
Abstract
The effect of Copy Number Variants (CNVs) on Type 2 Diabetes (T2D) remains little explored. The present study characterized large rare CNVs in 686 T2D and 194 non-T2D subjects of Mexican ancestry genotyped using the Affymetrix Genome-Wide Human SNP array 5.0. Rare CNVs with ≥ 100 kb length were identified using a stringent strategy based on merging CNVs calls generated using Birdsuit, iPattern and PennCNV algorithms. We applied three different strategies to evaluate the distribution of CNVs in the T2D and non-T2D samples: 1) Burden analysis, 2) Identification of CNVs in loci previously associated to T2D, and 3) Identification of CNVs observed only in the T2D group. In the CNV burden analysis, the T2D group showed a higher proportion of CNVs, and also a higher proportion of CNVs overlapping at least one gene than the non T2D group. Five of the six loci previously associated with T2D had duplications or deletions in the T2D sample, but not the non-T2D sample. A gene-set analysis including genes with CNVs observed only in the T2D group highlighted gene-sets related with sensory perception (olfactory receptors, OR) and phenylpyruvate tautomerase/dopachrome isomerase activity (MIF and DDT genes).
Similar content being viewed by others


Introduction
Type 2 Diabetes (T2D) is a complex metabolic disorder that constitutes a worldwide public health problem1. The evidence of a strong hereditary component in T2D has led to numerous efforts to study the genetic factors underlying the disease, mainly through genome-wide association studies (GWAS) focused on common variants2,3,4,5,6.
Copy Number Variants (CNVs), defined as DNA deletions or duplications above 1 kb in length, represent a significant proportion of human genetic variation and have been linked to many complex human diseases, but their implication in T2D remains little explored. In particular, large CNVs may encompass many genes and/or regulatory sequences and could be of particular relevance for T2D because of their potential pathological effects7.
The current Mexican population is an admixed population with a genetic background derived from European, Native American and to lesser extent, West African populations8. Mexico has one of the highest prevalences of T2D in the world9. In this study, we characterized large rare CNVs in T2D and non-T2D individuals (controls) of Mexican ancestry that were genotyped with the Affymetrix 5.0 array10. We then applied three different strategies in order to evaluate the distribution of CNVs in the T2D and control samples: 1) Burden analysis, 2) Identification of CNVs in loci previously associated to T2D, and 3) Identification of CNVs observed only in the T2D group.
Results
Subjects
A total of 686 T2D and 194 non-T2D control subjects were analyzed. As outlined in Table 1, the T2D group had more females and showed higher body mass index (BMI) and triglycerides but lower Multidimensional Scaling (MDS) ancestry vector 1 values than the control group (e.g. lower European ancestry). No differences were observed in relation to CNV size and deletion/duplication distribution among comparison groups [Supplementary Figure 4].
Rare CNV burden analysis
In the first comparative analysis [Fig. 1], the rare CNV burden analysis, the T2D group showed a higher CNV deletion rate (0.25 vs 0.19, corrected p-value = 0.029) and a higher proportion of rare genic CNVs (e.g. CNVs overlapping with at least one gene) than the control group (0.42 vs 0.31, corrected p-value = 0.044). We did not observe significant differences in CNV rate, genic CNV rate, CNV sample proportion, total CNV size and genic CNV enrichment between the T2D and control groups [Table 2]. In the genic CNV regional analysis, ZNF718 CNVs were found inversely associated with T2D (p = 0.048 for Duplication + Deletions). However, it is important to note that all the CNVs identified in this gene were present only in the control group [Table 3].

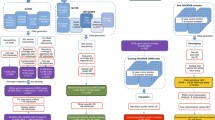
Analysis flowchart of the study’s aims.
CNVs in candidate genes
We evaluated if rare CNVs in our samples overlapped with a list of 129 loci previously described as being implicated in T2D [Supplementary Table 3]. In the T2D group, we found duplications in the genes AMY2B, LPP and RASGRP1 and deletions in the HFE and CDKAL1 genes. In the control group, we found a deletion in the ARL15 gene [Table 3].
CNVs seen in the T2D group but not in the control group
We observed a total of 76 genic CNVs in the T2D group that were not seen in the control group and also had frequencies below 0.2% in the Database of Genomic Variants (DGV). These CNVs overlapped with 123 genes, 59 of which are listed in the Online Mendelian Inheritance in Man database (www.omim.org ) [Supplementary Table 4].
Gene-sets analysis
A total of 130 genes were included in the gene-set analysis (one gene identified in the genic regional analysis, 6 genes from the candidate gene analysis, and 123 genes from the analysis of CNVs that are present in the T2D group but not in the control group). The gene-set analysis identified 27 gene-sets. Most of the gene-sets related to sensory perception (olfactory receptors –OR-). Two of the gene-sets pointed to phenylpyruvate tautomerase/dopachrome isomerase activity. Duplications are the most common type of CNV observed in these gene-sets [Fig. 2, Supplementary Table 5].

Results of the gene-set analysis. The map shows a network of gene sets (nodes) related by mutual overlap (edges). A total of 130 genes derived from the genic regional analysis, the analysis of CNVs in candidate genes and the analysis of genic CNVs seen in the T2D group but not in the control group were used as input for the gene-set analysis, which identified 27 gene-sets. Blue circles corresponded to gene-sets with duplications and yellow circles represented gene-sets with duplications and deletions.
Discussion
T2D is a complex disorder caused by genetic and environmental factors. The high concordance between twins and the disparities in rates of development of T2D from children of parents with and without the diabetes show the influence of genetic factors in the occurrence the disease11. GWAS have emerged as the most successful methodology to discover genetic variants associated with complex diseases4,5,6.
CNVs represent a significant proportion of human genetic variation. It has been estimated that 79.55% of the genome contains CNVs (http://dgv.tcag.ca/dgv/app/home). In the last few years, there has been an increased interest in the evaluation of the contribution of rare CNVs to complex diseases7,12,13,14. This type of genomic variation could be relevant for disease risk due to dosage or disrupting effects. For example, genic duplications may create imbalance due to the excess of products of duplicated genes7.
It is possible to characterize CNVs using microarray data. In this study, we characterized large rare CNVs in 686 T2D and 194 control subjects of Mexican ancestry genotyped using the Affymetrix Genome-Wide Human SNP array 5.0. In our comparative analyses, we found that the T2D group showed a higher proportion of CNVs overlapping with at least one gene than the control group. In our genic CNV regional analysis, we observed that CNVs (Duplications + Deletions) in the ZNF718 gene were associated to T2D status. However, all the duplications and deletions were observed in the control group, and this does not support a role of CNVs within this gene in the pathogenesis of T2D. We found CNVs in 6 loci previously associated with T2D (AMY2B, LPP, RASGRP1, HFE, CDKAL1, ARL15). With the exception of the deletion found in the ARL15 gene, all the other CNVs (duplications or deletions) were observed in the T2D group. Finally, we identified a list of 130 genes for further analysis, based on three different strategies: 1) Genic CNV regional analysis, 2) Identification of CNVs overlapping with previously reported T2D genes, and 3) Genic CNVs observed in the T2D but not in the control group. A gene-set analysis including these 130 genes identified 27 gene-sets. Most of the aforementioned gene-sets included OR genes. The remaining gene-sets included the genes MIF and DDT, which are involved in Dopachrome/phenylpyruvate isomerase activity.
Loss of smell has been associated with reductions in fat mass and insulin resistance15. However, it is important to note that the OR gene family is the largest vertebrate gene family and several studies have shown that OR loci are enriched in CNVs because they are often located in segmentally duplicated regions16,17,18,19,20. For example, in a high-resolution study using oligonucleotide tiling microarrays, Hasin et al.16 analyzed 851 OR gene and pseudogene loci, and reported the presence of CNVs in 93 OR genes and 153 pseudogenes. Our gene-set results have been primarily driven by the inclusion of genes with CNVs identified in the T2D group but absent in the control group. It is possible that the enrichment in OR genes observed in our analysis is the result of ORs being prone to the presence of CNVs, in combination with the unbalanced number of T2D and control samples in our study, instead of a functional effect of CNVs located within OR genes on T2D risk. Another gene-set identified in our analysis includes the gene MIF, which codes for the cytokine macrophage migration inhibitory factor that has been previously related with adipose tissue inflammatory processes and with T2D21,22. It would be important to carry out additional research to evaluate if CNVs affecting this gene may play a role in T2D risk.
Similarly to a recent study in Mexican Americans23, we found that duplications are on average larger in size than deletions. However, in contrast to Blackburn’s study, in which deletions were more frequent than duplications (deletion/duplication ratio between 1.56 to 2.75), in our study we observed a larger number of duplications than deletions (deletion/duplication ratio between 0.25 to 0.56). These differences may be due to the type of microarray assay, the CNV calling algorithms and/or the CNV length and selection protocols used in both studies. Our study also found that the T2D group had a higher CNV deletion rate in comparison with the control group. Our CNV deletion and duplication distribution was quite similar to that described in the T2D and control cohorts from the Welcome Trust Case-Control Consortium study24.
The contemporary Mexican population is the result of an admixture process that involved Native American, European and African populations25,26,27,28. The average ancestral contributions of this sample have been estimated as 60%, 35% and 5% respectively25. It is well known that the presence of population stratification can inflate the rate of false positives in association studies in admixed populations. One strategy to minimize this problem was to include ancestry proportions as covariates in the statistical analyses. In our study, we used the coordinates of the first MDS axis, which reflects the relative ancestral contributions from European and Native American populations. In addition to the MDS1 ancestry vector, the rare CNV burden analysis was also adjusted by sex, BMI, and triglycerides, given that significant differences for these variables were observed between the T2D and control groups in the initial comparative analysis. Unfortunately, due the small counts, the aforementioned adjustment was not possible neither for ZNF718 gene nor for the diabetes candidate gene analysis.
The lack of detailed clinical information like blood glucose and hemoglobin A1c concentrations, the relatively high percentage of samples dropped during the QC protocol, the limited sample size and the unbalanced number of T2D subjects and controls are the main limitations of our study. With respect to the number of samples removed, Grassi29 reported that 12.8% of the samples genotyped with the Affymetrix Genome-Wide Human SNP array 5.0 were eliminated using only two quality control criteria (gender incompatibilities, and genotyping rate <90%). The higher percentage of samples removed in our study (32.8%) could be explained by the rigorous four-step quality control analysis used to maximize our confidence in the CNV calls.
Despite the limited sample size and the unbalanced number of T2D and control samples, in a post-hoc analysis, the power to detect overall genic CNV proportion differences and ZNF718 gene differences among comparison groups were 0.799 and 0.646 respectively. However, the statistical power decrease to 0.483 when there was a CNV in a control subject but not in T2D and 0.003 when there was a CNV in a T2D subject but not in controls.
Conclusions
In this study conducted in Mexico we found that the T2D group showed a higher proportion of CNVs than the control sample, and also a higher proportion of CNVs overlapping at least one gene than the control group. We found duplications or deletions in six genes that have been previously associated with type 2 diabetes. Finally, gene-sets related with sensory perception (olfactory receptors) and phenylpyruvate tautomerase/dopachrome isomerase activity were enriched in the T2D group.
Methods
Study design and population
As outlined below, we analyzed a total of 686 T2D and 194 non-T2D control subjects selected from the initial sample comprised 1,310 individuals (967 T2D subjects and 343 controls) that were previously included in a GWAS for T2D10. Briefly, the T2D sample included subjects previously diagnosed according to the American Diabetes Association (ADA) criteria30 recruited from Family Medicine Clinics located at the southern part of Mexico City. The control group included healthy blood donors without family history of T2D and with normal glucose tolerance tests, according to the ADA criteria, that were recruited from the Central Blood Bank of the National Medical Center “Siglo XXI”. Both groups were sampled between 2000 and 2005. Information on sex, age, BMI, total cholesterol and triglycerides was available for all the samples. Ancestry proportions were estimated using a genome-wide panel of 3,500 Ancestry Informative Markers (AIMs) with the program ADMIXMAP [http://homepages.ed.ac.uk/pmckeigu/admixmap/index.html] and also a MDS approach based on genome-wide data for six parental population samples (Nahua and Maya from Mexico, Andeans and Aymara from South America, Spanish, CEPH European and Yoruba) and the Mexico City sample.
SNP genotyping
Genotyping was carried out at the Centre for Applied Genomics (Toronto, ON) using the Affymetrix Genome-Wide Human SNP array 5.0 (Affymetrix, Santa Clara, CA) and following standard protocols.
Quality control analysis and CNV analysis
A four-step quality control (QC) procedure was applied to the initial set of 1,310 samples to ensure that ascertainment of CNVs was consistent between comparison groups. Rare CNVs with ≥100 kb length were identified using a stringent strategy based on merging CNVs calls generated using Birdsuite (version 1.5.3)31, iPattern32,33 and PennCNV34 algorithms. At the end of the QC analysis, the dataset comprised 880 individuals (686 T2D subjects and 194 control subjects) and 657 rare large CNVs. [Supplementary Information, Supplementary Figures 1–3 and Supplementary Tables 1 and 2].
Comparative analysis of general characteristics
Comparisons of sex, age, BMI, total cholesterol, triglycerides, ancestry vectors, CNV size and deletion/duplications proportions between the T2D and control groups were carried out with the X2 and Mann–Whitney U tests.
Rare CNV burden analysis
The CNV burden analysis was carried out using PLINK v 1.07 (http://zzz.bwh.harvard.edu/plink/), considering two categories: rare CNVs and genic rare CNVs (e.g. CNVs that overlap with at least one gene). The global CNV burden in the T2D group compared to the control group was tested using seven measures: 1) CNV rate (number of CNVs per sample), 2) CNV sample proportion (proportion of samples with one or more CNVs), 3) total CNV size, 4) average CNV size, 5) genic CNV rate (number of regions/genes spanned by rare CNVs), 6) genic CNV proportion (proportion of rare CNVs overlapping at least one gene) and 7) genic enrichment (number of regions/genes per total CNVs kb size). Statistical significance was evaluated using an adaptive permutation procedure for one-sided tests (i.e., hypothesizing that T2D subjects will show greater burden of rare CNVs than controls). For each of 10,000 permutations, samples were randomly reassigned either T2D or control status. Rare measures were also adjusted by significant variables in the comparative group analysis: sex, BMI, triglycerides and ancestry (coordinates of the first MDS axis, which explains the relative proportion of Native American and European ancestry in the individuals analyzed). CNVs were considered rare if they were found at a frequency of <1% in the Mexican control sample set (n = 194), and showed an overlap of less than 50% of their length with other CNVs or segmental duplications found at a frequency >1%. Rare CNVs were classified as genic based on RefSeq annotations (UCSC, v. March 2006, NCBI v36, hg18).
In a second comparative analysis of burden of rare CNVs (genic regional analysis), we tested for specific genic regions associated with T2D, using Fisher’s exact test to assess for differences between the T2D and the control groups.
CNVs in candidate genes and CNVs seen in the T2D group but not in the control group
After the rare burden analysis and the genic regional analysis, we looked for CNVs present in a list of 129 loci previously described as being implicated in T2D [Supplementary Table 3], and we also looked for CNVs present in T2D subjects but not in the control group.
Gene-sets analysis
A final analysis was carried out to find gene-sets that may be implicated in T2D. Here we considered the results of the genic regional analysis, the analysis of CNVs in candidate genes and the analysis of genic CNVs seen in the T2D group but not in the control group. Enrichment Map plugin v2.2.1 (http://baderlab.org/Software/EnrichmentMap) and Cytoscape software v3.5.1 (http://cytoscape.org/) were used to draw a map of the gene-sets network, in which the nodes represented the gene-sets and the edges corresponded to the overlap between sets.
Ethics Statement
The research was approved by the National Ethical Committee of the Mexican Institute of Social Security (register number 2008-785-073) and by The Ethics Review Office at the University of Toronto. Written informed consent was obtained from each participant. All methods were performed in accordance with the ethical standards and regulations of the institutional research committees and national laws and with the 1964 Helsinki declaration and its later amendments.
Data Availability Statement
The datasets analysed during the current study are not publicly available due to ethical concerns, but are available from the corresponding author on reasonable request.
References
World Health Organization. Diabetes Key facts. http://www.who.int/mediacentre/factsheets/fs312/en/ (2017).
Stumvoll, M., Goldstein, B. J. & van Haeften, T. W. Type 2 diabetes: principles of pathogenesis and therapy. Lancet. 365(9467), 1333–1346 (2005).
Doria, A., Patti, M. E. & Kahn, C. R. The emerging genetic architecture of type 2 diabetes. Cell Metab. 8(3), 186–200 (2008).
McCarthy, M. I. & Zeggini, E. Genome-wide association studies in type 2 diabetes. Curr Diab Rep. 9(2), 164–171 (2009).
McCarthy, M. I. Genomics, type 2 diabetes, and obesity. N Engl J Med. 363(24), 2339–2350 (2010).
Grotz, A. K., Gloyn, A. L. & Thomsen, S. K. Prioritising Causal Genes at Type 2 Diabetes Risk Loci. Curr Diab Rep. 17(9), 76 (2017).
Lee, C. & Scherer, S. W. The clinical context of copy number variation in the human genome. Expert Rev Mol Med. 12, e8 (2010).
Stavenhagen, R. & Carrasco, T. La diversidad étnica y cultural. In: Florescano E: El patrimonio nacional de México, 1st ed. (Consejo Nacional para la Cultura y las Artes) 249–280 (1997).
International Diabetes Federation. IDF Diabetes Atlas Seventh edition. http://www.diabetesatlas.org/ (2015).
Parra, E. J. et al. Genome-wide association study of type 2 diabetes in a sample from Mexico City and a meta-analysis of a Mexican-American sample from Starr County, Texas. Diabetologia. 54(8), 2038–2046 (2011).
Barroso, I. Genetics of Type 2 diabetes. Diabet Med. 22(5), 517–535 (2005).
Cook, E. H. Jr. & Scherer, S. W. Copy-number variations associated with neuropsychiatric conditions. Nature. 455(7215), 919–923 (2008).
de Cid, R. et al. Deletion of the late cornified envelope LCE3B and LCE3C genes as a susceptibility factor for psoriasis. Nat Genet. 41(2), 211–215 (2009).
Diskin, S. J. et al. Copy number variation at 1q21.1 associated with neuroblastoma. Nature. 459(7249), 987–991 (2009).
Riera, C. E. et al. The Sense of Smell Impacts Metabolic Health and Obesity. Cell Metab. 26(1), 198–211 (2017).
Hasin, Y. et al. High-resolution copy-number variation map reflects human olfactory receptor diversity and evolution. PLoS Genet. 4(11), e1000249 (2009).
Young, J. M. et al. Extensive copy-number variation of the human olfactory receptor gene family. Am J Hum Genet. 83(2), 228–242 (2008).
Redon, R. et al. Global variation in copy number in the human genome. Nature. 444, 444–454 (2006).
Korbel, J. O. et al. Paired-End Mapping Reveals Extensive Structural Variation in the Human. Genome. Science. 318, 420–426 (2007).
Nozawa, M., Kawahara, Y. & Nei, M. Genomic drift and copy number variation of sensory receptor genes in humans. Proc Natl Acad Sci USA 104, 20421–20426 (2007).
Kim, B. S., Pallua, N., Bernhagen, J. & Bucala, R. The macrophage migration inhibitory factor protein superfamily in obesity and wound repair. Exp Mol Med. 47, e161 (2015).
Morrison, M. C. & Kleemann, R. Role of Macrophage Migration Inhibitory Factor in Obesity, Insulin Resistance, Type 2 Diabetes, and Associated Hepatic Co-Morbidities: A Comprehensive Review of Human and Rodent Studies. Front Immunol. 6, 308 (2015).
Blackburn, A. et al. Utilizing extended pedigree information for discovery and confirmation of copy number variable regions among Mexican Americans. Eur J Hum Genet. 21, 404–409 (2013).
Grozeva, D. et al. Reduced burden of very large and rare CNVs in bipolar affective disorder. Bipolar Disord. 15(8), 893–898 (2013).
Martinez-Marignac, V. L. et al. Admixture in Mexico City: implications for admixture mapping of type 2 diabetes genetic risk factors. Hum Genet. 120(6), 807–819 (2007).
Martinez-Fierro, M. L. et al. Ancestry informative markers and admixture proportions in northeastern Mexico. J Hum Genet. 54(9), 504–509 (2009).
Silva-Zolezzi, I. et al. Analysis of genomic diversity in Mexican Mestizo populations to develop genomic medicine in Mexico. Proc Natl Acad Sci USA 106(21), 8611–8616 (2009).
Galanter, J. M. et al. Development of a panel of genome-wide ancestry informative markers to study admixture throughout the Americas. PLoS Genet. 8(3), e1002554 (2012).
Grassi, M. A. et al. Genome-wide meta-analysis for severe diabetic retinopathy. Hum Mol Genet. 20(12), 2472–2481 (2011).
American Diabetes Association. Standards of medical care in diabetes 2017. Diabetes Care. 40 (Suppl1): S1–S135 (2017).
Korn, J. M. et al. Integrated genotype calling and association analysis of SNPs, common copy number polymorphisms and rare CNVs. Nat Genet. 40(10), 1253–1260 (2008).
Pinto, D. et al. Functional impact of global rare copy number variation in autism spectrum disorders. Nature. 466(7304), 368–372 (2010).
Pinto, D. et al. Comprehensive assessment of array-based platforms and calling algorithms for detection of copy number variants. Nat Biotechnol. 29(6), 512–520 (2011).
Wang, K. et al. PennCNV: an integrated hidden Markov model designed for high-resolution copy number variation detection in whole-genome SNP genotyping data. Genome Res. 17(11), 1665–1674 (2007).
Acknowledgements
We would like to thank to Dr. Chao Lu, Dr. Bhooma Thiruvahindrapuram and Dr. Zhuozhi Wang from The Centre for Applied Genomics for the microarray experiments and the Birdsuite and iPattern data processing. We thanks to staff of Biochemistry Research Unit for their excellent technical support. The project was supported by CONACYT SALUD-2008-785-073. IJAM is a recipient of a Consejo Nacional de Ciencia y Tecnología (CONACyT, Mexico) grant 72694.
Author information
Authors and Affiliations
Contributions
M.C. conceived the study question. A.V., E.J.P. and S.W.S. contributed to the analysis plan. I.J.A.M. and D.P. carried out statistical analyses, interpreted the data and drafted the manuscript. All authors reviewed the manuscript
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
de Jesús Ascencio-Montiel, I., Pinto, D., Parra, E.J. et al. Characterization of Large Copy Number Variation in Mexican Type 2 Diabetes subjects. Sci Rep 7, 17105 (2017). https://doi.org/10.1038/s41598-017-17361-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-17361-7
- Springer Nature Limited
This article is cited by
-
Molecular Variants in Human Trace Amine-Associated Receptors and Their Implications in Mental and Metabolic Disorders
Cellular and Molecular Neurobiology (2020)