed by others


Introduction
Yersinia pseudotuberculosis is a Gram-negative pathogen believed to be a direct evolutionary ancestor of Yersinia pestis, the causative agent of bubonic plague1,2. As a pathogen, Y. pseudotuberculosis causes disease and zoonotic infections in both humans and a variety of animals, where it is often transmitted via the fecal-oral route3,4. In humans, Y. pseudotuberculosis is associated with several medical conditions, including the gastrointestinal disease yersiniosis5, mesenteric lymphadenitis6, fatal septicemia7 and, at times, may lead to post-infectious reactive arthritis8.
This pathogen has evolved an exquisite method for delivering virulence factors and effectors into cells of the host, where they inhibit signaling cascades and block the cell’s response to infection8. One way it achieves this is through its type-three secretion system (TTSS)9, which plays a crucial role in effector translocation into host cells. This pathogen also expresses the invasin (INV), attachment invasion locus (AilA) and adhesion (YadA) proteins10,11, all of which mediate its tight adhesion to and entry into mammalian cells.
This is not to say that all strains of Y. pseudotuberculosis are created equal. For instance, some strains harbor a large (typically around 70-kb) virulence plasmid that encodes for the Yersinia outer proteins (Yops)12,13. This is true for Y. pseudotuberculosis YPIII, while Y. pseudotuberculosis ATCC 29833 does not appear to have this plasmid. Upon intimate contact with a target host cell, the Yops are delivered into the host cell via its Type III Secretion System (T3SS)14. In addition, not all strains of Y. pseudotuberculosis produce a functional cytotoxic necrotizing factor (CNFy)15. This toxin activates the host Rho GTPase16,17 by deamidating the Gln63 residue18 and contributes to the activity of the Yop proteins16,19. Although CNFy and its virulent mechanisms are well known, the means by which this protein is introduced into the host cells has thus far remained unclear. However, the study by Lockman et al. (2002) found cell-free supernatants from Y. pseudotuberculosis YPIII cultures retained the ability to induce multinucleation15, suggesting at the time that CNFy was secreted extracellularly.
Recent research, however, has established many bacterial strains, including both Gram-negative and Gram-positive, secrete membrane vesicles (MVs)20,21 to be used in a variety of functions, including in defense22, the horizontal transfer of genes23,24 and in pathogenesis25. This is also true for Y. pestis, where the MVs produced were found to contain a number of virulence factors that may contribute to this disease26. We report here that its relative, Y. pseudotuberculosis, also secretes MVs and that this pathogen uses them to mediate the transfer of the CNFy toxin to host cells.
Results and Discussion
Figure 1A shows Y. pseudotuberculosis ATCC 29833 secretes MVs, which were visible as membrane blebs on the surface of the cells, while Fig. 1B shows the MVs after they were purified. We also purified MVs from cultures of Y. pseudotuberculosis YPIII (Fig. 1C), a better characterized strain of this pathogen15,27. Although the diameters of both MVs were similar, ranging from around 70 to 150 nm, their proteomes were quite distinct from one another, as shown in Fig. 1D,E. The proteins present within both MV preparations were subsequently identified using tandem mass spectrometry and classified according to their localization within the cell using PSORTB v. 3.028. In total, the Y. pseudotuberculosis ATCC 29833 MVs contained 276 proteins, while 303 proteins were identified within the Y. pseudotuberculosis YPIII MVs (Fig. 1E). For both preparations, approximately half of the proteins were cytoplasmic (Fig. 1E). In fact, the top twenty cytoplasmic proteins, as defined by the number of hits, were found in both MV preparations (Table S1). The presence of cytoplasmic proteins in Gram negative MVs has been documented in several previous studies26,29,30. While the cytoplasmic protein content within Salmonella enterica and E. coli DH5α MVs was around 40%29,30, with Y. pestis the percentage of cytoplasmic proteins was 58%26, a level that affirms the results seen here with Y. pseudotuberculosis. The significant presence of cytoplasmic proteins implies these bacteria are generating double membrane vesicles, a type of MV that is produced by other Gram-negative bacterial species, including Shewanella vesiculosa31, Vibrio cholerae32 and Pseudalteromonas marina33, where they are also involved in transporting RNA and DNA. In the V. cholerae study, the authors found the larger MVs (>100 nm) were the ones that had double membranes while the smaller MVs tended to be single membrane32. As noted above, the average diameter of the MVs in this study were between 70 and 150 nm.

Y. pseudotuberculosis secretes MVs and the proteomes differ based on the strain. (A) TEM image showing MVs (arrows) budding off of Y. pseudotuberculosis. (B,C) TEM images of MVs purified from cultures of Y. pseudotuberculosis ATCC 29833 (B) and YPIII (C). (D,E) The proteomes of the two MVs differed based upon SDS-PAGE analyses (D) and LC-MS-MS (E). (D) Distinct bands can be seen in the respective MVs, hinting at differences in their proteomes. The full gel is shown in Fig. S1. (E) The LC-MS-MS analyses found a high degree of similarity in the MV proteomes but also some significant difference, particularly in the outer membrane fraction.
Although a majority of the proteins identified (nearly 70%) were shared by both MVs, clear differences were also seen. For instance, the MVs from YPIII had significantly more outer membrane proteins (OMPs) than those from ATCC 29833 (Figs 1E and S2). The greater number of OMPs in the YPIII MVs is further supported by Table 1, where the top 20 OMPs are listed based upon peptide hits. As listed in this table, all twenty of these proteins were present in the YPIII MVs, including several Yersinia outer proteins (Yops) and the CNFy toxin, while the ATCC 29833 MVs carried only four. The complete list of OMPs is provided in Table S1. One potential explanation for the differences between the two MV preparations is the large virulence plasmid that is found within Y. pseudotuberculosis YPIII12, but has never been reported in strain ATCC 29833. This plasmid encodes for many virulence factors, including YopD, YopE, YopH and YopN, four of the proteins that were found within the YPIII MVs but lacking in the ATCC 29833 MVs (Table 1). During an infection, YopD forms a translocation channel along with YopB within the host cell membrane to inject the other Yop proteins34, while YopH, a tyrosine phosphatase, modulates the immune response. It accomplishes this by targeting p130Cas and the focal adhesion kinase (FAK), leading to reduced phagocytosis of Yersinia by macrophages35, as well as by counteracting T-cell activation36. Although Yops are transported into the host cell via the Y. pseudotuberculosis T3SS37, the proteomic data here suggests they are also be transported via MVs.
Another virulence factor that was found only in the YPIII MVs is the cytotoxic necrotizing factor (CNFy, Table 1). This protein is an AB-type, pore-forming toxin produced by Y. pseudotuberculosis and pathogenic strains of E. coli38. Within Y. pseudotuberculosis YPIII this toxin is chromosomally encoded by the cnfy gene (3,045 bp in length) but is truncated (1.8 kb) in Y. pseudotuberculosis ATCC 2983315. As a virulence factor, CNFy has been identified within host cells during infection where it contributes to the delivery and activity of the Yops16,19, activates Rho GTPase16,17 and leads to multinucleation22. The mechanism used by Y. pseudotuberculosis YPIII to introduce CNFy into host cells, however, has not been established. The cytotoxic necrotizing factor produced by uropathogenic strains of E. coli (CNF1), however, is transferred in an active form via MVs39. Given the sequence similarity between the CNFy and CNF1 proteins15, we hypothesized that the same might be true of CNFy. Although this idea was proposed previously16, the authors did not provide any evidence to support this in their study.
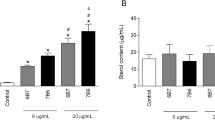
Although the results above show CNFy was present in the YPIII MVs, it was not clear if the toxin was active or if the MVs transport this toxin to host cells. To evaluate this, we exposed HeLa cells to MVs purified from either Y. pseudotuberculosis ATCC 29833 or YPIII cultures. The former was used since the truncated CNFy protein has a reduced activity and does not strongly induce multinucleation15. Figure 2A shows the HeLa cultures responses after an exposure to 10 mg/ml of either MV. Whereas the purified YPIII MVs led to both an extensive β-actin rearrangement and multinucleation within the HeLa cells, the ATCC 29833 MVs induced only mild cellular responses by comparison. This is supported by the results in Fig. 2B, which shows the degree of multinucleation increases as the concentration of MVs added increased. Based on these results, a minimum of 2.5 µg of YPIII MVs is needed to elicit a significant response.

CNFy is transported to host cells via MVs. (A) MVs from Y. pseudotuberculosis YPIII led to significantly greater responses from the HeLa cell cultures, including actin rearrangement and multinucleation, than those from Y. pseudotuberculosis ATCC 29833. (B) Percent of cells displaying multinucleation after treatment with the purified MVs or the addition of the his-tagged Y. pseudotuberculosis YPIII CNFy. (C) Semi-quantification of CNFy within the YPIII MVs using western blotting. All of the samples were probed on the same blot as shown. The results suggest there is approximately 0.05 to 0.1 µg per 10 µg of MVs. Treatment with proteinase K (PKMVs) removed this signal, implying this protein is present on the surface of the MVs. (D) Proposed mechanism used by Y. pseudotuberculosis YPIII to introduce its CNFy into host cells using MVs.
Focusing on the cell cultures exposed to 10 µg of MVs, a vast majority of the HeLa cells were multinucleated, proving the MV-bound CNFy is active. These results support the findings of Lockman et al. (2002), where filtered cell-free supernatants induced multinucleation of Hep-2 cells15. Given their diminutive size, bacterial MVs are not removed by typical filter sterilization methods and, thus, will remain in the media to induce the responses as seen in their study. In contrast, the ATCC 29833 MVs induced only very mild responses from the HeLa cells regardless of the concentration added. This was expected given the CNFy protein expressed by this strain is truncated, making it much less potent and unable to elicit the same level of cellular responses15. The YPIII MVs not only led to stronger cellular responses than their ATCC counterparts but were also toxic towards the HeLa cells as evidenced by the Live/Dead stain results in Fig. S3. Moreover, treatment of the YPIII MVs with either heat (95 °C for 10 min) or proteinase K eliminated their activities (Figs S3B and S4), implying the CNFy toxin is located on the surface of the MVs and exposed to the surrounding media.
To prove CNFy is truly associated with YPIII MVs, next we constructed and expressed a C-terminal his-tagged CNFy fusion gene within Y. pseudotuberculosis YPIII. When MVs purified from these cultures were probed using anti-his antibodies, CNFy was readily detected (Fig. 2C). A semi-quantitative analysis using purified CNFy suggested its concentration within the MVs was between 0.05 and 0.1 µg per 10 µg of total MV protein. Although the expression of CNFy by Y. pseudotuberculosis YPIII was initially reported to be constitutive15, a more recent study found this toxin is expressed more strongly when this pathogen is cultured at 37 °C rather than at 25 °C16,40. This is also true for the Yops mentioned above, the expression levels increasing between 7.2 and 28.4-fold40. As the temperature used in this study was 30 °C, the concentrations of the Yops and CNFy within the MVs will likely be greater under physiological conditions.
When purified CNFy was added to the HeLa cell cultures at the above concentrations, i.e., between 0.05 and 0.1 µg/ml, we found they induced similar levels of multinucleation (Figs 2B and S5), implying the activity of the MV-bound CNFy is comparable with that of the free toxin. Agreeing with the results in Figs S3B and S4, treatment of the MVs with proteinase K eliminated any signal in the western blots (Fig. 2C), further proving this toxin is present in the outer membrane of the MVs as listed in Table 1. Additional experiments also found the YPIII MVs associate with, and are likely taken up into, the HeLa cells (Fig. S6), implying they may be used by Y. pseudotuberculosis YPIII to introduce this toxin into host cells. This concept is illustrated in Fig. 2D.
In summary, this study shows different strains of Y. pseudotuberculosis secrete MVs harboring several different virulence factors. Based upon the genotype, with Y. pseudotuberculosis YPIII harboring a large virulence plasmid that is absent in Y. pseudotuberculosis ATCC 29833, the MV proteomes differed; the YPIII MVs carried many Yops and CNFy protein, while these were missing in the ATCC 29833 MVs. As such, this is the first study to demonstrate the CNFy toxin of Y, pseudotuberculosis can be transported to host cells via MVs and that they can induce cellular responses and death in the absence of whole pathogens.
Materials and Methods
Bacterial strains and growth conditions
Yersinia pseudotuberculosis ATCC 29833 was obtained from the Korean Agricultural Culture Collection (KACC) (http://genebank.rda.go.kr/eng/uat/uia/actionMain.do)) and Y. pseudotuberculosis YPIII was obtained from the Biodefense and Emerging Infections Research Resources Repository (BEI Resources, USA). The bacterial strains were maintained as frozen 20% glycerol stocks at −80 °C. Upon need, they were grown on Lysogeny broth (LB) agar plates (BD; Difco, USA) at 30 °C overnight. From this, a single colony was inoculated into 5 ml of LB broth and grown overnight at 30 °C with agitation (250 rpm).
Human cell cultures
The HeLa cells, which we purchased from the American Type Culture Collection (ATCC; www.atcc.org) were cultured in T75 flasks in Dulbecco’s Modified Eagles Medium (MEM, 1:1 with sodium bicarbonate (Life Technologies, USA)) supplemented with 10% heat inactivated fetal bovine serum and 100 μg/ml Normocin (Invivogen, USA).
Purification of the MVs
MVs were purified as mentioned previously26, with slight modification. Briefly, the 500 ml bacterial cultures were grown in LB medium at 30 °C and 250 rpm in 2-liter flasks for 24 h (OD600 of ∼2.3). The bacterial cells and debris were removed by low-speed centrifugation (5,000 × g, 30 min) and the medium was sterilized by passing it through a 0.2 μm filter (Millipore, USA). The cell-free medium was concentrated to ~5 ml using a 100-kDa MWCO Amicon filter (USA). The concentrated MVs were pelleted by ultracentrifugation (40000 rpm, 2 h, 4 °C) and suspended in 25 mM HEPES (pH 7.5). They were then mixed with OptiPrep (Sigma Aldrich, USA) to generate a 40% (vol/vol) OptiPrep solution in a total volume of 2 ml. The samples were loaded into a 13.2-ml ultracentrifuge tube, and lower-concentration OptiPrep solutions were layered on top (2 ml [40%], 2 ml [30%], 2 ml [25%], 2 ml [20%], 1 ml [15%] and finally 0.5 ml [0%]). The samples were centrifuged (100,000 × g, 16 h, 4 °C) in a swinging-bucket rotor, after which 1 ml fractions were collected sequentially from the top of the tube. The protein content in each fraction was examined by 10% Tris-glycine SDS-PAGE gels (Biorad, USA). Adjacent fractions with similar protein profiles were combined, diluted into 25 mM HEPES (pH 7.5) and recovered via ultracentrifugation (100,000 × g, 2 h, 4 °C). The recovered pellets were then resuspended in 25 mM HEPES (pH 7.5) and the MV concentration determined using the Bradford assay.
TEM analysis of the bacteria and MVs
For the TEM images, 2 µl of purified MVs or bacterial culture were placed on carbon-coated grids (Polysciences, Inc., USA). The samples were treated with 2.5% glutaraldehyde for 1 h, washed sequentially with increasing amounts of ethanol (40%, 50% 60% 80% 90% and 100%) and finally in absolute acetone. The samples were then negatively stained with 0.1% uranyl acetate and examined under a transmission electron microscope (JEOL 1200 EX, USA).
Proteomic analysis of Yersinia pseudotuberculosis MVs
To identify the proteins within the MVs, triplicate samples for both Y, pseudotuberculosis ATCC 29833 and YPIII cultures were subjected to in-gel digestion. For this 10 μg of the MVs (based on the Bradford assay) were separated on 10% Tris-glycine SDS-PAGE gels (Biorad, USA). After staining with coomassie blue, each lane was cut into six pieces and the proteins in each were subjected to in-gel tryptic digestion as described by Shevchenko and co-workers41. The resulting tryptic peptides were analyzed by LC-MS/MS. All the mass analyses were performed on an LTQ-Orbitrap (Thermo, Bremen, Germany) equipped with a nanoelectrospray ion source. To separate the peptide mixture, we used a C18 reverse phase HPLC column (150 mm × 75 um i.d.) using an acetonitrile/0.1% formic acid gradient from 10 to 24% for 90 min at a flow rate of 300 nl/min.
For the MS/MS analysis, the precursor ion scan MS spectra (m/z 400–2000) were acquired in the Orbitrap at a resolution of 60,000 at m/z 400 with an internal lock mass. The 20 most intensive ions were isolated and fragmented in the linear ion trap by collisionally induced dissociation (CID). All the MS/MS samples were analyzed using Sequest (Thermo Fisher Scientific, San Jose, CA, USA; version 1.4.1.14) and X! Tandem (The GPM, thegpm.org; version CYCLONE (2010.12.01.1)). Sequest and X! Tandem were set up to search the Y. pseudotuberculosis protein sequence database (9823 entries, UniProt (http://www.uniprot.org/)) assuming digestion by trypsin. Sequest and X! Tandem were searched with a fragment ion mass tolerance of 0.60 Da and a parent ion tolerance of 10.0 ppm.
Scaffold (version Scaffold_4.4.3, Proteome Software Inc., Portland,) was used to validate the MS/MS-based peptide and protein identifications. Protein identifications were accepted only if they could be established at greater than 99.0% probability to achieve a false detection rate of less than 1.0% and contained at least two identified peptides. Protein probabilities were assigned by the Protein Prophet algorithm. Proteins that contained similar peptides and could not be differentiated based on MS/MS analysis alone were grouped to satisfy the principles of parsimony. The proteins were annotated with their gene ontology terms, which were obtained from the NCBI database (downloaded Aug 1, 2016). Localization of the identified proteins were manually determined using PSORTB v. 3.0 bacterial protein subcellular localization prediction program28.
Cloning and expression of His-tagged CNFy
The gene encoding for the CNFy toxin within the genome of Y. pseudotuberculosis YPIII was amplified by PCR using primers CNF-F (5′-TATTCTAGATTTTACATGTAATAATAAATAGCAG-3′) and CNF-R (5′-ATACTCGAGAAAGTCTTTTTGTAAAACATTAAAC-3′). These primers introduced restriction sites for Xba1 and Xho1 (underlined regions in the primers, respectively). The resulting PCR product was digested using these enzymes, ligated into pET-31b+ to generate a 6X his-Tag fusion and transformed into E. coli DH5α. After confirming the insert was correct, this plasmid (pET31B + CNFy) was purified and transformed into Y. pseudotuberculosis YPIII.
Purification of CNFy
Y. pseudotuberculosis YPIII harboring pET31B + CNFy was grown in LB medium with 100 µg/ml ampicillin added. When the culture reached an OD of 1.0, the cells were collected by centrifugation at 5,752 × g (15 min), washed with PBS and then suspended in lysis buffer (50 mM Tris-HCl pH 7.5; 200 mM NaCl) supplemented with lysozyme (1 mg/ml). This suspension was kept on ice for 30 min before sonication (20% amplitude for 30 sec pulses). After sonication, the cell debris was removed by centrifugation at 5,752 × g for 30 min at 4 °C. The recombinant CNFy (rCNFy) was then purified by affinity chromatography using Ni-NTA agarose beads (Qiagen, USA) according to the manufacturer’s suggested protocol. The purified protein was then quantified using the Bradford assay. To confirm that the protein was pure, a sample was separated using a 10% SDS PAGE gel and visualized after staining with colloidal coomassie (Fig. S7). Expression of rCNFy within Y. pseudotuberculosis YPIII was also confirmed by western blotting with the Penta·His HRP Conjugate Kit from Qiagen (USA) (Fig. 2C).
Semi-quantitative analysis of CNFy in MVs using western blotting
Samples containing 10 μg of protein from whole cells or MVs from Yersinia pseudotuberculosis YPIII pET31b-CNFy were separated on 4–20% Tris-glycine SDS-PAGE gels (Biorad, USA). Samples of purified rCNFy (0.2 µg, 0.1 µg and 0.05 µg) was also run on the same gel. Subsequently, the proteins were electro-blotted onto a polyvinylidene difluoride (PVDF) membrane (GE health care, USA) using a semi-dry transblotter (Biorad, USA). The membrane was then incubated for 2 h in a blocking solution (5% wt/vol skim milk (Difco, USA) in Tris-buffered saline (TBS; 20 mM Tris-HCl buffer (pH 7.6) containing 137 mM NaCl). After three sequential washes with TBS, the membrane was probed with a Penta·His-HRP conjugate (Qiagen, USA) for 2 h using 5% skim milk in TBS. The blot was then washed three times with TBST (TBS containing 0.05% (vol/vol) Tween-20). The signals were then developed using the Supersignal West Femto kit (Thermo scientific, USA) according to the manufacturer’s suggested protocol, and captured using the LAS 200 System (GE Healthcare, USA).
Immunostaining and confocal microscopy
To inspect the HeLa cells for any morphological changes induced after being exposed to the MVs, confluent monolayers were prepared on Labtek™ II chambered cover glasses (Nunc, Germany). Before treatment, i.e., addition of the MVs, the HeLa cells were serum starved for at least 20 h. The cells were then treated with the purified MVs for 12 h, subsequently washed with DPBS and fixed with 3.7% paraformaldehyde (in PBS) for 20 min at room temperature. After fixing the cells, they were washed with DPBS and permeabilized with 0.1% Triton X-100 (Sigma Aldrich, USA) in DPBS for 5 min. The actin cytoskeleton was then stained with a phalloidin-rhodamine solution (0.5 μ g/ml in DPBS; Invitrogen, USA) for 30 min at room temperature. After washing the cells once more with DPBS, the cellular DNA was counter-stained with DAPI (1 μg/ml in PBS) (Life Technologies, USA) for 5 min at room temperature. These cells were then visualized using a laser confocal microscope LSM 780 (Carl Zeiss, Germany) and the captured images processed using Zen 2009 software.
Quantifying multinucleation
To determine the degree of multinucleation, HeLa cells were seeded into 96 well tissue culture plates (10,000 cells/well) (Corning, USA) and incubated for 24 h with 5% CO2. The cells were then incubated with the different concentrations of either the MVs or purified CNFy for 48 h under 5% CO2. After this incubation step, the cells were washed with DPBS (Sigma-Aldrich, USA) and fixed with 3.7% paraformaldehyde (in PBS) for 10 min at room temperature. After washing the fixed cells three times with DPBS, they were counterstained with DAPI (1 μg/ml in PBS) (Life Technologies, USA) for 5 min at room temperature. The cells were then visualized using a laser confocal microscope LSM 780 (Carl Zeiss, Germany) and the captured images processed using the Zen 2009 software. The number of multinucleated cells from confocal images were visually counted and used to calculate the percentage.
Visualization of MVs associated with the HeLa cells
MVs were purified from cultures of Y. pseudotuberculosis ATCC 29833 and YPIII as described above and then labeled with Octadecyl Rhodamine B Chloride (R18) (0.5 μg/ml in DPBS; Invitrogen, USA) for 20 min. After staining, any unbound dye was removed by ultracentrifugation at 40,000 rpm for 2 h. Monolayers of the HeLa cells were prepared in 8 well Nunc Lab-Tek II chambered cover glass plates (Thermo Scientific, USA) by seeding 2 × 104 HeLa cells/well and incubating them in a CO2 incubator at 37 °C for 24 h. To initiate the experiment, 0.5 ml of the rhodamine BR-18 stained MVs (10 µg/ml in MEM) were added to the wells and the plate was incubated for 2 h at 37 °C before the HeLa cells were washed with DPBS. The cells were then fixed with 3.7% paraformaldehyde (in PBS) for 20 min at room temperature, washed with PBS and permeabilized with a solution of 0.1% Triton X-100 (Sigma Aldrich, USA) in PBS for 5 min.
To visualize the HeLa cells, the actin cytoskeleton was stained using Alexa Fluor 488 phalloidin (0.5 μg/ml in DPBS; Thermo Scientific, USA) for 30 min at room temperature. After washing the cells with DPBS, the cellular DNA was counter-stained with DAPI (1 μ g/ml in PBS; Thermo Scientific, USA) for 5 min at room temperature. The cells and MVs were then visualized using a laser confocal microscope (LSM 780; Carl-Zeiss, Germany). The images were processed using Zen software (Carl-Zeiss, Germany).
Cell viability assays
The impact of the MVs on the viability of the cultured HeLa cells was determined using the MTT assay according to the manufacturer’s suggested protocol. Briefly, 1 × 104 cells were seeded into the wells of a 96-well plate and grown for 24 h. The cells were then exposed to the MVs from Y. pseudotuberculosis ATCC 29833 or YPIII by replacing the media within the wells with fresh media containing MVs. The cells were grown for an additional 24 h in the presence of the MVs. Afterwards, 5 μg/ml of 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazoliumbromide (MTT reagent, Life Technologies, USA) was added to each well and the plates were incubated at for an additional 4 h at 37 °C in the dark. After thoroughly removing the media, 400 μl of DMSO was added to each well and the plate was incubated with shaking (150 rpm) at room temperature for 15 min to allow the color to develop. The OD was measured at 550 nm and used as a proxy for the HeLa cell viability.
Live/Dead imaging
The live and dead cells were labeled using two dyes, calcein AM and ethidium homodimer-1 (EthD-1), respectively. For this, the HeLa cell cultures were prepared by seeding 1 × 104 cells/well in 96-well plates and grown as above. After 24 h, the cells were treated with either media alone or media containing MVs purified from either Y. pseudotuberculosis ATCC 29833 or YPIII cultures. After the cells were grown for an additional 24 h in the presence of the MVs, they were washed gently with 1X Dulbecco’s phosphate-buffered saline (DPBS) at room temperature. To each well, a 100 μl mixture of the Live/Dead Assay reagents (containing 1 μM calcein AM and 2 μM EthD-1 (Life Technologies, USA)) was added. After a 10 min incubation at room temperature, the cells were observed using confocal microscopy (LSM 700; Carl Zeiss, Germany).
Statistical analysis
The data is expressed as the mean (average) of at least three independent replicates with the standard deviations shown as the error bars. An unpaired Student’s t-test was used to analyze the data. P-values of <0.05 were considered statistically significant.
References
Achtman, M. et al. Yersinia pestis, the cause of plague, is a recently emerged clone of Yersinia pseudotuberculosis. P Natl Acad Sci USA 96, 14043–14048, https://doi.org/10.1073/pnas.96.24.14043 (1999).
Wang, X. Y. et al. Genomic comparison of Yersinia pestis and Yersinia pseudotuberculosis by combination of suppression subtractive hybridization and DNA microarray. Arch Microbiol 186, 151–159, https://doi.org/10.1007/s00203-006-0129-1 (2006).
Galindo, C. L., Rosenzweig, J. A., Kirtley, M. L. & Chopra, A. K. Pathogenesis of Y. enterocolitica and Y. pseudotuberculosis in Human Yersiniosis. J Pathog 2011, 182051, https://doi.org/10.4061/2011/182051 (2011).
Paglia, M. G. et al. Yersinia pseudotuberculosis septicemia and HIV. Emerg Infect Dis 11, 1128–1130, https://doi.org/10.3201/eid1107.041268 (2005).
Leino, R. et al. Yersiniosis as a Gastrointestinal-Disease. Scand J Infect Dis 19, 63–68, https://doi.org/10.3109/00365548709032379 (1987).
Jelloul, L., Fremond, B., Dyon, J. F., Orme, R. L. E. & Babut, J. M. Mesenteric adenitis caused by Yersinia pseudotuberculosis presenting as an abdominal mass. Eur J Pediatr Surg 7, 180–183, https://doi.org/10.1055/s-2008-1071088 (1997).
Deacon, A. G., Hay, A. & Duncan, J. Septicemia due to Yersinia pseudotuberculosis - a case report. Clin Microbiol Infec 9, 1118–1119, https://doi.org/10.1046/j.1469-0691.2003.00746.x (2003).
Cave, M. H., Macaleenan, F. A., Hunter, J., Bell, A. L. & Curran, R. Reactive Arthritis Following Yersinia-Pseudotuberculosis Infection. Ulster Med J 59, 87–89 (1990).
Ramamurthi, K. S. & Schneewind, O. Type III protein secretion in Yersinia species. Annu Rev Cell Dev Bi 18, 107–133, https://doi.org/10.1146/annurev.cellbio.18.012502.105912 (2002).
Tsang, T. M., Wiese, J. S., Felek, S., Kronshage, M. & Krukonis, E. S. Ail Proteins of Yersinia pestis and Y-pseudotuberculosis Have Different Cell Binding and Invasion Activities. PLoS One 8, https://doi.org/10.1371/journal.pone.0083621 (2013).
Eitel, J. & Dersch, P. The YadA protein of Yersinia pseudotuberculosis mediates high-efficiency uptake into human cells under environmental conditions in which invasin is repressed. Infect Immun 70, 4880–4891, https://doi.org/10.1128/Iai.70.9.4880-4891.2002 (2002).
Hammerl, J. A., Freytag, B., Lanka, E., Appel, B. & Hertwig, S. The pYV virulence plasmids of Yersinia pseudotuberculosis and Y. pestis contain a conserved DNA region responsible for the mobilization by the self-transmissible plasmid pYE854. Env Microbiol Rep 4, 433–438, https://doi.org/10.1111/j.1758-2229.2012.00353.x (2012).
Cornelis, G. R. et al. The virulence plasmid of Yersinia, an antihost genome. Microbiol Mol Biol R 62, 1315-+ (1998).
Aepfelbacher, M. & Heesemann, J. Modulation of Rho GTPases and the actin cytoskeleton by Yersinia outer proteins (Yops). Int J Med Microbiol 291, 269–276, https://doi.org/10.1078/1438-4221-00130 (2001).
Lockman, H. A., Gillespie, R. A., Baker, B. D. & Shakhnovich, E. Yersinia pseudotuberculosis produces a cytotoxic necrotizing factor. Infect Immun 70, 2708–2714, https://doi.org/10.1128/Iai.70.5.2708-2714.2002 (2002).
Schweer, J. et al. The Cytotoxic Necrotizing Factor of Yersinia pseudotuberculosis (CNFY) Enhances Inflammation and Yop Delivery during Infection by Activation of Rho GTPases. PLoS Pathog 9, https://doi.org/10.1371/journal.ppat.1003746 (2013).
Hoffmann, C., Pop, M., Leemhuis, J., Aktories, K. & Schmidt, G. The Yersinia pseudotuberculosis cytotoxic necrotizing factor (CNFY) selectively activates RhoA. N-S Arch Pharmacol 369, R52–R52 (2004).
Boquet, P. & Lemichez, E. Bacterial virulence factors targeting Rho GTPases: parasitism or symbiosis? Trends Cell Biol 13, 238–246, https://doi.org/10.1016/S0962-8924(03)00037-0 (2003).
He, Y. H. et al. RhoA activation by CNFy restores cell-cell adhesion in kindlin-2-deficient keratinocytes. J Pathol 233, 269–280, https://doi.org/10.1002/path.4350 (2014).
Im, H., Lee, S., Soper, S. A. & Mitchell, R. J. Staphylococcus aureus extracellular vesicles (EVs): surface-binding antagonists of biofilm formation. Mol Biosyst 13, 2704–2714, https://doi.org/10.1039/c7mb00365j (2017).
Perez-Cruz, C., Delgado, L., Lopez-Iglesias, C. & Mercade, E. Outer-Inner Membrane Vesicles Naturally Secreted by Gram-Negative Pathogenic Bacteria. PLoS One 10, https://doi.org/10.1371/journal.pone.0116896 (2015).
Manning, A. J. & Kuehn, M. J. Contribution of bacterial outer membrane vesicles to innate bacterial defense. BMC Microbiol 11, https://doi.org/10.1186/1471-2180-11-258 (2011).
Renelli, M., Matias, V., Lo, R. Y. & Beveridge, T. J. DNA-containing membrane vesicles of Pseudomonas aeruginosa PAO1 and their genetic transformation potential. Microbiol-SGM 150, 2161–2169, https://doi.org/10.1099/mic.0.26841-0 (2004).
Domingues, S. & Nielsen, K. M. Membrane vesicles and horizontal gene transfer in prokaryotes. Curr Opin Microbiol 38, 16–21, https://doi.org/10.1016/j.mib.2017.03.012 (2017).
Ellis, T. N. & Kuehn, M. J. Virulence and Immunomodulatory Roles of Bacterial Outer Membrane Vesicles. Microbiol Mol Biol R 74, 81-+, https://doi.org/10.1128/Mmbr.00031-09 (2010).
Eddy, J. L., Gielda, L. M., Caulfield, A. J., Rangel, S. M. & Lathem, W. W. Production of Outer Membrane Vesicles by the Plague Pathogen Yersinia pestis. PLoS One 9, e107002, https://doi.org/10.1371/journal.pone.0107002 (2014).
Pisano, F. et al. Influence of PhoP and Intra-Species Variations on Virulence of Yersinia pseudotuberculosis during the Natural Oral Infection Route. PLoS One 9, https://doi.org/10.1371/journal.pone.0103541 (2014).
Gardy, J. L. et al. PSORT-B: improving protein subcellular localization prediction for Gram-negative bacteria. Nucleic Acids Res 31, 3613–3617, https://doi.org/10.1093/nar/gkg602 (2003).
Bai, J., Kim, S. I., Ryu, S. & Yoon, H. Identification and characterization of outer membrane vesicle-associated proteins in Salmonella enterica serovar Typhimurium. Infect Immun 82, 4001–4010, https://doi.org/10.1128/IAI.01416-13 (2014).
Lee, E. Y. et al. Global proteomic profiling of native outer membrane vesicles derived from Escherichia coli. Proteomics 7, 3143–3153, https://doi.org/10.1002/pmic.200700196 (2007).
Perez-Cruz, C. et al. New type of outer membrane vesicle produced by the Gram-negative bacterium Shewanella vesiculosa M7T: implications for DNA content. Appl Environ Microbiol 79, 1874–1881, https://doi.org/10.1128/AEM.03657-12 (2013).
Sjostrom, A. E., Sandblad, L., Uhlin, B. E. & Wai, S. N. Membrane vesicle-mediated release of bacterial RNA. Sci Rep 5, 15329, https://doi.org/10.1038/srep15329 (2015).
Hagemann, S. et al. DNA-bearing membrane vesicles produced by Ahrensia kielensis and Pseudoalteromonas marina. J Basic Microbiol 54, 1062–1072, https://doi.org/10.1002/jobm.201300376 (2014).
Francis, M. S. & Wolf-Watz, H. YopD of Yersinia pseudotuberculosis is translocated into the cytosol of HeLa epithelial cells: evidence of a structural domain necessary for translocation. Mol Microbiol 29, 799–813, https://doi.org/10.1046/j.1365-2958.1998.00973.x (1998).
Persson, C., Carballeira, N., Wolf-Watz, H. & Fallman, M. The PTPase YopH inhibits uptake of Yersinia, tyrosine phosphorylation of p130Cas and FAK, and the associated accumulation of these proteins in peripheral focal adhesions. EMBO J 16, 2307–2318, https://doi.org/10.1093/emboj/16.9.2307 (1997).
Gerke, C., Falkow, S. & Chien, Y. H. The adaptor molecules LAT and SLP-76 are specifically targeted by Yersinia to inhibit T cell activation. J Exp Med 201, 361–371, https://doi.org/10.1084/jem.20041120 (2005).
Cornelis, G. R. Yersinia type III secretion: send in the effectors. J Cell Biol 158, 401–408, https://doi.org/10.1083/jcb.200205077 (2002).
Orden, J. A. et al. Necrotoxigenic Escherichia coli from sheep and goats produce a new type of cytotoxic necrotizing factor (CNF3) associated with the eae and ehxA genes. Int Microbiol 10, 47–55 (2007).
Kouokam, J. C. et al. Active cytotoxic necrotizing factor 1 associated with outer membrane vesicles from uropathogenic Escherichia coli. Infect Immun 74, 2022–2030, https://doi.org/10.1128/Ia1.74.4.2022-2030.2006 (2006).
Nuss, A. M. et al. Transcriptomic profiling of Yersinia pseudotuberculosis reveals reprogramming of the Crp regulon by temperature and uncovers Crp as a master regulator of small RNAs. PLoS Genet 11, e1005087, https://doi.org/10.1371/journal.pgen.1005087 (2015).
Shevchenko, A., Tomas, H., Havlis, J., Olsen, J. V. & Mann, M. In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nat Protoc 1, 2856–2860, https://doi.org/10.1038/nprot.2006.468 (2006).
Acknowledgements
Funding for this research was sponsored by the National Research Foundation (NRF) of Korea within the General Research Program (Grant No. 2016R1D1A1A09919912) and under Basic Research Program for Young Researchers (Grant No. 2017R1C1B1003495). We would like to thank them for the financial support. We also would like to thank the UOBC facilities at UNIST for use of the equipment.
Author information
Authors and Affiliations
Contributions
A.K.M., W.B. and R.J.M. designed the experiments. A.K.M. performed the experiments. R.J.M. supervised the experimental work. J.K.S. ran the LC/MS-MS. A.K.M. and J.K.S. analyze the mass spectrometry data. R.J.M. evaluated all the data. A.K.M., W.B. and R.J.M. wrote the manuscript.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Monnappa, A.K., Bari, W., Seo, J.K. et al. The Cytotoxic Necrotizing Factor of Yersinia pseudotuberculosis (CNFy) is Carried on Extracellular Membrane Vesicles to Host Cells. Sci Rep 8, 14186 (2018). https://doi.org/10.1038/s41598-018-32530-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-32530-y
- Springer Nature Limited
Keywords
This article is cited by
-
Wie RNA-Thermometer die Sekretion von Virulenzfaktoren kontrollieren
BIOspektrum (2024)