
Abstract
Evolutionary relationships in the black-lip pearl oyster Pinctada margaritifera which is highly valued for pearl production remain poorly understood. This species possesses an 18,000 km Indo-Pacific natural distribution, and its current description includes six subspecies defined exclusively on morphological characters. To evaluate its taxonomic identity using molecular data, 14 populations in both the Indian and Pacific Oceans (n = 69), and the congeneric taxa P. maxima and P. mazatlanica (n = 29 and n = 10, respectively) were sampled. Phylogenomic reconstruction was carried out using both 8,308 genome-wide SNPs and 10,000 dominant loci (DArTseq PAVs). Reconstructions using neighbour-joining (Nei’s 1972 distances), maximum likelihood and Bayesian approaches all indicate that the taxonomy of P. margaritifera is quite complex, with distinct evolutionary significant units (ESUs) identified within Tanzanian and Iranian populations. Contrastingly, phylogenies generated for Pacific Ocean oysters resolved a large monophyletic clade, suggesting little support for two current morphological subspecies classifications. Furthermore, P. mazatlanica formed a basal clade closest to French Polynesian P. margaritifera, suggesting it may be conspecific. Collectively, these findings provide evidence that P. margaritifera comprises a species complex, perhaps as a result of population fragmentation and increased divergence at range limits.
Similar content being viewed by others

Introduction
The black-lip pearl oyster Pinctada margaritifera is a marine bivalve mollusc that has a broad Indo-Pacific distribution spanning ~18,000 km (Fig. 1), and is highly valued for cultured pearl and pearl shell production1,2. While several studies have examined contemporary population structure and connectivity in P. margaritifera 3,4,5,6,7,8,9,10,11,12,13, none have investigated its range-wide evolutionary history and species identity using molecular data. An understanding of the taxomomic status and genetic structure of this bivalve is important for management of the species given its community and economic value, but also more broadly to elucidate the drivers of evolutionary relationships between widely separated populations of an extensively-distributed marine invertebrate14,15,16.

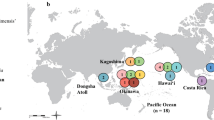
Natural distributions and sampling locations from where specimens of P. margaritifera (n = 69; solid black circles and black circles superimposed with yellow stars), P. maxima (n = 29; yellow circles and black circles superimposed with yellow stars) and P. mazatlanica (n = 10; solid red circle), were collected. The approximate known distributions of each species are presented in light grey (P. margaritifera), medium grey (P. maxima) and dark grey (P. mazatlanica) respectively; and adapted from Wada and Tëmkin2. Site codes represent the following locations; TAN: Mafia Island and Mtwara, Tanzania; IRN: Hendorabi Island, Iran; TAI: Checheng, Taiwan (P. margaritifera), HNI: Hainan Island, China (P. maxima); VNM: Nha Trang (P. margaritifera) and Phú Quốc (P. maxima), Vietnam; IND: Manado, Indonesia; ABR: Abrolhos Islands, Australia; BRM: Broome, Australia; TRS: Torres Strait, Australia; GBR/HB: Great Barrier Reef (P. margaritifera) and Hervey Bay (P. maxima), Australia; PNG: Kavieng, Papua New Guinea; SOL: Gizo Island, Solomon Islands; FJI: Kadavu, Savusavu, Lau and the Yasawa group, Fiji Islands; TON: Tongatapu, Tonga; CKI: Manihiki Atoll, Cook Islands; FRP: Arutua, French Polynesia and MEX: Guaymas, Mexico. This map was produced using ArcGIS release 10.276.
Species-level taxonomic relationships in the genus Pinctada had remained unresolved, particularly because many earlier descriptions either heavily, or exclusively relied on morphological descriptions of shells, which are now known to display considerable phenotypic, developmental and environmental plasticity2,17,18. With the increasing use of molecular tools to resolve both higher and lower level relationships in this taxon, clarity in the taxonomic identity of several species important for cultured pearl production is being established. An example of this which is still pending ultimate resolution, is the status of the P. fucata/martensii/radiata/imbricata (Akoya pearl oyster) species complex. It is currently recognised that this group may comprise one cosmopolitan, circum-globally distributed species, possessing a very high degree of intraspecific variation across its range1,2.
Given the phenotypic and adaptive plasticity, as well as morphological diversity present in Pteriid pearl oysters17,18,19, and that shell shape and size differences are apparent between populations of P. margaritifera 2,17,20, molecular data are required to elucidate Evolutionary Significant Units (ESUs)21 in this species. For the purpose of this study, we define ESUs after Funk et al.21 and Crandall et al.22, as being a classification of populations possessing substantial reproductive isolation and/or adaptive differences, so that the population represents a significant evolutionary component of the species. Due to the degree of molecular intraspecific variation documented in P. margaritifera, localised studies have suggested that it might constitute a species complex2,20,23,24. The term species complex is used to describe populations where a group of organisms may represent more than one species, and/or where species boundaries cannot be discerned with certainty25,26,27. In the case of P. margaritifera, the disagreement between current molecular14 and morphological2,17,20 information could indicate the presence of a species complex involving several discrete ESUs, which require identification and delimitation.
Particular studies which point to a P. margaritifera species complex include a thorough morphological and molecular characterisation of the superfamily Pterioidea by Tëmkin23, who reported that P. mazatlanica formed an unresolved clade with P. margaritifera, suggesting their conspecificity. In the Persian Gulf, Ranjbar, et al.20 using mitochondrial COI data discovered that P. m. var. persica formed a divergent ESU, and suggested its reclassification as P. persica, while P. m. var. zanzibarensis from Mauritius formed a basal clade to French Polynesian and Japanese specimens24.
Descriptions of P. margaritifera include a total of six subspecies16, along with a former seventh (P. margaritifera mazatlanica), that has since been elevated as a distinct species; P. mazatlanica (Hanley, 1856)2,24,28,29. The six subspecies of P. margaritifera are described exclusively on the basis of morphological characters16,28, and are closely associated with their geographic type locations (see Table 1 for a summary). In the Pacific basin, Hawaiian populations are known as P. margaritifera var. galstoffi, Cook Islands and French Polynesian individuals P. m. var. cummingi, and all Central and Western Pacific specimens P. m. var. typica. Indian Ocean populations are represented by P. m. var. persica (Persian Gulf), P. m. var. erythraensis (Red Sea) and P. m. var. zanzibarensis (East Africa, Madagascar and Seychelle Islands). Recent efforts to characterise distribution-wide population genetic structure in P. margaritifera 14 indicate a high degree of homogeneity within the Pacific basin, bringing into question the current subspecies classifications of P. m. var. typica and P. m. var. cummingi. Conversely, populations examined from the Indian Ocean displayed substantial vicariance from Pacific Ocean demes, possibly supporting the existence of distinct ESUs in that region.
The goals of the present study were to use genome-wide SNP and dominant marker data to resolve species-level phylogenetic relationships and the presence of ESUs within P. margaritifera, and provide information on its taxonomic identity. This investigation is the first of its kind to assess individuals spanning the distributional range of the black-lip pearl oyster, and the data generated has high utility for informing regional spatial marine management strategies for conservation and aquaculture efforts.
Results
Population and species-level relationships were reconstructed using neighbour-joining (NJ), maximum-likelihood (ML) and Bayesian inference approaches. Additionally, Nei’s (1973) minimum (D m) and unbiased (D, 1972) genetic distances were computed for the SNP and dominant marker (DArTseq PAV) datasets respectively (see methods).
Genotyping and SNP/PAV filtering
The raw SNP dataset contained a total of 23,599 SNPs genotyped across all 118 individuals, at call rates ranging from 20–100%. The first filtering step undertaken to remove duplicate SNPs within the same sequence tag resulted in the removal of 5,634 SNPs (24% loss), after which the dataset was filtered for call rate (>70%), average PIC (>1%), MAF (>2%) and average repeatability (>95%). A total of 2 loci were monomorphic within a single taxon, and subsequently removed. These steps collectively resulted in the retention of 8,308 SNPs, across 107 individuals. The raw PAV dataset contained a total of 42,159 variant scores across all individuals, at call rates ranging from 90–100%. No duplicate genomic loci were represented in the dataset, and following filtering for call rate (100%), MAF (>10%) and average reproducibility (>98%), 12,212 PAVs remained. No individuals were lost due to poor call rates as with the SNP loci, however the dataset was trimmed to 10,000 PAVs to maximise computational efficiency. All 118 taxa were included in the analyses, and Pt. penguin retained as the outgroup taxon.
Phylogenomic reconstruction
NJ and ML approaches
Both NJ and ML reconstructions (Figs 2 and 3) produced nearly identical topologies, demonstrating clear separation of P. margaritifera and P. maxima into species-level clades. Interestingly, within the larger monophyletic P. margaritifera clade, all individuals of P. mazatlanica formed a single basal group with short internal branch lengths, and node support for this division in both NJ and ML reconstructions was high (100). Similarly, all Tanzanian with Iranian P. margaritifera also formed a distinct monophyletic group, which was basal to the larger monophyletic clade resolved for all other P. margaritifera samples. As with the P. mazatlanica clade, high bootstrap node support (100) and short internal branch lengths were resolved for this division. Overall, the shallow levels of divergence documented among Pacific Ocean P. margaritifera populations suggests their conspecificity, and consequently does not support the current subspecies classifications of P. margaritifera var. typica and P. m. var. cummingii. Monophyly of P. mazatlanica, as well as of the combined Iranian and Tanzanian populations of P. margaritifera within the larger P. margaritifera cluster, is suggestive of discrete ESUs present in these locations. Examination of Iranian and Tanzanian specimens in isolation indicates a paraphyletic relationship between the two groups, and underscores the need for further work to resolve evolutionary relationships among Indian Ocean black-lip pearl oyster populations.

Neighbourjoining tree generated in MEGA660 using 8,308 SNPs based on Nei's 1972 genetic distances. P. maxima was used as the outgroup taxon. Values reported at nodes indicate bootstrap support at a threshold higher than 60 (1,000 bootstraps used). Clades for P. mazatlanica and Iranian with Tanzanian P. margaritifera are highlighted in blue and red respectively.

Maximumlikelihood tree generated using the SNPhylo package61, with 100,000 bootstraps and 8,308 SNPs. P. maxima was used as the outgroup taxon. Values reported at nodes indicate bootstrap support at a threshold higher than 65. Clades for P. mazatlanica and Iranian with Tanzanian P. margaritifera are highlighted in blue and red respectively.
Bayesian approach for PAV dataset
A total of 480,002 trees were sampled from both runs, and following discard of the burn-in set, 414,101 credible trees remained for calculation of posterior probabilities. The final average standard deviation of split frequencies achieved was 0.013, with an average potential scale reduction factor (PSRF) for parameter values of 1.000. Within the final set of credible trees, cut-off thresholds of individual (p = 0.000) and cumulative (P < 0.009) posterior probabilities were implemented, to select 1,141 highly credible trees with which to construct a consensus phylogram (Fig. 4). The reconstruction resolved three major groups, corresponding to established species-level divisions of P. fucata martensii with P. imbricata (Akoya species complex), P. margaritifera and P. maxima. As with the NJ and ML reconstructions, two distinct monophyletic groups were resolved within the larger P. margaritifera clade. However, for the sub-group which included P. mazatlanica; French Polynesian, Cook Islands, Fijian and Tongan P. margaritifera were found to nest together. Similarly, all Abrolhos Islands individuals clustered alongside the Tanzanian and Iranian individuals. It is possible that due to the lower discriminating power of the PAV dataset compared to SNPs at the species level, these more distal relationships were not able to be resolved. This may also explain the similar branch tip lengths observed for all taxa in the final phylogram.

Bayesian reconstruction generated using 10,000 DArTseq PAVs in MrBayes v.3.269. The consensus tree reported here was generated from 1,141 of the most credible set of trees (p = 0.01), using a 50% majority consensus rule. Posterior probabilities ≥0.50 for node support are indicated below the branches. Clades for P. mazatlanica and Iranian with Tanzanian P. margaritifera are highlighted in blue and red respectively. Representative specimens of P. margaritifera (A; Savusavu, Fiji Is.), P. mazatlanica (B; Guaymas, Mexico) and P. maxima (C; Bali, Indonesia), are shown to illustrate gross differences in shell morphology.
Assessment of genetic distances between population groups and species
Genetic distance estimates differed between the SNP and PAV datasets due to the different marker systems (Table 2), although overall trends for the majority of pairwise comparisons were similar. Nei’s (1973) minimum genetic distances (D m) values obtained from the SNP dataset indicated that P. maxima from all locations were significantly and substantially divergent from all P. margaritifera and P. mazatlanica populations, (D m > 0.22; p < 0.05). Among sampling sites for P. maxima, estimates ranged from 0.023 (Hervey Bay and Vietnam) to 0.069 (Hervey Bay and Solomon Islands), confirming their conspecificity. Pacific Ocean populations of P. margaritifera were largely homogenous (D m = 0.006–0.038; p < 0.05), and only showed signs of divergence when compared against Indian Ocean populations from Tanzania (D m = 0.076–0.089) and Iran (D m = 0.069–0.113). Interestingly, all P. mazatlanica estimates (D m = 0.057–0.133; p < 0.05), fell within the range limits of all between-site P. margaritifera estimates (D m = 0.006–0.113; p < 0.05).
Nei’s (1972) unbiased genetic distance (D) estimates also indicated clear separation of P. maxima from P. margaritifera (D = 0.126–0.296; p < 0.05), and P. mazatlanica (D = 0.165–0.307; p < 0.05) samples, in keeping with the trend observed for D m values. Similarly, broad-scale divergence between P. mazatlanica and P. margaritifera was not apparent, although the Abrolhos, Tanzanian and Iranian populations did display slightly increased values (0.051, 0.068 and 0.047, respectively), compared to the range estimated for Pacific populations (D = 0.034–0.038, p < 0.05), reflecting divergence between ocean basins.
Discussion
The current study represents the most comprehensive evaluation of evolutionary relationships in the black-lip pearl oyster, incorporating two independent high density genome-wide marker sets and sample representation from the entire species distribution. It is clear that the taxonomy of P. margaritifera is more complex than previously thought, suggesting that its status may require re-examination.
The presence of morphological differences17,28 between locations in P. margaritifera is unsurprising, considering that the species distribution spans over 18,000 km across heterogeneous habitats2,14, and that bivalve molluscs can display very high levels of phenotypic plasticity24,30. The high degree of morphological variation and overlapping geographical distributions of many bivalves due to high larval dispersal capability, however, can make the delimitation of population and species boundaries problematic, highlighting the need for molecular information to resolve these differences. The data presented in the current study indicates varying degrees of support for the current morphological subspecies classifications for P. margaritifera.
In the Pacific Ocean, the existence of the morphological subspecies P. m. var. typica and P. m. var. cummingii is questionable. Previous work14 to examine genetic structure in the Pacific basin supports this finding, as results showed very high gene flow between P. margaritifera populations separated by several thousands of kilometres. While five discrete Pacific Ocean stocks were identified in that study, populations remained sufficiently undifferentiated in the current study to resolve any subspecies groupings. It is possible that the morphological subspecies recorded in the Pacific17,28 may be the result of local adaptation and habitat differences. Lal et al.14 detected signatures of selection between all five stocks of P. margaritifera identified in the Pacific, and given the adaptive capacity of many bivalve taxa31,32, it is possible that local selective pressures may give rise to the morphological variability observed. This information was used as the basis for recommending separate regional fishery management of these populations, applying criteria established by Funk et al.21 and Crandall et al.14,21,22.
Despite considerable effort, it was not possible to obtain specimens of Hawaiian P. margaritifera for inclusion in the current study, and therefore examination of the status of the third and last Pacific Ocean subspecies described in the literature; P. m. var. galstoffi 2,33, remained out of reach. However, given the degree of genetic homogeneity between other Pacific populations across similar spatial scales, it is possible that Hawaiian populations could also display shallow levels of divergence, with the strength of migration between these locations determining their degree of isolation. A future study incorporating these populations will be able to address this question.
The discovery that P. mazatlanica specimens comprised a basal group nested within P. margaritifera is interesting, given the lack of an overlap in known species range limits2. In the light of the substantial trans-Pacific dispersal ability of this species as discussed earlier, it is entirely possible that recruits originating from eastern Pacific populations could find their way to the Gulf of California, thus maintaining gene flow. Early descriptions of P. mazatlanica using morphological characters had in fact classified it as a subspecies of P. margaritifera (P. margaritifera mazatlanica; and it was noted that its shell morphology appears to be an intermediate form between P. margaritifera and P. maxima 2,28,29,34).
Assessment of the population genetic structure of P. mazatlanica still remains unresolved. Arnaud, et al.35 discovered that mtDNA nucleotide divergence (COI and 12s rRNA) between locations ranged from 0.12–1.3%, while divergence from three individuals of P. margaritifera reached ~4%. Importantly, divergence at mtDNA loci across the broader distribution of P. margaritifera remains unknown. Subsequent examinations of species-level taxonomy within the genus Pinctada 23 using multiple nuclear and mitochondrial loci, found that P. mazatlanica formed an unresolved clade with P. margaritifera, in concordance with a later reconstruction by Cunha, et al.24, although in their study the latter authors state that results supported the present species-level classification. The genome-wide data presented here provides further strong evidence that these taxa might be conspecific. These results also support a further observation by Cunha, et al.24, which suggests that French Polynesian P. margaritifera are more closely related to P. mazatlanica than Indian Ocean specimens. This pattern of differentiation in the data also extended to populations located in the western Pacific in the data, indicating that a clinal effect related to geographic separation may be present, as seen in Crassostrea spp. oysters30.
The lower than expected levels of divergence separating P. mazatlanica from P. margaritifera, despite the morphological differences between the two taxa, raises several questions as to the processes driving their divergence, or maintaining genetic structure. One possibility is that P. mazatlanica is a distinct ESU within P. margaritifera (see criteria outlined by Funk et al.21 for evaluating ESUs), and that its morphological differences are a consequence of phenotypic and adaptive plasticity. Another scenario is that P. mazatlanica may be undergoing incipient speciation36, which has been documented in other broadcast spawning marine invertebrates, including bivalves37,38. To resolve these questions, a fine-scale study incorporating range-wide samples of P. mazatlanica, together with specimens from marginal eastern Pacific P. margaritifera populations (e.g. Hawaii, Kiribati and French Polynesia), is required.
All three phylogenetic reconstruction methods applied in the current study resolved a pattern of paraphyly for Iranian and Tanzanian specimens respectively, which corresponds with the morphological subspecies descriptions of P. m. var. zanzibarensis (Zanzibar, Madagascar and eastern African coastline) and P. m. var. persica (Persian Gulf only)28. This difference in evolutionary trajectories from Pacific Ocean specimens also explains the divergence observed in examination of range-wide population genetic structure in P. margaritifera 14. Separation of Persian Gulf populations as a distinct ESU was also detected by Ranjbar, et al.20, who suggested their reclassification as a species in its own right named P. persica. While the findings here do not indicate that specimens from Iran were sufficiently divergent to warrant this elevation in taxonomic rank (see criteria outlined by Crandall et al.22), further research is required to ascertain the degree of isolation of Persian Gulf populations from specimens living in the Red Sea (i.e. P. m. erythraensis), as well as the broader Indian Ocean.
It is clear that dense sampling of the Indian Ocean is required, as the present study was only able to assess three marginal populations sampled at its geographic limits. At the eastern extent of the Indian Ocean, specimens collected from the Abrolhos Islands (Western Australia) formed a weakly monophyletic clade, which nested closest to the Tanzanian and Iranian specimens, suggesting a restriction in gene flow between the Indian and Pacific Oceans14. This observation may also indicate the presence of an Indian-Pacific Ocean genetic break, however a future study incorporating a range of samples from both the Indian Ocean basin centre and periphery is required for confirmation. Additionally, Lal et al.14 recommended separate fishery management given the levels of divergence observed between these Indian Ocean populations, and the findings presented here add weight to the hypothesis that they have evolved as separate lineages. The criteria outlined by Crandall et al.22 are particularly useful for application of fishery management action for P. margaritifera, as they permit the preservation of adaptive diversity and evolutionary processes across the species’ range.
The possibility that P. margaritifera may constitute a species complex has been suggested by a number of studies, where distinct ESUs have been discovered during localised investigations of genetic structure2,20,24. The current study incorporating samples spanning the extent of the species distribution contributes further evidence to support this taxonomic classification, and highlights the need for further research to investigate segments of the species range that were not able to be sampled.
Of the five major pearl oyster species which are the focus of commercial aquaculture efforts1,2, the only species distributed over a natural range comparable to that of P. margaritifera, is the Akoya complex; P. fucata/martensii/radiata/imbricata. The Akoya complex is characterised by substantial intra- and interpopulation morphological variability, local geographic isolation of some populations, human introductions, hybridisation and inconsistent taxonomic practice2,23. Originally, three distinct species were recognised; P. imbricata (Röding, 1798; western Atlantic), P. radiata (Leach, 1814); eastern Indian Ocean and Red Sea) and P. fucata (Gould, 1850; Indo-Pacific). Japanese populations were recognised as a distinct species (P. martensii; Dunker, 1872) or subspecies (P. fucata martensii 2,39,40). These classifications were on the basis of questionable morphological characters, and subsequent molecular analyses (see Wada and Tëmkin2 and Tëmkin23 for summaries), revealed that Australian, south-east Asian and Japanese populations form a monophyletic group that is highly likely to be conspecific. Furthermore, mating experiments have supported the conspecificity of south-east Asian and Japanese populations41, and the current consensus is that the Akoya complex may be a cosmopolitan, globally distributed species, possessing substantial intraspecific variation2.
Given the similarities in intraspecific morphological variability and the extensive natural distribution of P. margaritifera when compared with members of the Akoya species complex, in the light of the current study, it is certainly feasible that P. margaritifera (as it is currently known) might comprise a species complex. Ultimate resolution of its taxonomic identity however, will require a large-scale, systematic, molecular and morphological characterisation of samples collected across the entire natural distribution.
The current study is the first range-wide examination of evolutionary relationships in the black-lip pearl oyster using genome-wide molecular markers. It identifies the presence of discrete ESUs within the species distribution, and presents evidence for the conspecificity of P. mazatlanica and P. margaritifera. Collectively, these findings provide early indications that P. margaritifera may constitute a species complex, and highlight the requirement for further range-wide investigations to fully resolve its taxonomic status. This information is valuable not only for the regional fishery management and aquaculture of P. margaritifera, as discrete ESUs require independent management14,22, but also for a broader understanding of the ecology and evolution of similarly wide-ranging marine invertebrates.
Methods and Materials
Specimen collection, tissue sampling and DNA extraction
Specimens between 7–18 cm in dorso-ventral measurement (DVM) were collected from several sites spanning the natural distributions of five Pteriid pearl oyster species (Fig. 1). Black-lip pearl oysters (Pinctada margaritifera; n = 69) were sampled from 14 sites, which for the Indian Ocean included five samples from each of two Tanzanian sites; (Mafia Island and Mtwara), and Post Office Island in the Abrolhos Islands group, Western Australia; with four samples from the Persian Gulf (Hendorabi Island, Iran). Five oysters each were also sampled from Western and Central Pacific Ocean populations, including Checheng, Taiwan; Nha Trang, Vietnam; Manado, Indonesia; Kavieng, Papua New Guinea; Gizo Island in the Solomon Islands; Arlington, Sudbury and Tongue Reef systems within the Great Barrier Reef (GBR), Australia; Kadavu, Savusavu, Lau and the Yasawa group, Fiji Islands; and Tongatapu, Tonga; respectively. In the Eastern Pacific, five oysters each were collected from Manihiki Atoll in the Cook Islands, and Arutua, French Polynesia respectively.
Silver-lip pearl oyster (P. maxima, n = 29) specimens were obtained from Hainan Island, China; Phú Quốc, Vietnam; Broome, Western Australia; Thursday Island in the Torres Straits, Australia; Hervey Bay, eastern Australia and Gizo Island in the Solomon Islands. Five samples were obtained from each of these sites, with the exception of Hervey Bay, where four oysters were collected. P. maxima was included here as it is the closest known relative of P. margaritifera 42. Panamanian pearl oyster (P. mazatlanica, n = 10) specimens were also collected from a single site at Guaymas, Mexico, while Akoya pearl oysters (P. fucata martensii, n = 1 and P. imbricata, n = 3) were collected from Okinawa, Japan and Port Stephens, Australia; respectively. These additional taxa were selected as they were recovered by Yu and Chu42 and Cunha, et al.24 as separate clades to P. margaritifera, and consequently included to provide a basis for comparison when using conserved dominant loci in the phylogenomic analysis. Penguin’s winged pearl oyster Pteria penguin was selected as the outgroup taxon, with specimens (n = 5) obtained from Savusavu, Fiji Islands.
Proximal mantle and adductor muscle tissues (3 and 6 cm respectively) were removed from each specimen and transferred to tubes containing 20% salt saturated dimethyl sulfoxide (DMSO-salt) preservative43. Genomic DNA was extracted using a modified cetyl trimethyl ammonium bromide (CTAB) chloroform/isoamyl alcohol protocol with a warm (30 °C) isopropanol precipitation44. To clean up all DNA extractions, a Sephadex G5045 spin column protocol was used prior to quantification with a Nanodrop 1000 Spectrophotometer (Thermo Scientific). All samples were subsequently normalised at 100 ng/μL in a 50 μL final volume, and submitted for DArTseq. 1.0 genotyping at Diversity Arrays Technology PL, Canberra, ACT, Australia.
DArTseq. 1.0 library preparation and sequencing
Diversity Arrays Technology (DArT PL) proprietary genotyping by sequencing (DArTseq) reduced-representation libraries were prepared as described by Kilian, et al.46 and Sansaloni, et al.47, with a number of modifications for P. margaritifera. Briefly, genome complexity reduction was achieved with a double restriction digest, using a PstI and SphI methylation-sensitive restriction enzyme (RE) combination, in a joint digestion-ligation reaction at 37 °C for 2 hr with 150–200 ng gDNA. Because P. margaritifera, like other bivalve species, is highly polymorphic12,48, highly repetitive genomic regions were avoided and low copy regions more efficiently targeted for sequence capture with the use of methylation-sensitive REs49.
Custom proprietary barcoded adapters (6–9 bp) were ligated to RE cut-site overhangs as per Kilian, et al.46, with the adapters designed to modify RE cut sites following ligation, to prevent insert fragment re-digestion. The PstI-compatible (forward) adapter incorporated an Illumina flowcell attachment region, sequencing primer sequence and a varying length barcode region46,50. The reverse adapter also contained a flowcell attachment region, and was compatible with the SphI cut-site overhang. Samples were processed in batches of 94, with 15% of all samples in a batch randomly selected for replication, to provide a basis for assessing region recovery and genotyping reproducibility. Target “mixed” fragments50, containing both SphI and NlaIII cut-sites were selectively amplified using custom designed primers for each sample, under the following PCR conditions: initial denaturation at 94 °C for 1 min, then 30 cycles of 94 °C for 20 s, 58 °C for 30 s and 72 °C for 45 s, followed by a final extension step at 72 °C for 7 min. Amplified samples were subsequently cleaned using a GenElute PCR Clean-up Kit (Sigma-Aldrich, cat.# NA1020-1KT), on a TECAN Freedom EVO150 automated liquid handler.
To examine fragment size concordance and digestion efficiency, all samples were visualised on a 0.8% agarose gel stained with EtBr, and quantified using the ImageJ software package51. Samples which did not appear to have undergone complete digestion and/or amplification were removed from downstream library preparation. A total of 288 samples were normalised and pooled using an automated liquid handler, at equimolar ratios for sequencing in single lanes on the Illumina HiSeq. 2500 platform. After cluster generation and amplification (HiSeq SR Cluster Kit V4 cBOT, cat.# GD-401-4001), 77 bp single-end sequencing was performed at the DArT PL facility in Canberra, Australia.
Sequence quality control, marker filtering and genotype calling at DArT PL
Raw reads obtained following sequencing were processed using Illumina CASAVA v.1.8.2 software for initial assessment of read quality, sequence representation and generation of FASTQ files. Filtered FASTQ files were then supplied to the DArT PL proprietary software pipeline DArTtoolbox, which performed further filtering, variant calling and generated final genotypes in sequential primary and secondary workflows. Within DArTtoolbox, the primary workflow first involved the package DArTsoft14 to remove reads with a quality score <25 from further processing, and apply stringent filtering to the barcode region of all sequences to increase confidence in genomic region recovery. Individual samples were then de-multiplexed by barcode, and subsequently aligned and matched to catalogued sequences in both NCBI GenBank and DArTdb custom databases to check for viral and bacterial contamination, with any matches removed from further processing.
The secondary workflow employed the DArTsoft14 and KD Compute packages along with the DArTdb database, to identify polymorphisms by aligning identical reads to create clusters across all individuals sequenced. These clusters were then catalogued in DArTdb, and matched against each other to create reduced-representation loci (RRL), based on their degree of similarity and size. SNP and reference allele loci were identified within clusters and assigned DArT scores (“0” = reference allele homozygote, “1” = SNP allele homozygote and “2” = heterozygote), based on their frequency of occurrence. To ensure robust variant calling, all monomorphic clusters were removed, SNP loci had to be present in both allelic states (homozygous and heterozygous) and a genetic similarity matrix produced using the first 10,000 SNPs called was used to assess technical replication error52. Gene duplications were eliminated by excluding clusters containing tri-allelic or aberrant SNPs and overrepresented sequences.
Presence-absence variant (PAV) markers (termed SilicoDArT loci) were also identified using restriction site-associated (RAD) fragments recovered in the sequence data. SilicoDArTs were scored in a binary fashion, with “1” = RAD fragment presence, “0” = RAD fragment absence and “-“ = insufficient counts re-classified as “1”; indicating a hemizygote state. DArTseq PAV markers can be considered to be genome-wide “dominant” markers53,54,55, and were called based on a minimum reproducibility of 95%. Once SNP and PAV markers had been confidently identified, each locus was assessed in the KD Compute package for homozygote and heterozygote call rate, frequency, polymorphic information content (PIC), average SNP count, read depth and repeatability, before final genotype scores were supplied by DArT PL.
Following the receipt of genotype data from DArT PL, the SNP dataset was initially filtered using a custom Python script (https://github.com/esteinig/dartQC). This script retained only a single informative SNP (determined by call rate) at each genomic locus, and then filtered all SNPs at a Minor Allele Frequency (MAF) of 2%. Final filtering of the SNP dataset was by call rate (>70%), read depth (>4) and reproducibility (>95%). A total of 11 individuals across 3 taxa; (a single P. margaritifera individual sampled from Iran, along with all Pt. penguin, P. imbricata and P. fucata martensii specimens), were excluded from the final dataset due to poor call rates (<1%). These taxa were the most divergent compared to all others considered for analysis, and likely experienced poor genotyping coverage due to severe allelic dropout. Consequently, all analyses using the SNP dataset were assigned P. maxima as the outgroup. PAV markers were filtered manually, first to retain the most informative marker at each genomic locus, and then in the order of call rate (100%), MAF (>9.6%), reproducibility (>98%) and read depth (>113).
Assessment of differentiation between population groups and species
Nei’s (1973) minimum genetic distances (D m) between populations were computed in Genetix v.4.05.2 with 1,000 permutations56 for the SNP dataset, while for the PAV dataset, Nei’s unbiased (1972) genetic distances (D) were calculated after Lynch and Milligan57, using the AFLP-SURV v.1.0 package and 10,000 permutations58,59. Estimates were calculated for both the SNP and PAV datasets, to permit comparison between the two marker types. Computations using the PAV dataset had to be limited to 5,000 loci, as this was the maximum number of sites able to be handled by the software. Random sub-sampling of different sets of 5,000 loci from the larger dataset, and recalculation of genetic distances ensured estimates remained unaffected.
Phylogenomic reconstruction
Population and species-level relationships were reconstructed using neighbour-joining (NJ), maximum-likelihood (ML) and Bayesian inference approaches (see Supplementary Figure 1 for a summary). The NJ tree was constructed in the MEGA6 software package60. A matrix of Nei’s (1972) genetic distances calculated in Genetix v.4.0556 was used as input, and the consensus tree generated over 1000 bootstraps using the SNP dataset. The outgroup taxon selected was P. maxima, as the more distant outgroup of Pt. penguin was highly divergent from P. margaritifera, and as a result received very low genotyping coverage in the SNP dataset (>99% missing data, see results).
Additionally, the SNPhylo61 and RAxML v8.262 packages were both used to perform ML analyses on the SNP dataset, with RAxML only for the PAV dataset. As the SNP dataset had already been filtered, for SNPhylo computations, the -r flag was selected which bypassed filtering options for low SNP sample coverage and missing data, and the -l flag set at 1.0 to ignore filtering for linkage disequilibrium (LD) for the final analysis. Preliminary runs were performed both with and without LD pruning to ascertain if this effected changes in tree topology, and no rearrangement of higher-level groups was observed. The transition/transversion ratio was set at 2.0 (−T flag), empirical base frequencies used (−F flag), and constant rate variation set among sites (−R flag). All other options remained at their default settings. SNPhylo first performs a multiple sequence alignment by MUltiple Sequence Comparison by Log-Expectation (MUSCLE; ref.63), and generates a ML tree using the DNAML program in the PHYLIP package64, with bootstrap support provided by the R package phangorn 65. The final tree was generated following 100,000 bootstraps, with P. maxima used as the outgroup taxon. RAxML analyses used the site-specific heterogeneity models66 ASC_GTRGAMMA[X] (for SNPs) and ASC_BINCAT[X] (for PAVs), with the following options selected: ascertainment bias correction set to ‘Lewis’, the rapid bootstrap algorithm with ‘autoMRE’ option enabled and retention of the best ML tree62,67,68. All RAxML reconstructions showed nearly identical topologies for the SNP dataset, and supported the relationships recovered by the Bayesian reconstruction for the PAV dataset.
Bayesian inference of phylogenetic relationships was carried out using only the PAV dataset, with the MrBayes v3.2 package69,70,71 and parameter settings were adapted after Koopman, et al.72. The analysis incorporated two runs of 60,000,000 generations each, with each run comprising 4 independent chains. A temperature of 0.10 was set for the heated chains, with a sampling frequency of 1000 and burn-in fraction of 20%. The burn-in threshold was selected on the basis that both independent runs had achieved convergence (i.e. stable log likelihood values reached for all sampled trees, gauged by the average standard deviation of split frequencies). Convergence was also independently assessed using Tracer v.1.673. The Dirichlet prior for state frequencies was set at (90, 10), matching the frequencies of “0” and “1” PAV scores present in the dataset. The outgroup taxon set incorporated all individuals of Pteria penguin, together with the additional taxa P. imbricata and P. fucata martensii. Inclusion of these samples became possible as the PAV dataset was more informative compared to the SNP dataset, for these more evolutionarily distant specimens.
The final trees for the Bayesian analysis were generated by selecting only those post-burn in trees found with the highest individual and cumulative posterior probabilities (p = 0.000 and P < 0.015 respectively), during Markov Chain Monte Carlo (MCMC) computations. A consensus tree was then constructed from this final credible set of trees using a 50% majority consensus rule in the Dendroscope 3.5.7 package74. All phylograms were visualised, inspected and edited in FigTree v.1.4.275.
Data availability
Genotypic data have been provided as supplementary datasets. Incorporated in text.
References
Southgate, P. C. et al. In The Pearl Oyster (eds Southgate, P. C. & Lucas, J. S.) Ch. 9, 303–355 (Elsevier, 2008).
Wada, K. T. & Tëmkin, I. In The Pearl Oyster (eds Southgate, P. C. & Lucas, J. S.) Ch. 2, 54–57 (Elsevier, 2008).
Arnaud-Haond, S., Bonhomme, F. & Blanc, F. Large discrepancies in differentiation of allozymes, nuclear and mitochondrial DNA loci in recently founded Pacific populations of the pearl oyster Pinctada margaritifera. Journal of Evolutionary Biology 16, 388–398, https://doi.org/10.1046/j.1420-9101.2003.00549.x (2003).
Arnaud-Haond, S. et al. Genetic structure at different spatial scales in the pearl oyster (Pinctada margaritifera cumingii) in French Polynesian lagoons: beware of sampling strategy and genetic patchiness. Marine Biology 155, 147–157, https://doi.org/10.1007/s00227-008-1013-0 (2008).
Arnaud-Haond, S. et al. Spatio-temporal variation in the genetic composition of wild populations of pearl oyster (Pinctada margaritifera cumingii) in French Polynesia following 10 years of juvenile translocation. Molecular Ecology 13, 2001–2007, https://doi.org/10.1111/j.1365-294X.2004.02188.x (2004).
Lemer, S. & Planes, S. Translocation of wild populations: conservation implications for the genetic diversity of the black-lipped pearl oyster Pinctada margaritifera. Molecular Ecology 21, 2949–2962, https://doi.org/10.1111/j.1365-294X.2012.05588.x (2012).
Lemer, S. & Planes, S. Effects of habitat fragmentation on the genetic structure and connectivity of the black-lipped pearl oyster Pinctada margaritifera populations in French Polynesia. Marine Biology 161, 2035–2049, https://doi.org/10.1007/s00227-014-2484-9 (2014).
Benzie, J. A. H. & Ballment, E. Genetic differences among black-lipped pearl oyster (Pinctada margaritifera) populations in the western Pacific. Aquaculture 127, 145–156, https://doi.org/10.1016/0044-8486(94)90421-9 (1994).
Durand, P. & Blanc, F. Genetic diversity in a tropical marine bivalve - Pinctada margaritifera (Linne, 1758). Bulletin De La Societe Zoologique De France - Evolution Et Zoologie 113, 293–304 (1988).
Durand, P., Wada, K. T. & Blanc, F. Genetic variation in wild and hatchery stocks of the black pearl oyster, Pinctada margaritifera, from Japan. Aquaculture 110, 27–40, https://doi.org/10.1016/0044-8486(93)90431-W (1993).
Lal, M. M., Southgate, P. C., Jerry, D. R., Bosserelle, C. & Zenger, K. R. A Parallel Population Genomic and Hydrodynamic Approach to Fishery Management of Highly-Dispersive Marine Invertebrates: The Case of the Fijian Black-Lip Pearl Oyster Pinctada margaritifera. PLoS ONE 11, e0161390, https://doi.org/10.1371/journal.pone.0161390 (2016).
Lal, M. M., Southgate, P. C., Jerry, D. R. & Zenger, K. R. Fishing for divergence in a sea of connectivity: The utility of ddRADseq genotyping in a marine invertebrate, the black-lip pearl oyster Pinctada margaritifera. Marine. Genomics 25, 57–68, https://doi.org/10.1016/j.margen.2015.10.010 (2016).
Guo, Q. Incorporating latitudinal and central-marginal trends in assessing genetic variation across species ranges. Molecular Ecology 21, 5396–5403, https://doi.org/10.1111/mec.12012 (2012).
Lal, M. M., Southgate, P. C., Jerry, D. R., Bosserelle, C. & Zenger, K. R. Swept away: ocean currents and seascape features influence genetic structure across the 18,000 Km Indo-Pacific distribution of a marine invertebrate, the black-lip pearl oyster Pinctada margaritifera. BMC Genomics 18, 66, https://doi.org/10.1186/s12864-016-3410-y (2017).
Eckert, C. G., Samis, K. E. & Lougheed, S. C. Genetic variation across species’ geographical ranges: the central–marginal hypothesis and beyond. Molecular Ecology 17, 1170–1188, https://doi.org/10.1111/j.1365-294X.2007.03659.x (2008).
Gervis, M. H. & Sims, N. A. The Biology and Culture of Pearl Oysters (Bivalvia: Pteriidae). 21 (ICLARM, 1992).
Allan, J. Australian Shells with Related Animals Living in the Sea, in Freshwater and on the Land. (Charles T. Branford Company, 1959).
Masaoka, T. & Kobayashi, T. Natural hybridization between Pinctada fucata and Pinctada maculata inferred from internal transcribed spacer regions of nuclear ribosomal RNA genes. Fish Sci 71, 829–836, https://doi.org/10.1111/j.1444-2906.2005.01034.x (2005).
Masaoka, T. & Kobayashi, T. Species identification of Pinctada imbricata using intergenic spacer of nuclear ribosomal RNA genes and mitochondrial 16S ribosomal RNA gene regions. Fish Sci 71, 837–846, https://doi.org/10.1111/j.1444-2906.2005.01035.x (2005).
Ranjbar, M. S. et al. Rising the Persian Gulf Black-Lip Pearl Oyster to the Species Level: Fragmented Habitat and Chaotic Genetic Patchiness in Pinctada persica. Evolutionary Biology 43, 131–143, https://doi.org/10.1007/s11692-015-9356-1 (2016).
Funk, W. C., McKay, J. K., Hohenlohe, P. A. & Allendorf, F. W. Harnessing genomics for delineating conservation units. Trends in Ecology & Evolution 27, 489–496, https://doi.org/10.1016/j.tree.2012.05.012 (2012).
Crandall, K. A., Bininda-Emonds, O. R., Mace, G. M. & Wayne, R. K. Considering evolutionary processes in conservation biology. Trends Ecol Evol 15, 290–295 (2000).
Tëmkin, I. Molecular phylogeny of pearl oysters and their relatives (Mollusca, Bivalvia, Pterioidea). BMC Evolutionary Biology 10, 1–28, https://doi.org/10.1186/1471-2148-10-342 (2010).
Cunha, R. L., Blanc, F., Bonhomme, F. & Arnaud-Haond, S. Evolutionary Patterns in Pearl Oysters of the Genus Pinctada (Bivalvia: Pteriidae). Marine Biotechnology 13, 181–192, https://doi.org/10.1007/s10126-010-9278-y (2011).
Davis, M. A., Douglas, M. R., Collyer, M. L. & Douglas, M. E. Deconstructing a Species-Complex: Geometric Morphometric and Molecular Analyses Define Species in the Western Rattlesnake (Crotalus viridis). PLOS ONE 11, e0146166, https://doi.org/10.1371/journal.pone.0146166 (2016).
De Queiroz, K. Species Concepts and Species Delimitation. Systematic Biology 56, 879–886, https://doi.org/10.1080/10635150701701083 (2007).
Sanders, M. T. et al. One for each ocean: revision of the Bursa granularis (Röding, 1798) species complex (Gastropoda: Tonnoidea: Bursidae). Journal of Molluscan Studies 83, 384–398 (2017).
Jameson, L. H. On the Identity and Distribution of the Mother-of-Pearl Oysters; with a Revision of the Subgenus. Margaritifera. Proceedings of the Zoological Society of London 70, 372–394, https://doi.org/10.1111/j.1469-7998.1901.tb08552.x (1901).
Hanley, S. C. T. & Wood, W. An illustrated and descriptive catalogue of recent bivalve shells. xviii (Williams and Norgate, 1842).
Zhong, X., Li, Q., Yu, H. & Kong, L. SNP Mining in Crassostrea gigas EST Data: Transferability to Four Other Crassostrea Species, Phylogenetic Inferences and Outlier SNPs under Selection. PLoS One 9, e108256, doi:10.1371/journal.pone.0108256 (2014).
Riginos, C. & Cunningham, C. W. INVITED REVIEW: Local adaptation and species segregation in two mussel (Mytilus edulis × Mytilus trossulus) hybrid zones. Molecular Ecology 14, 381–400, https://doi.org/10.1111/j.1365-294X.2004.02379.x (2005).
Takeuchi, T. et al. Bivalve-specific gene expansion in the pearl oyster genome: implications of adaptation to a sessile lifestyle. Zoological Letters 2, 1–13, https://doi.org/10.1186/s40851-016-0039-2 (2016).
Bartsch, P. A new pearl oyster from the Hawaiian Islands. Proceedings of the United States National Museum 79, 1–2 (1931).
Ranson, G. Missions dans le Pacifique récifs coralliens, huîtres perlières. (Lechevalier, 1961).
Arnaud, S., Monteforte, M., Galtier, N., Bonhomme, F. & Blanc, F. Population structure and genetic variability of pearl oyster Pinctada mazatlanica along Pacific coasts from Mexico to Panama. Conservation Genetics 1, 299–307 (2000).
Marques, D. A. et al. Genomics of Rapid Incipient Speciation in Sympatric Threespine Stickleback. PLoS Genetics 12, e1005887, https://doi.org/10.1371/journal.pgen.1005887 (2016).
Dawson, M. N. Incipient speciation of Catostylus mosaicus (Scyphozoa, Rhizostomeae, Catostylidae), comparative phylogeography and biogeography in south-east Australia. Journal of Biogeography 32, 515–533, https://doi.org/10.1111/j.1365-2699.2004.01193.x (2005).
Vierna, J. et al. Species delimitation and DNA barcoding of Atlantic Ensis (Bivalvia, Pharidae). Zoologica Scripta 43, 161–171, https://doi.org/10.1111/zsc.12038 (2014).
Hayami, I. In Marine Mollusks in Japan (ed Takashi Okutani) 879–883 (Tokai University Press, 2000).
Matsukuma, A. In Encyclopedia of Shellfish (ed Takashi Okutani) 288 (Sekaibunkasha, 2004).
Atsumi, T., Komaru, A. & Okamoto, H. Genetic relationship among the Japanese pearl oyster Pinctada fucata martensii and foreign pearl oysters. Fish Genetics and Breeding Science 33, 135–142 (2004).
Yu, D. H. & Chu, K. H. Phylogenetics of the common pearl oysters in the genus Pinctada: evidence from nrDNA ITS sequence. Biodiversity. Science 13, 315–323 (2005).
Dawson, M. N., Raskoff, K. A. & Jacobs, D. K. Field preservation of marine invertebrate tissue for DNA analyses. Molecular marine biology and biotechnology 7, 145–152 (1998).
Adamkewicz, S. L. & Harasewych, M. G. Systematics and biogeography of the genus Donax (Bivalvia: Donacidae) in eastern North America. American Malacological Bulletin 13, 97–103 (1996).
GE. Illustra AutoSeq G-50 and AutoSeq. 96 dye terminator removal. Data file 28-9175-28. Illustra AutoSeq G-50 documents, 1–4 https://www.gelifesciences.com/gehcls_images/GELS/Related%20Content/Files/1314774443672/litdoc28917528AA_20110831092850.pdf (2007).
Kilian, A. et al. Diversity arrays technology: A generic genome profiling technology on open platforms. Methods Mol Biol 888 (2012).
Sansaloni, C. et al. Diversity ArraysTechnology (DArT) and next-generation sequencing combined: genome-wide, high throughput, highly informative genotyping for molecular breeding of Eucalyptus. BMC Proceedings 5, P54–P54, https://doi.org/10.1186/1753-6561-5-S7-P54 (2011).
Harrang, E., Lapègue, S., Morga, B. & Bierne, N. A High Load of Non-neutral Amino-Acid Polymorphisms Explains High Protein Diversity Despite Moderate Effective Population Size in a Marine Bivalve With Sweepstakes Reproduction. G3: Genes|Genomes|Genetics 3, 333–341, https://doi.org/10.1534/g3.112.005181 (2013).
Elshire, R. J. et al. A Robust, Simple Genotyping-by-Sequencing (GBS) Approach for High Diversity Species. PLoS ONE 6, e19379, https://doi.org/10.1371/journal.pone.0019379 (2011).
Ren, R. et al. Construction of a high-density DArTseq SNP-based genetic map and identification of genomic regions with segregation distortion in a genetic population derived from a cross between feral and cultivated-type watermelon. Mol Genet Genomics 290, 1457–1470, https://doi.org/10.1007/s00438-015-0997-7 (2015).
Mateos, J. M. & Pérez, J. P. Image Processing with ImageJ. (Packt Publishing Ltd, 2013).
Robasky, K., Lewis, N. E. & Church, G. M. The role of replicates for error mitigation in next-generation sequencing. Nat Rev Genet 15, 56–62, doi:10.1038/nrg3655 http://www.nature.com/nrg/journal/v15/n1/abs/nrg3655html#supplementary-information (2014).
Przyborowski, J. A., Sulima, P., Kuszewska, A., Zaluski, D. & Kilian, A. Phylogenetic Relationships between Four Salix L. Species Based on DArT Markers. International Journal of Molecular Sciences 14, 24113–24125, https://doi.org/10.3390/ijms141224113 (2013).
Steane, D. A. et al. Population genetic analysis and phylogeny reconstruction in Eucalyptus (Myrtaceae) using high-throughput, genome-wide genotyping. Molecular Phylogenetics and Evolution 59, 206–224, https://doi.org/10.1016/j.ympev.2011.02.003 (2011).
Steane, D. A. et al. Genome-wide scans reveal cryptic population structure in a dry-adapted eucalypt. Tree Genetics & Genomes 11, 32–45, https://doi.org/10.1007/s11295-015-0864-z (2015).
Belkhir, K., Borsa, P., Chikhi, L., Raufaste, N. & Bonhomme, F. GENETIX 4.05, logiciel sous Windows TM pour la génétique des populations http://www.genetix.univ-montp2.fr/genetix/intro.htm (1996).
Lynch, M. & Milligan, B. G. Analysis of population genetic structure with RAPD markers. Molecular Ecology 3, 91–99, https://doi.org/10.1111/j.1365-294X.1994.tb00109.x (1994).
AFLP-SURV version 1.0 (Laboratoire de Génétique et Ecologie Végétale, Université Libre de Bruxelles, Belgium., Brussels, Belgium, 2002).
Vekemans, X., Beauwens, T., Lemaire, M. & Roldán-Ruiz, I. Data from amplified fragment length polymorphism (AFLP) markers show indication of size homoplasy and of a relationship between degree of homoplasy and fragment size. Molecular Ecology 11, 139–151, https://doi.org/10.1046/j.0962-1083.2001.01415.x (2002).
Tamura, K., Stecher, G., Peterson, D., Filipski, A. & Kumar, S. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Molecular Biology and Evolution doi:10.1093/molbev/mst197 (2013).
Lee, T.-H., Guo, H., Wang, X., Kim, C. & Paterson, A. H. SNPhylo: a pipeline to construct a phylogenetic tree from huge SNP data. BMC Genomics 15, 1–6, https://doi.org/10.1186/1471-2164-15-162 (2014).
Stamatakis, A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, doi:10.1093/bioinformatics/btu033 (2014).
Edgar, R. C. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Research 32, 1792–1797, https://doi.org/10.1093/nar/gkh340 (2004).
Baum, B. R. Phylip: Phylogeny Inference Package (Version 3.2). The Quarterly Review of Biology 64, 539–541 (1989).
Schliep, K. P. phangorn: phylogenetic analysis in R. Bioinformatics 27, 592–593, https://doi.org/10.1093/bioinformatics/btq706 (2011).
Leaché, A. D., Banbury, B. L., Felsenstein, J., de Oca, A. N.-M. & Stamatakis, A. Short Tree, Long Tree, Right Tree, Wrong Tree: New Acquisition Bias Corrections for Inferring SNP Phylogenies. Systematic Biology 64, 1032–1047, https://doi.org/10.1093/sysbio/syv053 (2015).
Lewis, P. O. & Olmstead, R. A Likelihood Approach to Estimating Phylogeny from Discrete Morphological Character Data. Systematic Biology 50, 913–925, https://doi.org/10.1080/106351501753462876 (2001).
Pattengale, N. D., Alipour, M., Bininda-Emonds, O. R. P., Moret, B. M. E. & Stamatakis, A. How many bootstrap replicates are necessary? Journal of Computational Biology 17, 337–354 (2010).
Ronquist, F. et al. MrBayes 3.2: Efficient Bayesian Phylogenetic Inference and Model Choice Across a Large Model Space. Systematic Biology 61, 539–542 (2012).
Drummond, A. J. & Rambaut, A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evolutionary Biology 7, 1–8, https://doi.org/10.1186/1471-2148-7-214 (2007).
Huelsenbeck, J. P. & Ronquist, F. MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics 17, 754–755, https://doi.org/10.1093/bioinformatics/17.8.754 (2001).
Koopman, W. J. M. et al. AFLP markers as a tool to reconstruct complex relationships: A case study in Rosa (Rosaceae). American Journal of Botany 95, 353–366 (2008).
Rambaut, A., Suchard, M. & Drummond, A. J. Tracer v.1.6 http://tree.bio.ed.ac.uk/software/tracer/ (2003).
Huson, D. H. et al. Dendroscope: An interactive viewer for large phylogenetic trees. BMC Bioinformatics 8, 1–6, https://doi.org/10.1186/1471-2105-8-460 (2007).
Rambaut, A. FigTree v.1.4.2: tree drawing tool http://tree.bio.ed.ac.uk/software/figtree/ (2014).
ArcGIS Desktop: Release 10 (Environmental Systems Research Institute, Redlands, California, United States of America, 2011).
Coeroli, M., De Gaillande, D. & Landret, J. P. & Aquacop. Recent innovations in cultivation of molluscs in French Polynesia. Aquaculture 39, 45–67, https://doi.org/10.1016/0044-8486(84)90258-8 (1984).
Galstoff, P. S. Pearl and Hermes Reef, Hawaii: hydrographical and biological observations. (Bernice P. Bishop Museum, 1933).
Gug, M. Mother-of-pearl industry in FrenchOceania. SPC Quarterly Bulletin 7, 19–21 http://www.spc.int/DigitalLibrary/Doc/FAME/InfoBull/SPB/SPB7_3_Gug.html (1957).
Hedley, C. In The Australian Museum Magazine 2 5–11 (The Australian Museum, Australia, 1924).
Saville-Kent, W. Torres Straits pearlshell fisheries. 1075–1078 (Brisbane, Australia, 1905).
Hynd, J. S. A Revision of the Australian Pearl-shells, Genus Pinctada (Lamellibranchia). Marine and Freshwater Research 6, 98–138, https://doi.org/10.1071/MF9550098 (1955).
Cahn, A. 357 (ed. Fish and Wildlife Service) 1–91 (United States Department of the Interior, 1949).
Elamin, E. M. & Elamin, S. E. M. Biometric relationships of the mother of pearl oyster (Pinctada margaritifera var erythraensis) from Dongonab Bay, Red Sea. International. Journal of Science, Environment 3, 1193–1204 (2014).
Acknowledgements
This study was conducted within the Australian Centre for International Agricultural Research (ACIAR) Project FIS/2009/057: “Pearl Industry Development in the Western Pacific” led by the University of the Sunshine Coast. The research was carried out during a John Allwright Fellowship awarded to MML. We wish to thank Eike Steinig, David Jones and Roger Huerlimann for advice on various statistical analyses and Litia Gaunavou for generation of the sampling site map. For either providing or assisting with collection of oyster tissue samples, our gratitude also extends to Héctor Acosta-Salmón, Douglas McLaurin-Moreno, Gustaf Mamangkey, Naomi Gardiner, Ismail Saidi, Samantha Nowland, Rowan McIntyre, Steve Warden, Jo Buckee, Tina Weier, Georgia Langdon, Tevainui Frogier-Ellis, Hoc Tan Dao, Hứa Thái Tuyến, Samad Jahangard, Hossein Rameshi, Mehdi Doroudi, Max Wingfield, Laura Simmons, Gregory Bennett, Philippa Cohen, Waghon Lalao, Yu Wen Chiu, Eric Gan, Cherie Morris, Shirleen Bala, Justin Hunter, Pranesh Kishore, Adi Dionani Salaivanua, Kelly Brown, Jerome Taoi, Epeli Loganimoce, Albert Whippy, Bai Whippy, Toga Whippy, Isimeli Loganimoce, Marilyn Vilisoni, Babitu Rarawa, Ilitomasi Nuku, Samisoni Rakai, Patrick Fong, Nepoci Raleve and Claude Prévost. Logistic support for fieldwork in the Fiji Islands was provided by project partners the Secretariat of the Pacific Community (SPC) and the University of the South Pacific (USP).
Author information
Authors and Affiliations
Contributions
M.M.L. carried out all tissue collections, laboratory benchwork, participated in the investigation design and conceptualisation, developed modifications to the genotyping protocol, performed all data analyses and drafted the manuscript. P.C.S. developed the broad project concept, participated in the investigation design, provided supervisory support, all project funding, advice on pearl oyster biology and ecology, and edited the manuscript. D.R.J. participated in the investigation design, provided supervisory support and edited the manuscript. K.R.Z. participated in design and conceptualisation of the project, provided statistical advice and technical input on investigation design, developed modifications to the genotyping protocol, provided supervisory support and edited the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lal, M.M., Southgate, P.C., Jerry, D.R. et al. Genome-wide comparisons reveal evidence for a species complex in the black-lip pearl oyster Pinctada margaritifera (Bivalvia: Pteriidae). Sci Rep 8, 191 (2018). https://doi.org/10.1038/s41598-017-18602-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-18602-5
- Springer Nature Limited