
Abstract
In many human disorders mitochondrial dysfunction is central to degeneration of retinal ganglion cells. As these cells do not regenerate, vision is irreversibly lost. Here we show reversal of visual dysfunction by a mitochondrially targeted adeno associated virus in transgenic mice harboring a G11778A mutation in the ND4 subunit of complex I persists longterm and it is associated with reduced loss of RGCs and their axons, improved oxidative phosphorylation, persistence of transferred ND4 DNA and transcription of ND4 mRNA.
Similar content being viewed by others

Introduction
Visual loss from optic neuropathy is estimated to affect more than 50 million people1, and glaucoma is the leading cause. In this disease, once retinal ganglion cells (RGCs) whose axons comprise the optic nerve are gone, visual loss is irreversible2. Recent evidence suggests that mitochondrial dysfunction drives RGC loss and that this sequelus can be prevented by modulating mitochondrial function3. Leber hereditary optic neuropathy (LHON) is an uncommon cause of optic neuropathy, but it is one of the most common diseases caused by mutations in the mitochondrial genome4. Unlike most other hereditary optic neuropathies and glaucoma that are slowly progressive, visual loss in LHON is typically acute in onset, bilateral and severe. Over 95% of all cases are due to pathogenic mutations in one of three mitochondrial genes that encode complex I subunits of the respiratory chain: ND1G3460A, ND4G11778A or ND6T14484C5. These mutations impair oxidative phosphorylation and promote death of RGCs. Effective treatments that reverse the visual loss of mitochondrial diseases have yet to be realized6,7. Here we utilize a mitochondrially targeted adeno associated viral vector (MTS-AAV) to deliver the wild-type human ND4 gene into RGC mitochondria8 of mitomice that transmit the human ND4G11778A gene as a mitochondrial episome9 and demonstrate the long term persistence of reversal of the adverse effects of the mutant gene on the visual system.
Results
Wild-type ND4 mediates prompt and persistent reversal of visual function loss in mutant ND4 mitomice
To determine whether visual loss of G11778A mitomice is reversible, both eyes of 3 month old G11778A mitomice (n = 14) with low PERG amplitudes were injected with mitochondrially targeted AAV containing the wild-type (WT) human ND4 (MTSAAV-hND4) (Supp. Figure 1). In addition, both eyes of littermates with low PERG amplitudes (n = 10) were injected with mitochondrially targeted AAV carrying the mCherry gene in the mitochondrial genetic code (MTS-AAV-mCherry) (Supp. Figure 1, open circles or square). Gene therapy with wild-type ND4 reversed loss of PERG amplitudes with an increase of 17% at one month post injection (p = 0.007) that improved further to 42% at three months post injection (p = 0.007) (Fig. 1A). In contrast, PERG amplitudes of mCherry injected eyes remained low (Fig. 1B). While encouraging, it is unclear whether initial benefits to visual function return would diminish as seen in Leber Congenital Amaurosis (LCA2), a non-mitochondrial disease caused by recessive mutations in the RPE65 gene, where topographical maps of visual sensitivity in retinal regions treated by AAV delivery of the normal RPE65 cDNA had progressive diminution of the areas of improved vision 4–6 years after treatment10.

(A) Scatterplots of 3-month old transgenic mice with low PERG amplitudes at baseline (Bl) show amplitudes rise 1 (1 M), 3 (3 M), and 13 months (13 M) after intravitreal injection of MTSAAV containing the wild-type human ND4(MTSAAV-hND4). (B) Scatterplots of the littermate controls with low PERG amplitudes at baseline remain low 1, 3, and 13 months after intravitreal vector injection of MTSAAV carrying mCherry. (C) Representative PERG waveforms of MTSAAV-hND4 treated mice and untreated mice (D) with the x axis in 100-ms increments. Flash ERGs of MTSAAV-hND4 treated mice (E) and MTSAAV-mCherry injected controls (F) show no differences. (G) OCT 2D imaging of an MTSAAV-hND4 treated mouse eye 12 months postinjection shows the retinal ganglion cell layer plus inner plexiform layer appears thicker than an MTSAAV-cherry injected control (H). ONH = optic nerve head, RGC = retinal ganglion cell layer, IPL = inner plexiform layer, OPL = outer plexiform layer, ONL = outer nuclear layer, RPE = retinal pigment epithelial layer. (I) Representative 3D thickness maps of an MTSAAV-hND4 treated mouse and (J) an MTSAAV-mCherry injected control 12 months after injection. (K) Scatterplots show average thickness measurements of the RGC + IPL 12 months after intraocular injections.
Next, we tested for treatment persistence. Longer-term follow up of our hND4 treated mice showed stable visual function with a 46% increase in the PERG amplitude (Fig. 1A) relative to mCherry injected controls (Fig. 1B), p = 0.0276, 13 months postinjection. Relative to baseline values obtained prior to hND4 injections PERG amplitude increased 64%, p = 0.005. Representative waveforms of hND4 vector treated mutant ND4 mitomice mice at each time point (Fig. 1C) illustrated that hND4 vector improved visual function. PERG amplitudes of individual mice improved in most (Supp. Figure 2A–K), but not all eyes (Supp. Figure 2L–O). In contrast, PERG amplitudes of most (Supp. Fig. 3A–E), but not all (Supp. Fig. 3F–I) untreated mice eyes deteriorated (Fig. 1D). Flash ERGs, a measure of global retinal function, showed no changes (Fig. 1E,F) confirming that the reversal of visual dysfunction measured by the PERG was due to effects on RGCs and not photoreceptors11.
Wild-type ND4 reduces loss of RGCs and optic nerve axons
Next, we used optical coherence tomography (OCT) to look for loss of RGCs in the retinas of live affected mice. Unlike human LHON where OCT can identify changes in thickness of the RGC layer12, in the mouse the RGC layer itself is beyond the dependable resolution of the instrument, so here we measured the RGC + inner plexiform (IPL) layers. Representative OCTs of treated (Fig. 1G) and untreated (Fig. 1H) eyes at 12 months of age show a thicker RGC + inner plexiform (IPL) layer in hND4 vector injected mice. Segmentation revealed that differences in thickness maps of RGC + IPL thickness of hND4 treated mice (93 ± 5 μm; mean ± SD) (Fig. 1I) and mcherry control mice (83 ± 6 μm; mean ± SD) (Fig. 1J) were statistically significant (p = 4.41e−5) (Fig. 1K). While this finding may suggest MTSAAV-hND4 treatment attenuated loss of RGCs, it could also suggest it caused swelling of the RGC + IPL layers.
To determine whether the increase in thickness of treated mice might represent swelling, as seen initially in LHON patients13, or true preservation of RGCs we next examined ocular histopathology. Light microscopy of the retina performed 15 months after intraocular injections, confirmed that hND4 vector treated mice had more cells in the RGC layer and a thicker IPL (Fig. 2A) than the untreated mice (Fig. 2B). Quantitation of cells in the RGC layer revealed a 63% loss in cell numbers of control mice (5230 ± 494 cells/mm2; mean ± SD) relative to hND4 vector treated mice (8286 ± 196 cells/mm2; mean ± SD), p = 0.006 (Fig. 2C).

(A) Longitudinal retinal sections of an MTSAAV-hND4 treated mouse and (B) an untreated mouse injected with MTSAAV-mCherry at 15 months postinjection. Scale bars = 30 μm; (C) Bar plot of cell numbers in the RGC layer. (D) Transmission electron micrographs of the retrobulbar optic nerve of an MTSAAV-hND4 treated mouse and (E) an MTSAAV-mcherry nerve. Ax = axon, arrows = electron dense debris in cystic spaces were axons are missing. Scale bars = 2 μm. (F) A bar plot showing axon counts in MTSAAV-hND4 treated nerves and MTSAAV-mcherry controls. (G) A histogram of the percentage distribution of optic nerve axon diameters in control and treated mice (bin size, 0.1 μm). The distribution of axonal diameters in the MTSAAV-hND4 treated mice shows preservation of small diameter axons with a shift to larger diameter axons in MTSAAV-mcherry injected controls. (H) Quantile scatterplots of ATP production in the optic nerves of MTSAAV-hND4 treated and MTSAAV-mcherry controls. (I) Quantile scatterplots of ATP production in the retinas of MTSAAV-hND4 treated and MTSAAV-mCherry controls. (J) Agarose gel electrophoresis of an undigested 803 bp PCR amplification product spanning the G11778A point mutation (lanes 1 and 3) and MaeIII digested (lanes 2 and 4) PCR products from laser captured mtDNA of RGCs from MTSAAV-hND4 treated affected mice. MaeIII cuts wild-type ND4 into three bands of 527 bp, 255 bp and 21 bp, and cuts the mutant ND4 into 4 bands of 527 bp, 124 bp, 131 bp and 21 bp (see illustrations). Sanger sequencing confirmed that the 255 bp band was wild-type human ND4 and the 131 bp band was the mutant human ND4. An alignment shows the homology of the respective sequences between mutant and wild-type ND4. RT-PCR products from laser captured1RNA of RGCs from MTSAAV-hND4 treated affected mice show MaeIII cuts wild-type ND4 mRNA derived cDNAs into three bands of 527 bp, 255 bp and 21 bp, and cuts the mutant ND4 cDNAs into 4 bands of 527 bp, 124 bp, 131 bp and 21 bp (see lane 3 RT-PCR agarose panel right, lane 1 is undigested cDNA).
Furthermore, ultrastructural analysis revealed that the optic nerves of wild-type ND4 treated mice had numerous axons (Fig. 2D), while control animals had many cystic spaces and electron dense debris where axons were lost (Fig. 2E). Quantitative analysis revealed 51% more optic nerve axons in MTSAAV-hND4 treated mitomice than in controls (p = 0.0049) (Fig. 2F). Analysis of the distribution of axon diameters revealed treated mitomice had more small axons (≤1 μm) than control eyes that had axons larger than 1 μm (Fig. 2G). These data suggest that delivered wild type ND4 prevented the demise of small axons that are preferentially lost in human LHON14.
Respiratory chain function in mutant ND4 mice
Because RGCs are adversely impacted by the mutant G11778A ND4 and they are highly dependent on oxidative phosphorylation, we measured the rate of ATP synthesis by chemiluminescence with a modified luciferin-luciferase assay in freshly extracted digitonin-permeabilized optic nerve and retinal homogenates using malate and pyruvate that provide sources of NADH to complex I. We found that the rate of complex I-dependent ATP synthesis was significantly more robust in the optic nerves of wild-type ND4 injected mice than in mock treated eyes of transgenic mice (Fig. 2H) (p = 0.002). Thus, the energy deficit in transgenic mutant ND4 optic nerves was improved with wild-type hND4 treatment. The reduction in ATP synthesis in untreated eyes was not statistically significant in the retina, perhaps because RGCs comprise a small fraction of the cells (<6%) (Fig. 2I).
Wild-type human ND4 DNA persisted in mice 15 months after injection
Next, we wished to determine the longevity of the MTS-AAV transferred DNA. To do so we tested for mutant and wild type human ND4 DNA by laser microdissection of RGCs of MTSAAV-hND4 treated mice followed by extraction of mitochondrial DNA and PCR amplification of an 803 bp fragment spanning the G11778A point mutation. We then digested the PCR products with MaeIII, which cuts wild-type human ND4 into three bands of 527 bp, 255 bp and 21 bp, and cuts the mutated ND4 into 4 bands of 527 bp, 124 bp, 131 bp and 21 bp (Fig. 2J). Sanger sequencing confirmed that the 255 bp band was wild-type human ND4 and the 131 bp band was the mutant human ND4. Quantitative PCR of the ratio of human ND4 to mouse ND4 in RGCs was 31% but lower in the inner nuclear layer (2%) and outer nuclear layer (5%). As qPCR could not distinguish between mutant human and wild-type human ND4 densitometry of the MaeIII digested bands revealed the ratio of wild-type to mutant human ND4 DNA levels in RGC was 2.23. Thus transferred wild-type human ND4 persisted in RGCs for most of the lifespan of transgenic mutant ND4 mitomice.
Wild-type human ND4 mRNA is transcribed in mice 15 months after injection
Lastly, we tested for transcription of the injected wild-type ND4 as well as that of the transgenic mutant ND4 by laser microdissection of RGCs of MTSAAV-hND4 treated mice followed by extraction of mitochondrial RNA and RT-PCR amplification of an 803 bp fragment spanning the G11778A point mutation. We then digested the RT-PCR products with MaeIII (see RT-PCR panel right in Fig. 2J), which cuts wild-type human ND4 into three bands of 527 bp, 255 bp and 21 bp, and cuts the mutated ND4 into 4 bands of 527 bp, 124 bp, 131 bp and 21 bp (Fig. 2J). The 255 bp band was wild-type human ND4 cDNA and the 131 bp band was the mutant human ND4 cDNA. Quantitative RT-PCR revealed that the ratio of human to mouse ND4 cDNA was 245% in RGC’s but undetectable in the inner nuclear and outer nuclear layers. As qRT-PCR could not distinguish between mutant human and wild-type human ND4 densitometry of the MaeIII digested bands revealed the ratio of wild-type to mutant human ND4 DNA levels in RGC was 2.76. Thus, both wild-type and mutant human ND4 were transcribed longterm 15 months after intravitreal injections suggesting it may likely do so in human LHON patients.
Discussion
In our PERG experiments here we set the stimulus gratings to the largest size (0.05 c/deg = 2.77 logMAR7,8,9) and most contrast to detect severe visual deficits of our mice, as is also characteristic of human LHON, and to demonstrate reversal of them. The PERG amplitude of at least one eye of each transgenic mutant ND4 mouse randomized for treatment was at noise levels (2–3 uV), thus similar to blind G11778A LHON patients. The long lasting improvements in the PERG with treatment suggest these severe visual deficits were reversed for the most of the lifespan of our mice providing the preclinical data that similar improvements may be expected in LHON patients treated with this mitotargeted AAV containing wild-type human ND4 in future gene therapy trials.
Our work here complements the series of publications where we demonstrated that a mitochondrial-targeted AAV delivered the human wild-type or mutant ND4 to mitochondria where the gene expresses episomally without integration into the endogenous host mtDNA8,9,15. Using this approach we were able to correct the deleterious effects of the G11778A LHON mutation in human mutant ND4 cybrid cells. We then addressed the RGC and optic nerve degeneration of mice allotopically expressing the mutant human G11778A ND4 in retinal ganglion cells8. We then generated a mouse that stably expresses episomally the human mutant G11778A ND4 by delivering the mitotargeted AAV in the zygote that transmitted the mutant human G11778A ND4 mutant episome maternally over many generations. Another group has independently developed an MTS-AAV showing import and expression of the transgene in mitochondria16. Taken together, our data show that the severe visual loss induced by a mitochondrial disease may be reversed for most of the lifespan of laboratory mice where it reduced loss of RGCs and their axons, improved oxidative phosphorylation with persistence of transferred ND4 DNA and transcription of ND4 mRNA. thus supporting mitotargeted AAV gene therapy as a long-term platform for treatment of human mitochondrial optic neuropathies such as LHON.
Materials and Methods
Plasmids and AAVs
scHSP-hND4FLAG + mCherry was constructed as previously described8.9. In brief, human ND4FLAG and mitochondrial-encoded mCherry were cloned into self-complementary (sc) AAV serotype 2 backbones under the control of the human mitochondrial heavy strand promoter (HSP), where ND4FLAG is followed by mCherry with a stop codon between two genes. Meanwhile, mCherry cloned in the same scAAV2 backbone was used as control (scHSP-mCherry). The plasmids were amplified and purified using the Qiagen endotoxin free maxiprep and then packaged with the VP2COX8GFP plus VP1, VP3 and helper plasmid PXX6 (3-fold excess) into recombinant virus: MTSAAV-hND4 and MTSAAV-mCherry.
Polymerase chain reaction (PCR) and reverse transcriptase–polymerase chain reaction (RT-PCR)
PCR
Mitochondria were isolated from micro-dissected cells from the retinal ganglion cell layer, inner nuclear layer and outer nuclear layer. DNA was extracted using DNeasy blood and tissue kit (Qiagen). For RNA extraction, mitochondrial were treated with 10 u DNase (Invitrogen) at 37 °C for 30 min followed by 75 °C 15 min. Then, RNA was extracted using the RNeasy mini kit (Qiagen) and used for reverse transcription (iScript cDNA Synthesis kit, Bio-Rad). PCR was performed with primers F11124: TCGAAACCACACTTATCC and R11926: TGATCAGGAGAACGTGG. PCR products were purified with QIAquick gel extraction kit (Qiagen) and digested with MaeIII. The interest fragments were recovered with the same kit listed above and sent for sequencing(Genewiz). Quantitative PCR assay was performed with oligonucleotides hND4F: 5′-CCTACTCATCGCACTAAT-3′ and hND4R 5′-TCATATTAAGTTATTGGCTCAG-3′: for human ND4; oligonucleotides mND4F: 5′-TTCATCCTTCTCTCCCTA-3′ and mND4R: 5′-ATTATTAGTATTGTTGCTCCTAT-3′ for mouse ND4 using Light cycle96 (Roche).
Animals
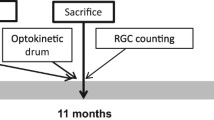
All animal procedures were performed in accordance with the National Institutes of Health Guide for Care and Use of Laboratory Animals and the ARVO Statement for the use of Animals in Ophthalmic and Vision Research, with approval from the University of Miami Institutional Animal Care and Use Committee. For the intraocular injection of recombinant AAV, 24 mitomice of three months of age were sedated by inhalation with 1.5% to 2% isofluorane. A local anesthetic (proparacaine HCl) was applied topically to the cornea, and then a 32-gauge needle attached to a Hamilton syringe was inserted through the pars plana. Fourteen mice in the treated group were injected into both eyes with 1 µl of MTS-AAV carrying the WT human ND4 (MTSAAV-hND4, titer = 4.4e11 vg/ml). Littermates (n = 10) in the control group were injected into both eyes with 1 µl of MTS-AAV carrying mCherry (MTSAAV-mCherry, titer = 3.08e11 vg/ml). PERGs were performed at baseline, and then again at 1, 3, and 13 months after intravitreal injections.
PERG and OCT
In brief, mice were weighed and anesthesized with IP injections of a mixture of ketamine (80 mg/kg body weight) and xylazine (10 mg/kg body weight) and were restrained by using a bite bar and a nose holder that allowed unobstructed vision. The animals were kept at a constant body temperature of 37.6 °C with a feedback-controlled heating pad. In these conditions, the eyes of mice were wide open and in a stable position with undilated pupils pointing laterally and upward. The ERG electrode had a diameter of 0.25 mm, made of silver wire configured to a semicircular loop of 2-mm radius, was placed on the corneal surface by means of a micromanipulator and positioned in such a way as to encircle the pupil without limiting the field of view. Reference and ground electrodes were stainless-steel needles inserted under the skin of the scalp and tail, respectively. After setting the mice on the stage and before recording, a small drop of balanced saline was topically applied on the cornea to prevent drying. To detect the presence of absence of vision a visual stimulus of contrast-reversing bars (field area, 50° × 58°; mean luminance, 50 cd/m2; spatial frequency, 0.05 cycles/degree; contrast, 100%; and temporal frequency, 1 Hz) was aligned with the projection of the pupil at 20 cm distance. Eyes were not refracted for the viewing distance, because the mouse eye has a large depth of focus because of the pinhole pupil. Retinal signals were amplified (10,000-fold) and band-pass filtered (1–30 Hz). Three consecutive responses to 600 contrast reversals each were recorded. The responses were superimposed to check for consistency and then averaged. The PERG is a light-adapted response. To have a corresponding index of outer retinal function, a light-adapted flash ERG (FERG) was also recorded with non-dilated pupils in response to strobe flashes of 20 cd/m2/s superimposed on a steady background light of 12 cd/m2 and presented within a Ganzfeld bowl. Under these conditions, rod activity is largely suppressed while cone activity is minimally suppressed. Averaged PERGs and FERGs were automatically analyzed to evaluate the major positive and negative waves by Sigma Plot (Systat software Inc., San Jose, CA). Retinal images were visualized with in vivo spectral-domain (SD) OCT (Bioptigen, Inc.) and then were analyzed with semiautomated custom software written using MATLAB software (MathWorks, Inc.).
Histology and Ultrastructure
At the appropriate time points rodents inoculated with the AAV vectors were overdosed with sodium pentobarbital. They were then perfused by cardiac puncture with PBS and then with fixative consisting of 4% paraformaldehyde in 0.1 M PBS buffer (pH 7.4). The eyes with attached optic nerves were dissected out and further processed by immersion fixation in 2.5% glutaraldehyde, postfixed in 1% osmium tetroxide, 0.1 M sodium cacodylate-HCl buffer (pH 7.4), 7% sucrose in the cold, and then dehydrated through an ethanol series to propylene oxide, infiltrated, and embedded in epoxy resin that was polymerized at 60 °C overnight. For immunocytochemistry, tissue specimens were postfixed in 5.0% acrolein, 0.1 M sodium cacodylate-HCl buffer (pH 7.4) and 7% sucrose and then dehydrated through an ethanol series and embedded in LR White (Ted Pella, Redding, CA) that was polymerized at 50 °C overnight. Examinations were made with a FEI CM10 transmission electron microscope, operating at 80 kV.
Optic Nerve Diameters and Axons Counts
Fifteen months after intraocular injections, optic nerves of approximately 5 mm in length were dissected from 1 mm behind the globe to the optic chiasm. For axon counts, an average of 10 transmission electron micrographs were photographed by a masked observer at a magnification of 4600× for each optic nerve specimen. The diameter and number of axons were then manually counted. Axons were identified by a clear axoplasmic cytoskeleton or surrounding electron dense myelin sheath. A total of 2972 axons were counted for MTSAAV-mcherry controls and 4904 axons for MTSAAV-hND4 treated mice.
Statistical Analysis
Statistical analyses were performed using the Graphpad Prism software. Two groups were compared using two- tailed t tests, with P values of < 0.05 considered significant. Values were expressed as means ± standard deviation (SD).
References
Quigley, H. A. & Broman, A. T. The number of people with glaucoma worldwide in 2010 and 2020. Br J Ophthalmol. 90, 262–267 (2006).
Laha, B., Stafford, B. K. & Huberman, A. D. Regenerating optic pathways from the eye to the brain. Science. 356, 1031–1034 (2017).
Williams, P. A. et al. Vitamin B3 modulates mitochondrial vulnerability and prevents glaucoma in aged mice. Science 355, 756–760 (2017).
Yu-Wai-Man, P. et al. Inherited mitochondrial optic neuropathies. J Med Genet. 46, 145–158 (2009).
Yu-Wai-Man, P. & Chinnery, P. F. Leber Hereditary Optic Neuropathy. In: Adam M. P. et al., editors. SourceGeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; (1993).
Yu-Wai-Man, P., Griffiths, P. G. & Chinnery, P. F. Mitochondrial optic neuropathies - disease mechanisms and therapeutic strategies. Prog Retin Eye Res 30, 81–114 (2011).
Carelli, V., La, M. C. & Sadun, A. A. Mitochondrial dysfunction in optic neuropathies: animal models and therapeutic options. Curr Opin Neurol. 26, 52–58 (2013).
Yu, H. et al. Gene delivery to mitochondria by targeting modified adenoassociated virus suppresses Leber’s hereditary optic neuropathy in a mouse model. Proc Natl Acad Sci USA 109(20), E1238–47 (2012).
Yu, H. et al. Consequences of zygote injection and germline transfer of mutant human mitochondrial DNA in mice. Proc Natl Acad Sci USA 112, E5689–E5698 (2015).
Jacobson, S. G. et al. Improvement and decline in vision with gene therapy in childhood blindness3. N Engl J Med. 372, 1920–1926 (2015).
Banitt, M. R. et al. Progressive loss of retinal ganglion cell function precedes structural loss by several years in glaucoma suspects. Invest Ophthalmol Vis Sci. 54, 2346-2352 (2013).
Kernstock, C., Friebe, K. & Tonagel, F. Applications of optical coherence tomography (OCT) in neuro-ophthalmology. Klin Monbl Augenheilkd. 230, 1097–1105 (2013).
Lam, B. L. et al. Trial end points and natural history in patients with G11778A Leber hereditary optic neuropathy: preparation for gene therapy clinical trial. JAMA Ophthalmol. 132, 428–436 (2014).
Sadun, A. A. et al. Leber’s hereditary optic neuropathy differentially affects smaller axons in the optic nerve. Trans Am Ophthalmol Soc. 98, 223–232 (2000).
Yu, H. et al. Mutant NADH dehydrogenase subunit 4 gene delivery to mitochondria by targeting sequence-modified adeno-associated virus induces visual loss and optic atrophy in mice. Mol Vis. 18, 1668–1683 (2012).
Araujo Zin, E. et al. Optimization of AAV-mediated Gene Delivery for Targeting Mitochondria in the Outer Retina and Müller glia. IOVS Supplr ARVO meeting Baltimore USA Abstract Number: 4090–B0090 (2017).
Acknowledgements
Supported by R01 EY017141, EY012355, P30-EY014801 and unrestricted grant to Bascom Palmer Eye Institute from Research to Prevent Blindness, Inc.
Author information
Authors and Affiliations
Contributions
Hong Yu performed experiments and data analysis. Vittorio Porciatti assisted in the interpretation of the electrophysiology. Alfred Lewin edited the manuscript. William Hauswirth packaged AAVs and edited the manuscript. John Guy helped perform the data analysis, wrote the manuscript and prepared the figures.
Corresponding author
Ethics declarations
Competing Interests
John Guy has a conflict in that he owns the intellectual property for the mito-targeted AAV. William Hauswirth has financial interest in a company AGTC that commercializes AAV vectors.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yu, H., Porciatti, V., Lewin, A. et al. Longterm Reversal of Severe Visual Loss by Mitochondrial Gene Transfer in a Mouse Model of Leber Hereditary Optic Neuropathy. Sci Rep 8, 5587 (2018). https://doi.org/10.1038/s41598-018-23836-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-23836-y
- Springer Nature Limited