
Abstract
The interaction of hC5a with C5aR, previously hypothesized to involve a “two-site” binding, (i) recognition of the bulk of hC5a by the N-terminus (NT) of C5aR (“site1”), and (ii) recognition of C-terminus (CT) of hC5a by the extra cellular surface (ECS) of the C5aR (“site2”). However, the pharmacological landscapes of such recognition sites are yet to be illuminated at atomistic resolution. In the context, unique model complexes of C5aR, harboring pharmacophores of diverse functionality at the “site2” has recently been described. The current study provides a rational illustration of the “two-site” binding paradigm in C5aR, by recruiting the native agonist hC5a and engineered antagonist hC5a(A8). The hC5a-C5aR and hC5a(A8)-C5aR complexes studied over 250 ns of molecular dynamics (MD) each in POPC bilayer illuminate the hallmark of activation mechanism in C5aR. The intermolecular interactions in the model complexes are well supported by the molecular mechanics Poisson–Boltzmann surface area (MM-PBSA) based binding free energy calculation, strongly correlating with the reported mutational studies. Exemplified in two unique and contrasting molecular complexes, the study provides an exceptional understanding of the pharmacological divergence observed in C5aR, which will certainly be useful for search and optimization of new generation “neutraligands” targeting the hC5a-C5aR interaction.
Similar content being viewed by others

Introduction
Complement component fragment 5a receptor (C5aR) is one among the two chemoattractant receptors known in the rhodopsin family of G-protein coupled receptors (GPCR)1. C5aR is known to be stimulated by the hC5a2, one of the most potent inflammatory modulator of the complement system, driving the host-defense mechanism. However, the protecting shield is often weakened or lost due to the aberrant stimulation of C5aR, exposing the host to variety of inflammatory, autoimmune and neurological disorders3,4. Though, understanding the hC5a-C5aR interaction for therapeutic intervention appears lucrative, clinical breakthroughs remains largely limited, apparently due to the lack of atomistic understanding of the molecular interactions, between the hC5a and C5aR. Thus, for realizing better and improved complement therapeutics for future clinical practices, it is highly imperative to obtain a rational picture of the molecular complexation between hC5a and C5aR, no matter how crude it may appear at this stage. Driven by large scale mutagenesis studies, the molecular complexation is hypothesized to involve two discrete sites5: (i) interaction between the NT peptide of C5aR with the bulk of hC5a (site1) and (ii) interaction between the ECS of C5aR with the CT peptide of hC5a (site2). It is apparently clear from the literature that the interactions at the “site1” play the anchorage function to arrest the hC5a, whereas the interactions at the “site2” trigger the cellular responses of C5aR. Interestingly, such “two-site” binding paradigm has recently been structurally exemplified in few peptide or protein binding GPCRs of rhodopsin family6,7. Nevertheless, no such structural studies or refined molecular models illustrating the intermolecular interactions at both the “site1” and “site2” are currently available for hC5a and C5aR.
In our quest to understand the hC5a-C5aR interaction better, we recently generated unique structural models of C5aR8 and subsequently illustrated the plausible orthosteric “site2” on its ECS9, by recruiting a variety of functionally diverse small molecule ligands, including the CT peptide (64NISHKDMQLGR74) of hC5a. In the current study, we subjected the modeled C5aR to pilot experimental scrutiny, involving biophysical techniques and further screened the model against the native agonist hC5a2 (74 amino acids) and the engineered antagonist (73 amino acids) hC5a(A8)10. Objective was to decipher the plausible orthosteric “site1” on the NMR derived NT peptide11, grafted to the modeled C5aR9 for generating the first set of distinct model molecular complexes, precisely illustrating the pharmaceutical landscape of the “two-site” binding paradigm in C5aR. Though, both hC5a and hC5a(A8) share ~90% sequence identity, hC5a(A8) competitively binds to the C5aR, albeit weakly (IC50 ~ 35 nM) compared to hC5a (IC50 ~ 3 nM) for reasons clearly not described12. Structurally hC5a(A8) appears to be an allosteric conformer of hC5a, that imparts the antagonistic effect on C5aR, due to its engineered CT (64NISFKRSLLR73) sequence. Interestingly, several single point mutations on the CT of hC5a(A8) has also been described that can reverse the antagonism of hC5a(A8) to agonism12. However, the mechanism of such action is still unclear in structural terms. In continuation to our earlier reports8,9,13, the comparison of the hC5a-C5aR, hC5a(A8)-C5aR model structural complexes, including the CT peptide variants of hC5a(A8) presented in the study provide the necessary rationalization important for understanding the observed antagonism and the switching of antagonism to agonism at the “site2” of C5aR. Moreover, the native agonist (hC5a-C5aR) and the engineered antagonist (hC5a(A8)-C5aR) bound model complexes, respectively presented in the current study rationalize a large set of point mutation based binding and signaling data12,14,15,16,17,18,19,20, by estimating the residue specific energetic contribution toward overall binding in structural terms. The model complexes, thus appear as a useful template for structure-based drug design, by illuminating the intermolecular interactions at atomistic resolution, highly essential for modeling and discovery of potential disruptive pharmacophores targeting the hC5a-C5aR interactions.
Results
Validating the model structure of C5aR
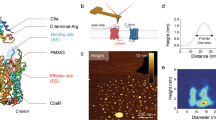
The topologically unique model of C5aR described earlier8,9, presented in Fig. 1 illustrates a modestly folded β-hairpin like structure with ~30% residues in ordered β-sheet conformation, as estimated from the in silico folding studies of the predicted extended extracellular loop 2 (ECL2) polypeptide [Ac-Y174-RVVREEYFPPKVLC188GVDYSHDKR-R198-NH2]8. The C5aR model (Fig. 1) also feature an unordered NT peptide, mostly derived from the previously reported NMR studies11. Given the known structure of many GPCRs8, it is highly unlikely that individual transmembrane (TM) domains of C5aR will demonstrate a structure other than α-helix, though their topological arrangement as a 7 TM bundle may slightly vary in real experimental conditions from the modeled C5aR (Fig. 1), which is a matter of future detailed structural studies. Further, structural analysis of the loop structures in known GPCRs evidence that the ECL2 peptide is longest among all other loops, and demonstrates structural diversity8. Thus, we decided to probe the conformational state of the predicted ECL2 peptide in various solvent conditions using circular dichroism (CD) and 1H-NMR spectroscopy. The ECL2 peptide was synthesized over solid phase with C188/S to avoid the unwanted aggregation in solution. Further, serine being isostructural to cysteine may not drastically alter the possible conformation of the ECL2 peptide in solution. Interestingly, in agreement with our folding simulation studies reported for the extended ECL2 peptide8, the synthetic ECL2 peptide with ≥95% purity (Fig. S1) demonstrated a CD signature (Fig. 2) reminiscent of a highly twisted short-stranded β-sheet conformation (Fig. 1b), frequently observed for βII-class of proteins21. Addition of 10–40% trifluoroethanol (TFE), a hydrogen bond promoting solvent22 to the PBS buffer, did not alter the overall CD signature but enhanced the intensity of the observed CD signature of the ECL2 peptide. Even in 100% methanol, the peptide demonstrated a similar CD signature with highest intensity, indicating the role of solvent dielectric on the overall conformation of the ECL2 peptide22. A detailed comparison of the CD intensities at 215–218 nm, 222 nm and 208 nm indicated that addition of TFE perhaps enhances the % β-sheet content in the peptide (Fig. 2b). In support, estimation of [θ]222/[θ]208 provided a value of 0.65, indicating the presence of a 310-turn23, and addition of TFE also did not change the estimated % helicity (~3%) further24. In further support to CD spectra, the 1H-NMR spectra of the ECL2 peptide in 10% D2O-water (Fig. S2) appeared well dispersed, indicating the presence of an ordered conformation of the peptide in solution. Nevertheless, the ECL2 being one of the major structural component in the ECS of C5aR that harbors the orthosteric “site2”, a separate detailed structural study can be undertaken later. Though inconclusive, prima facie, the pilot biophysical studies are in sync with the modeled conformation of the ECL2, which partially validate the presented model of C5aR (Fig. 1a), providing the necessary impetus to probe the “site1” on the modeled C5aR toward establishing a plausible “two site” binding interaction involving hC5a and C5aR.

(a) The model structure of C5aR illustrating the probable structure of ECL2 peptide (red) and the NMR derived structure of NT peptide (green). The conserved ECL2-TM3 disulfide bond is also highlighted in yellow. (b) The highly twisted short stranded β-sheet conformation of the ECL2 peptide derived from the in silico folding studies, illustrating the C188 in the loop region.

(a) Signature CD spectra demonstrating the effect of different solvent conditions on the secondary structure of the ECL2 peptide. (b) Effect of TFE concentration on the CD intensities in the 215–218 nm β-sheet region.
Structural complex of hC5a with C5aR
In our prior studies9, we have illustrated the interaction of hC5a-CT peptide at the “site2” of the modeled C5aR with minimum interference from the NT peptide of C5aR. However, to illustrate a “two-site” binding interaction between hC5a and C5aR, it is highly essential to understand the molecular interaction at the “site1” involving the bulk of hC5a and the NT peptide of the C5aR. Thus, to decipher the molecular interaction at the “site1”, the NMR-derived NT-peptide11, grafted to the C5aR model [Ac-D2-SFNYTTPDYGHYDDKDTLDLNTPVD-K28-NH2] (Fig. 1a) was subjected to a sequential buildup docking studies combined with energy minimization against the most populated conformer of hC5a, evolved over 50 ns of molecular dynamics (MD) study13. The docking protocol benchmarked against the CHIPS protein complex (Fig. S3) yielded an estimated Ki ~ 5.33 nM (−11.29 kcal/mol) for the best conformer of the NT peptide of C5aR complexed to hC5a (Fig. S4), illuminating the most plausible “site1” on C5aR. The molecular complex gauged over 100 ns of MD studies appears to be stable in the explicit water at 300 K, suggesting that the modeled interactions depicted at the “site1” are physically viable (Fig. S5). Further, the most populated conformer of the “site1” complex evolved over the MD (Fig. S4) was subjected to structural assembly with the previously described C5aR complexed to hC5a-CT9 at the “site2” for generating the complete hC5a-C5aR complex (Figs 3a and S6). The resulting “two-site” binding structural complex of hC5a-C5aR was carefully inserted into the POPC bilayer (Fig. 3a) as described8,9 and subjected to one quarter of a microsecond MD studies at 300 K. The “hot-spot” residues participating in variety of intermolecular interaction (Fig. 4) mainly hydrophobic, hydrogen bonding and salt bridge interactions (Fig. S7) at both the “site1” and “site2” of hC5a-C5aR complex are schematically illustrated in the Fig. 3b. Sustainability of many such important residue specific intermolecular interactions at both “site1” and “site2” over the duration of MD are summarized in Fig. 4 (Fig. S7), indicating the physical viability of the interactions under experimental conditions, overall molecular stability and atomistic nature of the modeled hC5a-C5aR complex.

(a) Illustration of “two-site” binding in the model complex of hC5a-C5aR inserted into the POPC bilayer. (b) Interaction map of hC5a illustrating the “hot-spot” residues, respectively at the “site1” and “site2” of C5aR. “Hot-spot” residues that are known to modulate both binding and signaling upon mutation are highlighted in red.

Summary of the specific intermolecular interactions monitored both at the “site1” and “site2” of hC5a-C5aR complex over 250 ns of MD at 300 K in POPC bilayer. Interacting residues that are not superscripted represents C5aR and the residues that are superscripted represents hC5a. The solid grey lines indicate the cut-off distance. (a) Moderate salt bridge interaction monitored between the D2 and H67. (b) Strong “cation-π” interaction observed between Y11 and R37. (c) Stable “π-π” interaction observed between Y14 and F51. (d) Strong hydrogen bonding noted between the side chain of D16 and the backbone NH of N29. (e) Strong hydrogen bond noted between the backbone carbonyl of D18 and the side chain NH of R62. (f) Stable salt bridge interactions observed between the terminal NH3+ of M1 and side chain of D21 and (g) D27. (h) Strong “π-π” interaction observed between F182 and H67. (i) Moderate salt bridge interaction noted between the side chain of D191 and the terminal CO2− of R74. (j) Strong hydrogen bond between side chain of S193 and terminal CO2− of R74. (k) Very strong hydrogen bond interaction between side chain of E269 and the backbone NH of I65. (l) Stable “cation-π” interaction observed between F275 and K68.
Structural complex of hC5a(A8) with C5aR
The hC5a(A8)10 is an engineered protein derived from hC5a, which has been described to act as a potent antagonist (ID50 ~ 22 nM) against C5aR due to its engineered CT (64NISFKRSLLR73) sequence12. Interestingly the protein is also described to switch its function from antagonist to agonist by introducing a point mutation at R69 of its CT. Recent structural studies indicate that hC5a(A8) is structurally different from native hC5a (IC50 ~ 3 nM), and competitively bind weakly to C5aR (IC50 ~ 35 nM)12. Thus, we decided to probe the molecular interaction of hC5a(A8) with C5aR, by subjecting the previously described C5aR model8,9 into action. Initially, we subjected the CT peptide of hC5a(A8) and some of its variants to automated docking studies against the “site2” of C5aR, as described for hC5a-CT9. Surprisingly, the A8, A8Δ71–73, and A8R69D CT peptides of hC5a(A8), respectively with an estimated Ki ~ 970 nM (−8.20 kcal/mol), Ki ~ 2.36 μM (−7.68 kcal/mol), and Ki ~ 117 μM (−5.36 kcal/mol), perfectly blocked the “site2” on C5aR (Fig. S8), surrounded by a cluster of hydrophobic residues. Further analysis revealed that the F67 on the CT peptide of both A8 and A8Δ71–73 is involved in a “π-π” interaction25 with F275 at the “site2” of the modeled C5aR (Fig. S9), as observed previously for PMX53 and NDT9. In contrast, the K68 on the CT peptide of A8R69D demonstrated a similar “cation-π” interaction26 involving the F275 (Fig. S9), as observed previously for hC5a-CT peptide9. Interestingly, the estimated affinity of the A8R69D (Ki ~ 117 μM; −5.36 kcal/mol) CT peptide of the hC5a(A8) is apparently in sync with our earlier estimation for hC5a-CT peptide (Ki ~ 35 μM, −6.08 kcal/mol) that is known to demonstrate binding affinity of ~150 μM toward C5aR in PMNL membranes27. Interestingly, the interactions observed for the CT-peptide variants of hC5a(A8) in the C5aR complexes, remained intact over 100 ns of MD (Fig. S9) in POPC bilayer, suggesting the physical viability of the modeled interactions and stability of the overall complex (Fig. S10).
As described for hC5a, the NT-peptide of C5aR was also subjected to systematic stepwise docking against the hC5a(A8), and the resultant complex illustrating the interaction at “site1” (Fig. S11) yielded an estimated Ki ~ 113 μM (−5.38 kcal/mol), compared to the interaction at “site1” for hC5a (Ki ~ 5.33 nM; −11.29 kcal/mol). The observation is broadly in sync with experiments and can be attributed to overall structural difference between hC5a and hC5a(A8). The resultant molecular complex of hC5a(A8) remained stable over 100 ns of MD in explicit water at 300 K, suggesting that the modeled interactions observed for bulk of hC5a(A8) at the “site1” of C5aR are physically viable (Fig. S12). Further, by applying requisite geometrical constraints, the hC5a(A8) complexed to NT-peptide of C5aR (site1; Fig. S11) was subjected to structural assembly with the C5aR complexed to CT peptide of hC5a(A8) at the “site2” (Fig. S10) for generating the complete hC5a(A8)-C5aR complex (Figs 5a and S13). The modeled complex was further subjected to MD studies in POPC bilayer (Fig. 5a) at 300 K over one quarter of a microsecond. The “hot-spot” residues involved in the “two-site” binding interaction between hC5a(A8) and C5aR are schematically illustrated in Fig. 5b. The various intermolecular interactions observed between the hot-spot residues of the complex are also sustained over the duration of MD (Figs 6 and S14), indicating the overall stability of the complex at par with the hC5a-C5aR complex.

(a) Illustration of “two-site” binding in the model complex of hC5a(A8)-C5aR inserted into the POPC bilayer. (b) Interaction map of hC5a(A8) illustrating the “hot-spot” residues, respectively at the “site1” and “site2” of C5aR. “Hot-spot” residues that are known to modulate both binding and signaling upon mutation are highlighted in red. Residues of hC5a(A8) whose mutation are known in hC5a are shown in red and underlined.

Summary of the specific intermolecular interactions monitored both at the “site1” and “site2” of hC5a(A8)-C5aR complex over 250 ns of MD at 300 K in POPC bilayer. Interacting residues that are not superscripted represents C5aR and the residues that are superscripted represents hC5a(A8). The distance cut-offs are shown as solid grey lines. (a) Very strong hydrogen bond interaction observed between the backbone CO of D10 and the backbone NH of S42. (b) Stable hydrogen bonding noted between the backbone CO of Y14 and the side chain of K20. (c) Stable salt bridge interactions observed between the head group of K20 and side chain of D15 and (d) D16. (e) Moderate salt bridge interaction observed between the side chain of D18 and R27 and (f) between the side chain of D21 and K19. (g) The strong hydrophobic interaction between Cγ2 of T19 and the centroid of Y23. (h) Stable “cation-π” interaction observed between the centroid of Y174 and the side chain of R73. (i) Strong hydrogen bonding noted between the backbone NH of F182 and the backbone CO of R69. (j) Very strong hydrogen bond interaction between the side chain of K185 and the backbone CO of L72. (k) Stable “π- π” interaction observed between F275 and F67. (l) Strong hydrogen bonding noted between the backbone CO of N279 and the side chain of S66.
Estimation of the energetic contribution of “hot-spot” residues
The molecular mechanics Poisson-Boltzmann surface area (MM-PBSA/MM-GBSA) has been a useful tool for estimating binding free energies of various protein-ligand complexes28,29,30 in remarkable correlation with the experimental results31,32, though application of MM-PBSA calculation to membrane proteins still remains tricky for various reasons33. Nevertheless, we decided to recruit the method for estimating an apparent binding energy of hC5a/hC5a(A8)-C5aR complexes in a moderate dielectric medium, by randomly selecting 150 conformers each from the most populated cluster (Fig. S15), evolved over the duration of the respective MD trajectories. This decision of modulating the dielectric was somehow influenced from our CD studies on ECL2 peptide in different solvent gradients. As presented in Fig. 3 (Fig. S6) and Fig. 5 (Fig. S13), the binding of hC5a or hC5a(A8) is largely influenced by the solvent exposed ECS and NT residues of C5aR. Thus, to avoid the complexity of the overall calculation involving the lipid bilayer, only the interacting residue pairs from C5aR and hC5a/hC5a(A8) were subjected to MM-PBSA calculations, respectively for estimating the binding free energy. Under such conditions, the hC5a-C5aR complex provided an estimated average binding energy of ~−16.12 ± 4.2 kcal/mol (Ki ~ 1.65 × 10−12 M) compared to ~−24.71 ± 8.7 kcal/mol (Ki ~ 1.06 × 10−18 M) for hC5a(A8)-C5aR complex, indicating that hC5a(A8) may be a better binder to the modeled C5aR (Table S1). Further, recruitment of the C5aR: N-terminus (1–37) and ECS (38–40, 94–108, 170–201, 261–280) residues, respectively against hC5a: residues (1–74), and hC5a(A8): residues (1–73) provided higher estimated binding free energy of −276 ± 26 kcal/mol for hC5a-C5aR complex and −315 ± 26 kcal/mol for hC5a(A8)-C5aR complex, which clearly indicates the existence of a strong binding interaction between the modeled C5aR and the ligands. However, it is worth mentioning that the estimated binding free energies presented for the complexes are indicative of strong binding only and may not be straightaway translated to experimental binding affinities. Decomposition of the overall binding energy provided the non-bonded interaction energy (summation of van der Waals and electrostatic) for specific “hot-spot” residues, respectively participating at both “site1” and “site2” of hC5a/C5aR and hC5a(A8)/C5aR complexes. The pairwise energy contribution of such “hot-spot” residues for hC5a-C5aR and hC5a(A8)-C5aR complexes are respectively presented in Fig. 7 and Fig. 8. Considering the cationic nature of hC5a34, it is clear that the binding at the plausible “site1” is strongly driven by the interaction with a set of anionic residues at the NT, such as D2, D16, D18, D21, and D27 of C5aR. In addition, Y11 and Y14 at the NT of C5aR also contribute significantly toward the binding affinity at the “site1”. This trend is consistent even in case of hC5a(A8)-C5aR complex, where D10, D15, D16, D18, D21 and Y14 make significant contribution toward the binding affinity at the “site1” of C5aR. Further, the K68 of hC5a makes significant contribution in contrast to F67 of hC5a(A8) at the “site2” of C5aR, matching to our earlier hypothesis that suggests “cation-π” interaction triggers agonism, and “π-π” interaction triggers antagonism at the “site2” of C5aR9.

Decomposition of the MM energy of “hot-spot” residues contributing toward overall binding free energy of hC5a-C5aR complex. Energetic contribution of hC5a and C5aR residues are respectively shown in grey and black. hC5a residues are indicated with superscripts.

Decomposition of the MM energy of “hot-spot” residues contributing toward overall binding free energy of hC5a(A8)-C5aR complex. Energetic contribution of hC5a(A8) and C5aR residues are respectively shown in grey and black. hC5a(A8) residues are indicated with superscripts.
Discussion
Understanding the molecular basis and conformational dynamics35,36,37,38 of receptor-ligand interaction is a valuable aspect in the field of pharmacology and drug design39,40. While most of the rhodopsin family GPCRs bind to small molecule ligands at a discrete site within the ECS/transmembrane segment41,42,43, there are few GPCRs such as C5aR that bind to small protein ligands like hC5a, which are too bulky to fit completely into the ECS/transmembrane region of C5aR. Thus, early biochemical studies had mapped the high-affinity binding of hC5a to more than one site on C5aR5,44, a testable hypothesis that need to be illustrated at an atomistic resolution. Over the years, with advance in structural biology techniques, few GPCRs in complex with small protein ligands have been obtained6,7,45 recently, illustrating the idea of “two-site” or multiple site binding models46,47,48,49. In addition, molecular modeling coupled with biophysical, pharmacological and MD studies have also provided an alternative support to the idea of “two-site” binding models in many GPCRs29,30,36,50. Nevertheless, no such highly refined structural complex is currently available for C5aR in the literature. Thus, it remains unclear how hC5a is arrested by the C5aR or what changes does it bring to the C5aR for triggering the activation and downstream signaling.
In our quest to understand the interaction of hC5a with C5aR better, we recently reported a unique model structure of C5aR in complex with hC5a-CT, PMX53 and NDT9. The model complexes indicate that the NT may have a minimal role in binding of small peptide or organic ligands at the “site2” on C5aR, but surely have a potential role in capturing the major part of hC5a. This assumption is well supported by several studies that implicate the role of C5aR NT in high affinity binding of hC5a18,19,20,51, whereas CT peptide of hC5a has been shown to be essential for triggering activation and downstream signaling52. Thus, a plausible step wise binding of hC5a to C5aR is hypothesized in this study, which is illustrated in Fig. 9. Briefly, in step-1, the NT (site1) of C5aR wraps around the allosteric region of hC5a13 with high affinity, triggering local conformational change both at the ECS of C5aR and at the CT of hC5a. This hypothesis is based on the structure of des-Arg74-hC5a53, and hC5a(A8)10, whose CT demonstrates an extended β-structure compared to the native hC5a2. Interestingly, during the MD simulation over 50 ns13, the CT of hC5a also adopts an extended β-structure, deviating from its native α-turn structure. Subsequently in step-2, the conformationally altered CT of hC5a is docked at the ECS (site2) of C5aR, triggering global conformational change in the overall complex, and further opening the intracellular face of C5aR for binding of G-protein or β-arrestin (unpublished data). It is noteworthy that the unique structural illustration of the “two-site” binding in hC5a-C5aR model complex (Fig. 3) finds great support from several studies that interrogated the interaction of hC5a with C5aR by recruiting site-directed mutagenesis studies15,16,17,18,19,20,51. For instance, the high affinity binding at the “site1” of the hC5a-C5aR (Fig. 3) is mainly driven by several salt bridge interactions between D2-H67, D21-NH3+ (M1), D27-NH3+ (M1) and hydrogen bond interactions between D16-N29, D18-C27, D18-R62, and T24/T32-M1, including several hydrophobic contacts, sustained over 250 ns of MD in POPC bilayer at 300 K (Figs 4 and S7). Literature evidences that both single or (double) mutation of anionic amino acids such as D10N and D27N (D21N, D27N) on NT of C5aR does not affect the binding of hC5a, whereas serial mutations such as (D10N, D15N, D16N) and (D10N, D15N, D16N, D21N, D27N) completely abrogates the binding of hC5a to C5aR20. In a separate study, it is also suggested that serial mutations such as (D15A, D16A, D18A, D21A) induces ~42-fold and (D10A, D15A, D16A, D18A, D21A) induces ~140-fold decrease in binding of hC5a51, whereas point mutations of D15A, and D18A trigger a remarkable loss in C5aR signaling19. Moreover, in the model complex, both Y11 and Y14 also participate in a strong “cation-π” and a strong “π-π” interaction, respectively involving R37 and F51 of hC5a (Fig. 4). In addition, both Y11 and Y14, including Y6 are also involved in hydrogen bonding, respectively with E35, D24, and N64 of hC5a (Fig. S7). It is evidenced that both Y11 and Y14 undergo sulfation and mutation of Y11F results in complete loss of binding, whereas mutation of Y14F confers ~50% loss in binding affinity of hC5a18. Even the low affinity binding of the CT region of hC5a at the “site2” experience numerous interactions with C5aR residues (Figs 4 and S7), whose mutation is known to affect both binding and signaling of hC5a15,16,17. For instance, P184, P270, F275 and N279 of C5aR are involved in strong hydrophobic, hydrogen bonding, and “cation-π” interaction with the K68 of hC5a. Similarly, both D191 and S193 are involved in anchoring the R74, whereas F182 is involved in a “π-π” interaction with H67 of hC5a. Interestingly, the “cation-π” and hydrogen bond interactions of K68 with F275 of C5aR remained stable up to 200 ns of MD, whereas the salt bridge and hydrogen bond interactions between R74 and D191 of C5aR remained stable up to 170 ns of MD. Its noteworthy that mutation of many such residues of hC5a54,55 implicated in the binding of C5aR (Fig. 3b) has been shown to affect both binding and signaling activity of C5aR significantly.

Illustrating the “two-site” binding paradigm of hC5a to C5aR. Step1: Wrapping of NT of C5aR (site1) around the allosteric region of hC5a with high affinity, triggering local conformational change both at the ECS of C5aR and at the CT of hC5a. Step2: Docking of the conformationally altered CT of hC5a at the ECS (site2) of C5aR triggering global conformational change in the overall complex, and further opening of the intracellular face of C5aR for downstream signaling.
Despite having an altered structure, the engineered antagonist hC5a(A8) also experiences similar level of molecular interactions at both the “site1” and “site2” of C5aR (Fig. 5) with few notable exceptions. The interaction at “site1” involves several hydrogen bonds, salt bridge and hydrophobic interactions that are stable over 250 ns of MD in POPC bilayer and have been summarized, respectively in Fig. 6 and Fig. S14. However, exceptions in interactions are noted at the “site2” of C5aR, where instead of K68, F67 of hC5a(A8) is engaged in a strong “π-π” interaction with F275 within a cage formed by several strong hydrophobic residues such as F182, P184, and P270 of C5aR (Figs S14 and S8). This differs from the interaction of hC5a at the “site2”, but strongly supports the previously observed interaction of the antagonist PMX53 and inverse agonist NDT at the “site2” of C5aR9. Further, this interaction is reversed in case of A8R69D variant of the hC5a(A8), where instead of F67, K68 is involved in a strong “cation-π” interaction with F275 of C5aR at the “site2”, strongly agreeing with the interactions described for hC5a. Interestingly, mutation of R69D in hC5a(A8) has been described to completely switch the antagonistic action of hC5a(A8) to agonism (IC50 ~ 5 nM), at par with hC5a12.
Moreover, both the agonist (hC5a) and antagonist (hC5a(A8) bound complexes illuminate important hallmark information about the activation process of C5aR, in agreement with several rhodopsin family GPCRs reported in the literature6,7,45. It is postulated that the breaking of the “ionic lock switch” between R3.50 (TM3) and E6.30 (TM6) is the hallmark of activation in many rhodopsin family GPCRs56,57. In addition, tryptophan (W6.48) rotamer toggling at a relatively conserved region (CWxPx) on TM658,59, recently renamed as the “transmission switch” (W6.48 and F6.44) and the “tyrosine toggle switch” (Y7.53) at a conserved region (NPxxY) on TM7 are also known to participate in receptor activation59. In our previous studies, we have hypothesized that activation of C5aR in model structures involve movement of almost all the TMs with a higher magnitude of movement noted between TM3 and TM68,9. Since, C5aR lacks an “ionic lock switch”60, we shifted our attention to relatively more conserved common activation switches in GPCRs such as the “transmission switch” and the “tyrosine toggle switch” for understanding the effect of hC5a and hC5a(A8) on the model structure of C5aR. As presented in Fig. 10, the concerted rotameric movement of W6.48, F6.44 (TM6: transmission switch) and Y7.53 (TM7: tyrosine toggle switch) at the respective conserved region of C5aR in response to the binding of hC5a (meta-active) and hC5a(A8) (inactive) correlate strongly with the experimental data49 of other rhodopsin family GPCRs41,42,43,61,62,63,64,65 (Fig. S16). Further, it appears that transition from inactive to meta-active or pseudo active state in noted GPCRs involve modulation of “π-π” interaction between W6.48 and F6.44, triggered via swift rotameric transitions (Fig. S16). The extent of “π-π” modulation is directly dependent on the type of neighboring residues. It is interesting to note that unlike the noted GPCRs41,42,43,61, C5aR lacks an “ionic lock switch”60, but demonstrates similar trend in rotameric transitions at its conserved region (Figs 10 and S16) on complexation with hC5a and hC5a(A8). Such correlation with experimental observations is surely encouraging, which favorably support the quality of the model complexes presented in the study and further seeds hope that the unique model complexes will withstand the future structural and biophysical scrutiny for further establishing the activation mechanism of C5aR.

(a) Comparative illustration of rotamer toggling (distribution of χ dihedral angles) observed at the conserved regions of C5aR complexed to agonist hC5a and engineered antagonist hC5a(A8) with respect to the other active-inactive pair of GPCRs41,42,43,61,62,63,64,65. (b) Comparison of rotamer toggling observed in C5aR on binding to hC5a with respect to free unbound C5aR. Rotamer toggling for F6.44, W6.48 of “transmission switch” and Y7.53 of “tyrosine toggling switch” are illustrated. Active GPCRs are marked in asterisk. C5aRI: free or unbound C5aR; C5aR: bound to hC5a(A8); C5aR*: bound to hC5a.
Conclusion
The study provides a partial validation of the unique C5aR model through pilot biophysical studies, illustrating a “two-site” binding interaction of C5aR with two established, contrasting pharmacological counterparts, such as hC5a and hC5a(A8). The presented model complexes illuminate energetically competent inter molecular interactions, largely in sync with reported experimental studies, highlighting the plausible activation mechanism of C5aR. In summary, the model complexes emerge as a significant development in the field for garnering further valuable insights into simple or extended ternary complexes, respectively involving C5aR, hC5a and heterotrimeric G-protein or β-arrestin, which can potentially serve as a template for search and design of disruptive pharmacophores, targeting the chronic inflammation induced malaises.
Materials and Methods
Data sets
The NMR structure of hC5a (PDB ID: 1KJS)2, crystal structure of hC5a(A8) (PDB ID: 4P39)10 and the following rhodopsin family GPCRs (1F8861; rhodopsin, 3DQB62; opsin*, 2RH143; β2AR, 3P0G63; β2AR*, 3REY42; A2AR, 3QAK64; A2AR*, 3UON41; M2R, 4MQS65; M2R*) were downloaded from www.rcsb.org. Visualization and presentation of C5aR complexes, including the other GPCRs were performed using DS 4.0 (Accelrys). PyMOL (The PyMOL Molecular Graphics System, Version 1.1r1. Schrödinger, LLC) was used for proper orientation and translation of ligands within the proximity of the extracellular surface (ECS) of C5aR. The starting model of C5aR TM residues were numbered following Ballesteros–Weinstein system66. The “cation-π” and “π-π” interaction angles were calculated using our in-house program as described elsewhere9. Data were plotted using GraphPad Prism (version 6 for Windows, GraphPad Software, La Jolla California USA, www.graphpad.com).
Biophysical studies on the ECL2 peptide
The predicted sequence of the ECL2 peptide [Ac-Y174-RVVREEYFPPKVLC188/SGVDYSHDKR-R198-NH2] of C5aR was prepared using standard Fmoc chemistry over solid phase, by recruiting the services of Genscript (NJ, USA). The analytical HPLC performed over AlltimaTM C18 (4.6 × 250 mm) column, using acetonitrile–water gradient in presence of 0.05–0.065% trifluoroacetic acid (TFA) indicates that the peptide is ≥95% pure (Fig. S1). The integrity of the peptide [Theoretical MW: 3137.52] was confirmed from the presence of the molecular ion peaks at 785.35 for MH+ [Observed MW: 3137.40], as observed in ESI-MS. The peptide was completely soluble in water and thus, the secondary structure of 100 µM peptide was analyzed in PBS buffer (pH ~ 7.4) as well as in 100% methanol, by recruiting the JASCO J-815 Circular Dichroism spectropolarimeter. Further, 10–40% of trifluoroethanol (TFE) was added to the PBS buffer to check the effect of TFE on the overall conformation of the peptide. Data were collected at 298 K in 1 cm path length quartz cell with 1 nm bandwidth in Far-UV (200–260 nm) range. Scanning at 50 nm/min with 1.0 s time constant in 1 nm steps, five scans were averaged after baseline correction for solvent. The observations in millidegrees were converted to residue ellipticity [θMRW] with a reported relation67. 1H-NMR of the peptide was recorded by using a Bruker 800 MHz instrument equipped with a cryoprobe at 298 K in 90% H2O/10% D2O at pH ~ 5. Solvent was suppressed using standard Watergate sequence as provided by Bruker.
Construction of hC5a/hC5a(A8) complexes of C5aR
Modeling of C5aR has been extensively detailed in our earlier studies8,9. The inactive C5aR (C5aRI) data and the meta-active C5aR model used in this study has been taken straight from the reported data to build the hC5a-C5aR or hC5a(A8)-C5aR complex further. The starting structure of the NT peptide of the C5aR was obtained from the NMR structure of CHIPS complex (PDB ID: 2K3U)11, which was further amino-terminally elongated by adding five amino acids to it using DS 4.0. The modified NT peptide harboring the “site1” was then subjected to flexible automated docking against the central conformer of hC5a populated over 50 ns of MD13, by recruiting AutoDock 4.268 and energy minimization by GROMACS in tandem with a carefully designed sequential build-up approach. The best conformer of the NT peptide complexed to the hC5a was subjected to MD over 100 ns at 300 K in presence of explicit water by recruiting the GROMACS package69. Further, the most populated conformer of the complex (Fig. S4) bound to the NT peptide of C5aR (site1) was harnessed and the hC5a-CT peptide (64NISHKDMQLGR74) was removed from the complex. By applying requisite geometrical constraints, the above truncated complex was further subjected to structural assembly with the previously generated central conformer of the major cluster, populated for hC5a-CT complexed to C5aR9, resulting the complete hC5a-C5aR complex (Fig. S6). Similar approach was also followed for the construction of hC5a(A8) complex of C5aR. Briefly, the NT of C5aR was docked to hC5a(A8) and continuously refined until no further change in binding energy was noticed (Fig. S11). The CT-peptide of hC5a(A8) and its variants were generated from the parent structure of hC5a(A8) and subsequently subjected to automated docking against the previously described meta-active structure of C5aR9. The C5aR complexed to the CT-peptide variants of hC5a(A8) at the “site2” were subjected to MD in POPC bilayer over 100 ns each. The most populated conformers of the major clusters were respectively harnessed (Fig. S10) and further subjected to structural assembly with the bulk of hC5a(A8) complexed to NT of C5aR, resulting the complete hC5a(A8)-C5aR complex (Fig. S13).
Molecular dynamics studies
The hC5a-C5aR and hC5a(A8)-C5aR complexes were inserted into POPC [1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine] bilayer using InflateGRO70 and further subjected to independent MD simulations for 250 ns each at 300 K by recruiting the GROMACS package69, as described previously8,9. Both hC5a-C5aR and hC5a(A8)-C5aR systems were charge neutralized by randomly placing 19 and 21 chloride ions respectively, in presence of 16724 and 16586 water molecules. Both the systems were equilibrated twice, first for 5 ns under NVT, followed by 50 ns under NPT conditions prior to the MD studies. Conformational clustering was performed as described with a time interval of 20 ps8. The utility programs available in GROMACS were implemented for detailed analysis of all the MD trajectories.
Estimation of binding free energy
Molecular mechanics Poisson-Boltzmann surface area (MM-PBSA) method, as implemented in g_mmpbsa program71 was used for calculating the binding free energies of both hC5a-C5aR and hC5a(A8)-C5aR complexes. Briefly, the binding free energies were estimated using implicit water by Poisson Boltzmann (PB) approaches. The solvation energy of the solute was estimated by using a dielectric continuum to account for the electrostatic as well as the non-polar contribution. The dielectric constant for the solvent and the solute were taken as 80 and 20 respectively for polar calculation. The non-polar contribution to solvation free energy term was calculated from solvent-accessible surface area (SASA). The grid spacing was set to 0.5 Å. Probe radius for SASA estimation was set to 1.4 Å. 150 conformers, randomly selected each from the first major cluster, populated for hC5a-C5aR (Fig. S15a) and hC5a(A8)-C5aR (Fig. S15b) complexes, respectively over 250 ns of MD in lipid bilayer were subjected to MM-PBSA based binding free energy calculation, by recruiting the interacting residues only, as illustrated, respectively in Fig. 3b (hC5a-C5aR) and Fig. 5b (hC5a(A8)-C5aR). MM-PBSA and the energy contribution of individual amino acids toward overall binding free energy of the complex were respectively calculated by utilizing the “MmPbSaStat.py”, and “MmPbSaDecomp.py” scripts.
Structural Note
While this manuscript was under peer review, the crystal structure of a thermostabilized C5aR with 11 mutations both in TMs and in Loops (StaR; PDB ID: 5O9H) bound to an inverse agonist appeared in the literature72. On comparison, it was noted that the StaR shares an incredible structural similarity with our previously modelled C5aR8 (backbone RMSD ~ 5 Å), which is used for generating the current structural complex. Our truncated model structure of the native C5aR, respectively lacked 26 residues on N-terminus and 34 residues on C-terminus. Interestingly, the StaR also lacks 29 residues from the N-terminus and 17 residues from the C-terminus. More importantly, we had hypothesized that the ECL2 peptide [Ac-Y174-RVVREEYFPPKVLCGVDYSHDKR-R198-NH2] could be an independently folded unit, which is most likely to adopt a β-hairpin like conformation in solution, as also evidenced in the current study. Indeed, as predicted, the StaR illustrates a β-hairpin structure exactly in the same region. Further, our inactive model C5aR illustrated TM1: P36-F64 (StaR: V35-A66); TM2: F75-Q98 (StaR: I69-Q98); TM3: I111-F139 (StaR: G106-F139); TM4: W154-F172 (StaR: A150-Y174); TM5: E199-R228 (StaR: R197-S231); TM6: L241-V260 (StaR: K239-S266); TM7: F275-V302 (StaR: T274-A303) and TM8: F307-L315 (StaR: P316-L323), which is in excellent agreement with the experimentally derived model structure of StaR. In addition, we had even observed a single α-turn in the ICL2 of our modelled C5aR, in contrast to the two-turn α-helical structure observed in StaR, which collectively provides a direct evidence supporting the excellent quality of our model.
References
Gerard, N. P. & Gerard, C. The chemotactic receptor for human C5a anaphylatoxin. Nature 349, 614–617 (1991).
Zhang, X., Boyar, W., Toth, M. J., Wennogle, L. & Gonnella, N. C. Structural definition of the C5a C terminus by two-dimensional nuclear magnetic resonance spectroscopy. Proteins 28, 261–267 (1997).
Guo, R. F. & Ward, P. A. Role of C5a in inflammatory responses. Annu. Rev. Immunol. 23, 821–852 (2005).
Ricklin, D. & Lambris, J. D. Complement-targeted therapeutics. Nat. Biotechnol. 25, 1265–1275 (2007).
Siciliano, S. J. et al. Two-site binding of C5a by its receptor: an alternative binding paradigm for G protein-coupled receptors. Proc. Natl. Acad. Sci. 91, 1214–1218 (1994).
Qin, L. et al. Structural biology. Crystal structure of the chemokine receptor CXCR4 in complex with a viral chemokine. Science (New York, NY) 347, 1117–1122 (2015).
Zheng, Y. et al. Structure of CC Chemokine Receptor 5 with a Potent Chemokine Antagonist Reveals Mechanisms of Chemokine Recognition and Molecular Mimicry by HIV. Immunity 46, 1005–1017 (2017).
Rana, S. & Sahoo, A. R. Model structures of inactive and peptide agonist bound C5aR: Insights into agonist binding, selectivity and activation. Biochem. Biophys. Rep. 1, 85–96 (2015).
Rana, S., Sahoo, A. R. & Majhi, B. K. Structural complexes of the agonist, inverse agonist and antagonist bound C5a receptor: insights into pharmacology and signaling. Mol. BioSyst. 12, 1586–1599 (2016).
Schatz-Jakobsen, J. A. et al. Structural and functional characterization of human and murine C5a anaphylatoxins. Acta Crystallogr. Sect. D. 70, 1704–1717 (2014).
Ippel, J. H. et al. Structure of the tyrosine-sulfated C5a receptor N terminus in complex with chemotaxis inhibitory protein of Staphylococcus aureus. J. Biol. Chem. 284, 12363–12372 (2009).
Otto, M. et al. C5a mutants are potent antagonists of the C5a receptor (CD88) and of C5L2: position 69 is the locus that determines agonism or antagonism. J. Biol. Chem. 279, 142–151 (2004).
Rana, S., Sahoo, A. R. & Majhi, B. K. Allosterism in human complement component 5a ((h)C5a): a damper of C5a receptor (C5aR) signaling. J. Biomol. Struct. Dyn. 34, 1201–1213 (2016).
Mery, L. & Boulay, F. Evidence that the extracellular N-terminal domain of C5aR contains amino-acid residues crucial for C5a binding. Eur. J. Haematol. 51, 282–287 (1993).
Klco, J. M., Nikiforovich, G. V. & Baranski, T. J. Genetic analysis of the first and third extracellular loops of the C5a receptor reveals an essential WXFG motif in the first loop. J. Biol. Chem. 281, 12010–12019 (2006).
Kolakowski, L. F. Jr., Lu, B., Gerard, C. & Gerard, N. P. Probing the “message:address” sites for chemoattractant binding to the C5a receptor. Mutagenesis of hydrophilic and proline residues within the transmembrane segments. J. Biol. Chem. 270, 18077–18082 (1995).
Klco, J. M., Wiegand, C. B., Narzinski, K. & Baranski, T. J. Essential role for the second extracellular loop in C5a receptor activation. Nat. Struct. Mol. Biol. 12, 320–326 (2005).
Farzan, M. et al. Sulfated tyrosines contribute to the formation of the C5a docking site of the human C5a anaphylatoxin receptor. J. Exp. Med. 193, 1059–1066 (2001).
Scola, A. M. et al. The role of the N-terminal domain of the complement fragment receptor C5L2 in ligand binding. J. Biol. Chem. 282, 3664–3671 (2007).
Mery, L. & Boulay, F. The NH2-terminal region of C5aR but not that of FPR is critical for both protein transport and ligand binding. J. Biol. Chem. 269, 3457–3463 (1994).
Sreerama, N. & Woody, R. W. Structural composition of β(I)- and β(II)-proteins. Protein Sci. 12, 384–388 (2003).
Buck, M. Trifluoroethanol and colleagues: cosolvents come of age. Recent studies with peptides and proteins. Q. Rev. Biophys. 31, 297–355 (1998).
Andersen, N. H., Liu, Z. & Prickett, K. S. Efforts toward deriving the CD spectrum of a 3(10) helix in aqueous medium. FEBS lett. 399, 47–52 (1996).
Chen, Y. H., Yang, J. T. & Chau, K. H. Determination of the helix and β form of proteins in aqueous solution by circular dichroism. Biochemistry 13, 3350–3359 (1974).
Leitgeb, B. & Tóth, G. Aromatic–aromatic and proline–aromatic interactions in endomorphin-1 and endomorphin-2. Eur. J. Med. Chem. 40, 674–686 (2005).
Ma, J. C. & Dougherty, D. A. The Cation-π Interaction. Chem. Rev. 97, 1303–1324 (1997).
Kawai, M. et al. Structure-function studies in a series of carboxyl-terminal octapeptide analogs of anaphylatoxin C5a. J. Med. Chem. 35, 220–223 (1992).
Tamamis, P. & Floudas, C. A. Elucidating a Key Component of Cancer Metastasis: CXCL12 (SDF-1α) Binding to CXCR4. J. Chem. Inf. Mod. 54, 1174–1188 (2014).
Xu, L., Li, Y., Sun, H., Li, D. & Hou, T. Structural basis of the interactions between CXCR4 and CXCL12/SDF-1 revealed by theoretical approaches. Mol. BioSyst. 9, 2107–2117 (2013).
Jiang, S. J. et al. Peptides derived from CXCL8 based on in silico analysis inhibit CXCL8 interactions with its receptor CXCR1. Sci. Rep. 5, 18638 (2015).
Hou, T., Wang, J., Li, Y. & Wang, W. Assessing the performance of the molecular mechanics/Poisson Boltzmann surface area and molecular mechanics/generalized Born surface area methods. II. The accuracy of ranking poses generated from docking. J. Comput. Chem. 32, 866–877 (2011).
Genheden, S. & Ryde, U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Discov. 10, 449–461 (2015).
Botello-Smith, W. M. & Luo, R. Applications of MMPBSA to Membrane Proteins I: Efficient Numerical Solutions of Periodic Poisson–Boltzmann Equation. J. Chem. Inf. Mod. 55, 2187–2199 (2015).
Monk, P. N., Scola, A. M., Madala, P. & Fairlie, D. P. Function, structure and therapeutic potential of complement C5a receptors. Br. J. Pharmacol. 152, 429–448 (2007).
Gadhe, C. G. & Kim, M. H. Insights into the binding modes of CC chemokine receptor 4 (CCR4) inhibitors: a combined approach involving homology modelling, docking, and molecular dynamics simulation studies. Mol. BioSyst. 11, 618–634 (2015).
Liou, J. W. et al. In silico analysis reveals sequential interactions and protein conformational changes during the binding of chemokine CXCL-8 to its receptor CXCR1. PloS one 9, e94178 (2014).
Leguebe, M. et al. Hybrid molecular mechanics/coarse-grained simulations for structural prediction of G-protein coupled receptor/ligand complexes. PloS one 7, e47332 (2012).
Saleh, N., Ibrahim, P., Saladino, G., Gervasio, F. L. & Clark, T. An Efficient Metadynamics-Based Protocol To Model the Binding Affinity and the Transition State Ensemble of G-Protein-Coupled Receptor Ligands. J. Chem. Inf. Mod. 57, 1210–1217 (2017).
Wacker, D., Stevens, R. C. & Roth, B. L. How Ligands Illuminate GPCR Molecular Pharmacology. Cell 170, 414–427 (2017).
Congreve, M. & Marshall, F. The impact of GPCR structures on pharmacology and structure-based drug design. Br. J. Pharmacol. 159, 986–996 (2010).
Haga, K. et al. Structure of the human M2 muscarinic acetylcholine receptor bound to an antagonist. Nature 482, 547–551 (2012).
Dore, A. S. et al. Structure of the adenosine A(2A) receptor in complex with ZM241385 and the xanthines XAC and caffeine. Structure (London, England: 1993) 19, 1283–1293 (2011).
Cherezov, V. et al. High-resolution crystal structure of an engineered human beta2-adrenergic G protein-coupled receptor. Science (New York, N.Y.) 318, 1258–1265 (2007).
Ward, P. A. The dark side of C5a in sepsis. Nat. Rev. Immunol. 4, 133–142 (2004).
Burg, J. S. et al. Structural biology. Structural basis for chemokine recognition and activation of a viral G protein-coupled receptor. Science (New York, N.Y.) 347, 1113–1117 (2015).
Booth, V., Clark-Lewis, I. & Sykes, B. D. NMR structure of CXCR3 binding chemokine CXCL11 (ITAC). Protein Sci. 13, 2022–2028 (2004).
Gustavsson, M. et al. Structural basis of ligand interaction with atypical chemokine receptor 3. Nat. Commun. 8, 14135 (2017).
Kleist, A. B. et al. New paradigms in chemokine receptor signal transduction: Moving beyond the two-site model. Biochem. Pharmacol. 114, 53–68 (2016).
Ziarek, J. J. et al. Structural basis for chemokine recognition by a G protein-coupled receptor and implications for receptor activation. Sci. Signal. 10, eaah5756 (2017).
Raucci, R., Polo, A., Budillon, A., Colonna, G. & Costantini, S. Conformational analysis of the human chemokine receptor CXCR3. Mol. Immunol. 92, 76–86 (2017).
DeMartino, J. A. et al. The amino terminus of the human C5a receptor is required for high affinity C5a binding and for receptor activation by C5a but not C5a analogs. J. Biol. Chem. 269, 14446–14450 (1994).
Finch, A. M. et al. Biologically active conformer of the effector region of human C5a and modulatory effects of N-terminal receptor binding determinants on activity. J. Med. Chem. 40, 877–884 (1997).
Cook, W. J., Galakatos, N., Boyar, W. C., Walter, R. L. & Ealick, S. E. Structure of human desArg-C5a. Acta Crystallogr. Sect. D 66, 190–197 (2010).
Toth, M. J. et al. The pharmacophore of the human C5a anaphylatoxin. Protein Sci. 3, 1159–1168 (1994).
Mollison, K. W. et al. Identification of receptor-binding residues in the inflammatory complement protein C5a by site-directed mutagenesis. Proc. Natl. Acad. Sci. USA 86, 292–296 (1989).
Ballesteros, J. A. et al. Activation of the β2-Adrenergic Receptor Involves Disruption of an Ionic Lock between the Cytoplasmic Ends of Transmembrane Segments 3 and 6. J. Biol. Chem. 276, 29171–29177 (2001).
Mahalingam, M., Martínez-Mayorga, K., Brown, M. F. & Vogel, R. Two protonation switches control rhodopsin activation in membranes. Proc. Natl. Acad. Sci. 105, 17795–17800 (2008).
Shi, L. et al. β2 Adrenergic Receptor Activation: Modulation of the proline kink in transmembrane 6 by a rotamer toggle switch. J. Biol. Chem. 277, 40989–40996 (2002).
Trzaskowski, B. et al. Action of molecular switches in GPCRs–theoretical and experimental studies. Curr. Med. Chem. 19, 1090–1109 (2012).
Rana, S. & Baranski, T. J. Third extracellular loop (EC3)-N terminus interaction is important for seven-transmembrane domain receptor function: implications for an activation microswitch region. J. Biol. Chem. 285, 31472–31483 (2010).
Palczewski, K. et al. Crystal structure of rhodopsin: A G protein-coupled receptor. Science (New York, N.Y.) 289, 739–745 (2000).
Scheerer, P. et al. Crystal structure of opsin in its G-protein-interacting conformation. Nature 455, 497–502 (2008).
Rasmussen, S. G. et al. Structure of a nanobody-stabilized active state of the beta(2) adrenoceptor. Nature 469, 175–180 (2011).
Xu, F. et al. Structure of an agonist-bound human A2A adenosine receptor. Science (New York, N.Y.) 332, 322–327 (2011).
Kruse, A. C. et al. Activation and allosteric modulation of a muscarinic acetylcholine receptor. Nature 504, 101–106 (2013).
Visiers, I., Ballesteros, J. A. & Weinstein, H. In Methods Enzymol. 343, 329–371 (Academic Press, 2002).
Greenfield, N. J. Using circular dichroism spectra to estimate protein secondary structure. Nat. Protoc. 1, 2876–2890 (2006).
Morris, G. M. et al. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 30, 2785–2791 (2009).
Hess, B., Kutzner, C., van der Spoel, D. & Lindahl, E. GROMACS 4: Algorithms for Highly Efficient, Load-Balanced, and Scalable Molecular Simulation. J. Chem. Theory Comput. 4, 435–447 (2008).
Kandt, C., Ash, W. L. & Tieleman, D. P. Setting up and running molecular dynamics simulations of membrane proteins. Methods (San Diego, Calif.) 41, 475–488 (2007).
Kumari, R., Kumar, R. & Lynn, A. g_mmpbsa - a GROMACS tool for high-throughput MM-PBSA calculations. J. Chem. Inf. Model. 54, 1951–1962 (2014).
Robertson, N. et al. Structure of the complement C5a receptor bound to the extra-helical antagonist NDT9513727. Nature 553, 111–114 (2018).
Acknowledgements
This research is supported by the SERB (EMR/2016/000681), DST, New Delhi. Use of BRAF facility at CDAC, Pune is highly appreciated. Use of CD spectrophotometer at ACTREC, Mumbai and Bruker 800 MHz NMR at TIFR, Mumbai is highly acknowledged.
Author information
Authors and Affiliations
Contributions
S.R. conceived the project and designed the approach for executing the project. S.R. and A.R.S. performed the computational modeling. S.R. accumulated the computational data and both S.R. and A.R.S. analyzed the data. S.R. and R.M. accumulated the experimental data and analyzed. S.R. wrote the manuscript. S.R. provided necessary support to both A.R.S. and R.M. in preparing the figures. A.R.S and R.M. did the referencing.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sahoo, A.R., Mishra, R. & Rana, S. The Model Structures of the Complement Component 5a Receptor (C5aR) Bound to the Native and Engineered hC5a. Sci Rep 8, 2955 (2018). https://doi.org/10.1038/s41598-018-21290-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-21290-4
- Springer Nature Limited