
Abstract
Time-dependent density functional theory (TDDFT) and atoms in molecules (AIM) theory are combined to study the photoinduced excited state intramolecular proton transfer (ESIPT) dynamics for eight anthraquinones (AQs) derivatives in solution. The calculated absorption and emission spectra are consistent with the available experimental data, verifying the suitability of the theory selected. The systems with the excited-state exothermic proton transfer, such as 1-HAQ, 1,5-DHAQ and TFAQ, emit completely from transfer structure (T), while the reactions for those without ESIPT including 1,4-DHAQ and AAAQ appear to be endothermic. Three reaction properties of three systems (1,8-DHAQ, DCAQ and CAAQ) are between the exothermic and endothermic, sensitive to the solvent. Energy scanning shows that 1,4-DHAQ and AAAQ exhibit the higher ESIPT energy barriers compared to 1-HAQ, 1,5-DHAQ and TFAQ with the “barrierless” ESIPT process. The ESIPT process is facilitated by the strengthening of hydrogen bonds in excited state. With AIM theory, it is observed that the change in electrons density ρ(r) and potential energy density V(r) at BCP position between ground state and excited state are crucial factors to quantitatively elucidate the ESIPT.
Similar content being viewed by others

Introduction
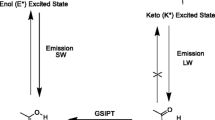
Due to the significance in modern photophysics, photochemistry and biochemistry, such as Green Fluorescent Protein (GFP)1,2, organic light emitting diodes (OLEDs)3,4,5 and fluorescent chemosensors6, the excited state intramolecular proton transfer (ESIPT) phenomenon have attracted numerous experimental and computational interests7,8,9,10,11,12,13,14,15,16. Researches of recent decades have shown that the molecules with ESIPT properties exist in the enol form in the ground state, stabilized by the intramolecular hydrogen bonding interactions. Upon photoexcitation, the molecules experience an ultrafast intramolecular proton transfer, giving rise to an excited state keto tautomeric form. The equilibrium between these two forms lead to various intriguing fluorescence properties for the molecules, including dual emission spectra, double proton transfer and back ESIPT process15,17,18,19,20,21,22,23.
The anthraquinone (AQs) derivatives (Fig. 1), including hydroxyl/dihydroxy-anthraquinone (HAQ/DHAQ) and 1-(acylamino)-anthraquinones (AYAAQs)17,18,24,25,26,27,28, exhibit the unique ESIPT properties, thus presenting emerging applications in fluorescence probes, dyes and drugs29,30,31,32,33. The experimental ESIPT evidences have been confirmed for 1-HAQ34,35,36,37, 1,5-DHAQ37,38,39, DCAQ and TFAQ40,41,42,43. However, 1,4-DHAQ37,38 and AAAQ40,41,42,43 with the similar molecular structures don’t show the ESIPT progress. More interestingly, 1,8-DHAQ25,37,38,39,44,45,46,47 and CAAQ40,41,42,43 switch their properties in different solvents. Blank et al. found that the long wavelength emissions (LWE) corresponding to the tautomeric (T) in excited state were dominant for DCAQ and TFAQ in dichloromethane solvent. Only short wavelength emissions (SWE) were observed for HPAQ which the R group is n-heptyl. The emission of CAAQ was mainly SWE and slight LWE43. When the solvent was changed to ethanol, LWE in CAAQ and DCAQ decreased greatly40. The substituent effect on ESIPT for AQs have been explained by nodal-plane model proposed in 199648, which have also been widely explored and proved to be a reliable theory to study the ESIPT systems49,50,51. However, this model only qualitatively describes ESPIT process based on the molecular skeletons and functional groups, without the quantitative descriptions of the dual fluorescence distribution and energy barriers, limiting an accurate understanding of ESIPT process.

Molecular structures of HAQ, DHAQs and AYAAQs related in this work.
In this work, the DFT/TDDFT methods were used to investigate two series of AQs in both ground and excited states. Analysis of the hydrogen bonding interactions, electronic transition energies and infrared vibrations provides the atomic insight into their ESIPT processes. The structures in ground state (S0(T)) were not stable with the evidence of optimized potential energy surface (PES) except for compound 1,4-DHAQ in which both the single and double protons transfer structures are stable. The AAAQ presented the unstable structure upon ESIPT, consistent with experiments40,43. Other seven compounds showed the stable structures at both S1(N) and S1(T), resulting in various ESIPT properties. The PES in excited state confirmed the exothermic reaction and “barrierless” ESIPT process in 1-HAQ, 1,5-DHAQ, DCAQ and TFAQ with the dominant LWE in dichloromethane. The ESIPT processes for other four compounds are endothermic with different energy barriers. 1,4-DHAQ has the highest energy barrier in S1 state, similar to AAAQ. The alterable ESIPT properties of 1,8-DHAQ, CAAQ and DCAQ came from their medium energy barriers. Hydrogen bond strengthening in excited state is confirmed by the redshift of vibration frequencies. AIM theory was used to investigate the relationship between ESIPT progress and property of BCP. The changes of electrons density ρ(r) and potential energy density V(r) at BCP position in ground state and excited state are significant for the ESIPT process. More importantly, the V(r) at BCP position is better than ρ(r) as a reference for hydrogen bond dynamics.
Theoretical methods
The calculations in this work were performed by the DFT/TDDFT method52,53,54,55 with the Becke’s three parameter hybrid exchange functional with Lee–Yang–Parr gradient-corrected correlation (B3LYP) functional56,57,58 and 6–311 + G(d, p) basis set59,60 by Gaussian 09 program61. The graph of FMO isosurfaces is drawn by Chemcraft 62. The vertical transition energies and geometric optimization in the excited state were calculated by TDDFT method. Vibrational frequencies of both ground and excited state were computed to ensure that the geometries indeed correspond to a minimum confirmed by no imaginary frequencies. The conductor-like polarizable continuum model (CPCM) was employed to describe the implicit solvent effect (dichloromethane)63,64. The PES of the S0 and S1 states were calculated to explore the transfer barrier and thermodynamics effect, revealing ESPIT mechanisms.
The atoms in molecules (AIM) theory proposed by Bader is used for analyzing property of wave function and other real space functions65,66,67. The bond critical point (BCP) generally appears between attractive atom pairs, which property is closely related to the bond or interaction strengthening68,69,70. Multiwfn was used to study the character of BCP71. More attention are paid to the electron density ρ(r) and potential energy density V(r) at BCP between the hydrogen bond72.
Results and Discussion
Hydrogen Bonding Dynamics
Figure 1 shows the molecular structures for AQs studied in this work. The n-heptyl group is replaced by the methyl group, as to simplify the DFT calculations. In the optimized structures in the ground and excited states, the atoms involve in ESIPT progress are in the same plane with the benzene rings. The geometric parameters of eight compounds in the ground and excited states are listed in Table 1, as to illustrate the changes in the hydrogen bonding interactions upon photoexcitation. The calculated systems are not stable in ground states (S0(T)), except for 1,4-DHAQ whose stable structures are successfully obtained for both single proton and double protons transfer in ground state (Fig. 2). Comparison of the hydrogen bond lengths in ground and excited states showed that the hydrogen bonds enhanced in the sequence of 1-HAQ > 1,5-DHAQ > 1,8-DHAQ > 1,4-DHAQ. The hydrogen bond distances between donor O and H atoms in S1(N) and S1(T) are 1.008 Å and 1.459 Å for 1,8-DHAQ, and 1.014 Å and 1.540 Å for 1,4-DHAQ respectively, suggesting that the proton transfer of 1,4-DHAQ experienced a larger distance compared to 1,8-DHAQ. The changes in hydrogen bond distance of AYAAQs result from its electron-withdrawing ability. AAAQ did not present a stable structure for S1(T), consistent with the absence of LWE in the experiments. On the other hands, these molecules show the similar OD(ND)-H bond weakening tendency, suggesting that strengthening of the hydrogen bond promoted the ESIPT process.

Stable structures of 1,4-DHAQ and 1,5-DHAQ in ground states and excited states for ESIPT tautomers.
The role of dispersion interaction in hydrogen bonding interaction are considered by calculating the eight ground states structures with B3LYP-D373. The data and comparison with B3LYP are listed in Table S1. The difference is negligible, which mostly result from the strong hydrogen bond in such systems. The B3LYP-D3 assuredly improved the result. While considering the minor differences and the comparion with excited state, we used the B3LYP for the calculation.
Absorption/emission spectra and FMO analysis
The electronic excitation energies and corresponding fluorescence emission spectra of low-lying excited states for eight compounds were calculated by TDDFT method. The data are list in Table 2, which are consistent with the available experimental data. Generally, the energies of charge transfer states are underestimated in traditional functional compared with the long-range correction (LC) methods74,75. Then, we use the electron−hole analysis function to estimate the degree of charge transfer and calculate the vertical excitation energy with LC-BLYP. As shown in Tables S2 and S3, we think that except 1,5-DHAQ all other compounds have some portion of CT excitation. Compared with the B3LYP functional, the LC-BLYP functional significantly overestimate the excitation energy and show larger deviation with the experiment. While the B3LYP functional underestimates the energies, it give more accurate result. The molecules except for AAAQ are stable after proton transfer in excited state, with the different conformations. The relative energies of S1(T) were higher than S1(N) for 1,4-DHAQ, 1,8-DHAQ and CAAQ in ESIPT progress (Figs 2 and S1). 1,4-DHAQ and 1,5-DHAQ exist double ESIPT phenomenon with different properties in the ground and excited states (Fig. 2). 1,4-DHAQ and 1,5-DHAQ presented the S1 energies in the order of the T1 > T2 > N and T2 > N > T1, respectively. Thus, a fast ESIPT process is expected for 1,5-DHAQ and 1,4-DHAQ with the double ESIPT process. Furthermore, the energy of 1,8-DHAQ became slightly higher after ESIPT (Figure S1), giving rise to the solvent-sensitive ESIPT process for 1,8-DHAQ25.
The AYAAQs can be classified into three categories by their ESIPT proprieties (Figure S1). AAAQ failed to get a stable structure after ESIPT in the TDDFT calculation, consistent with the experiments. After ESIPT, the energy become higher for CAAQ and slightly lower for DCAQ, verifying the double emission in dichloromethane and acetonitrile. In TFAQ, ESIPT is an intense exothermic progress, leading to a “barrierless” reaction.
Eight compounds have uneven HOMO distributions concentrating on the functional groups, and the evenly distributed LUMO (Fig. 3). The hydroxyl groups in different compounds show a stronger influence on HOMO compared to LUMO. The π-orbital can be observed at the carbonyl groups of 1-HAQ and 1,4-DHAQ in HOMO, while no distribution appeared in 1,5-DHAQ and 1,8-DHAQ. The 1,4-DHAQ has the a small energy gap, resulting in the longest absorption wavelength in all DHAQs76. The properties of compounds 1,5-DHAQ and 1,8-DHAQ are similar. The FMO plots of AYAAQs show the identical HOMO and LUMO, suggesting that their properties depend on the functional groups, in other words the electron-withdrawing ability. The energy levels of HOMO and LUMO decline from AAAQ to TFAQ, with the gap increased which is consistent with the fact that blue shift of the absorption wavelength in experiments40,43.

The schematic diagram of HOMOs and LUMOs for eight compounds and orbital energy levels (in eV).
Potential energy surface
Potential energy surfaces (PESs) were scanned for molecules at S0 and S1 states. Figure 4 shows the PESs in S1 state for the eight compounds, while that in S0 state are showed in Figure S2. For HAQ and DHAQs, the energy barriers for ESIPT follow an order of 1,4-DHAQ > 1,8-DHAQ > 1,5-DHAQ ≈ 1-HAQ. The energy barrier is up to 10 kJ/mol for 1,4-DHAQ with the energy of S1(T) much higher than S1(N). 1-HAQ and 1,5-DHAQ exhibit the energy barriers lower than 2 kJ/mol, suggesting a “barrierless” ESIPT process. 1,4-DHAQ and 1,8-DHAQ have different OD-H bond lengths at S1(T), i.e., 1.540 Å and 1.459 Å for 1,4-DHAQ and 1,8-DHAQ, respectively. The shorter distance of proton transfer for 1,8-DHAQ is expected to be benificial for the ESIPT. The decreased energy barrier and enhanced exothermic effects from AAAQ to TFAQ are consistent with the experiments. CAAQ has the energy barrier of 13 kJ/mol, higher than DCAQ and TFAQ. Thus, the electron-withdrawing groups in proton donor part promote the ESIPT process by strengthening the hydrogen bond in excited state.

The scan of PES of S1 state of eight compounds as a function of OD(ND)–H bond length. The energy of stable structure in S1 state after structure optimization is set as zero point.
Excited state hydrogen bonding dynamics and AIM analysis
The comparison in the vibrational frequencies of O–H or N-H stretching modes have been proved as a very reliable method to monitor the hydrogen bond strengthening and weakening from ground to s15,21,77,78,79. The calculated vibrational frequencies are presented in Fig. 5. The redshift of O–H or N-H stretching modes suggests the strengthening of hydrogen bond. For HAQ and DHAQs, the vibrational frequency is located at 3400–3600 cm−1 in ground state and widely disperse from 2200 to 3000 cm−1 in excited state. The DHAQs have two O–H stretching modes, i.e., anti-symmetry (as) mode and symmetry (s) mode, the intensity presents large difference and one mode are hardly distinguished from the IR spectra. Larger intensity O–H stretching in excited state appeared in s mode for 1,4-DHAQ and as mode for 1,5-DHAQ and 1,8-DHAQ. The largest shift is nearly 1000 cm−1 for 1-HAQ, suggesting a strongest hydrogen bond strengthening. The shift of 1,4-DHAQ and 1,8-DHAQ is relatively small, especially for 1,4-DHAQ without ESIPT process. The hydrogen bond of AYAAQs are strengthened to different extent. The N-H stretching mode densely distributed around 3400 cm−1 in ground state and dispersed form 2500 to 3000 cm−1 in excited state. The corresponding redshift is 548 cm−1 for AAAQ and 952 cm−1 for TFAQ. The sequence of strengthening is coincident with the degree and rate of ESIPT (TFAQ > DCAQ > CAAQ > AAAQ).

The calculated IR spectra of eight compounds in the spectral region of both O–H or N-H stretching modes in the S0 and S1 states (the solid line and dash line show the corresponding vibrational modes in S0 and S1 states, respectively).
The bond critical point (BCP) generally appears between attractive atom pair. The value of real space functions at BCP have great significance to analyze the weak interaction80,81,82. For example, the value of ρ(r) at BCP is closely related to bond strengthening in analogous bond type, and the V(r) at BCP has been shown to be highly correlated with hydrogen bond energies. The relationship between hydrogen bond energy EHB and V(r) at corresponding BCP can be approximately described as72:
The extent of the hydrogen bond strengthening after photoexcitation is calculated the change of ρ(r) and V(r) at BCP in ground states and excited states (Tables 3 and 4). Upon photoexcitation, all the hydrogen bonds are enhanced. Density of all electrons ρ(r) at BCP position show the similar tendency. For clear comparison, we define the change of ρ(r) and V(r) as Δρ% and ΔV% as Δρ% = (ρES − ρGS)/ρGS × 100% and ΔV% = (VES − VGS)/VGS × 100%. The three DHAQs have double hydroxyls while the calculated ρ(r) and V(r) are only characterized at single BCP. The molecules that present dominant ESIPT fluorescence emission in different solvents have a lager degree strengthening, such as 1-HAQ, 1,5-DHAQ, DCAQ and TFAQ and their ΔV% are higher than 104%. The 1,4-DHAQ and AAAQ which without ESIPT progress have lowest range of strengthening which the ΔV% are lower than 72%. Those with solvent-depended ESIPT progress have a moderate strengthening, such as CAAQ and 1,8-DHAQ. While the Δρ% of AAAQ and 1,8-DHAQ are same but with different properties. Thus, the ΔV% is a better reference than Δρ% when conjecture the property of ESIPT.
Conclusion
DFT/TDDFT methods were employed to investigate the ESIPT process of eight AQs compounds. By analyzing the geometric structures, absorption/fluorescence spectra, infrared vibration and AIM, the ultrafast ESIPT processes for eight molecules were systemically studied. Hydrogen bond strengthening in excited state verified by the decreased hydrogen bond lengths, redshift of O–H or N-H stretching vibration modes and the increase of ρ(r) and V(r) at BCP are the driving forces for proton transfer in the excited states. The electron-withdrawing groups play the role in strengthening the hydrogen bondof OA…H. The exothermic reaction and “barrierless” ESIPT process are observed for 1-HAQ, 1,5-DHAQ and TFAQ with the barrier lower than 2 kJ/mol. 1,4-DHAQ have the highest transfer barrier hindering the occurrence of ESIPT. The alterable ESIPT property of 1,8-DHAQ, DCAQ and CAAQ mainly owes to their medium barrier and similar energy of S1(N) and S1(T). The change of electrons density ρ(r) and potential energy density V(r) at BCP position in ground state and excited state are the important indicators for the ESIPT process. The V(r) at BCP position is the general reference for various kinds of hydrogen bonds.
References
Hsu, Y. H. et al. Locked ortho- and para-Core Chromophores of Green Fluorescent Protein; Dramatic Emission Enhancement via Structural Constraint. J. Am. Chem. Soc. 136, 11805–11812 (2014).
Stavrov, S. S., Solntsev, K. M., Tolbert, L. M. & Huppert, D. Probing the decay coordinate of the green fluorescent protein: Arrest of cis-trans isomerization by the protein significantly narrows the fluorescence spectra. J. Am. Chem. Soc. 128, 1540–1546 (2006).
Yao, D. D. et al. Hydroxyphenyl-benzothiazole based full color organic emitting materials generated by facile molecular modification. J. Mater. Chem. 21, 3568–3570 (2011).
Furukawa, S., Shono, H., Mutai, T. & Araki, K. Colorless, Transparent, Dye-Doped Polymer Films Exhibiting Tunable Luminescence Color: Controlling the Dual-Color Luminescence of 2-(2′-Hydroxyphenyl)imidazo[1,2-a]pyridine Derivatives with the Surrounding Matrix. Acs. Appl. Mater. Inter. 6, 16065–16070 (2014).
Tang, K. C. et al. Fine Tuning the Energetics of Excited-State Intramolecular Proton Transfer (ESIPT): White Light Generation in A Single ESIPT System. J. Am. Chem. Soc. 133, 17738–17745 (2011).
Reja, S. I., Kumar, N., Sachdeva, R., Bhalla, V. & Kumar, M. d-PET coupled ESIPT phenomenon for fluorescent turn-on detection of hydrogen sulfide. RSC Adv. 3, 17770–17774 (2013).
Ono, S. et al. A study of hydrogen-bonding of amino acids, peptides and polypeptides in the solid state as a function of temperature by static (2) HNMR method. Journal of Molecular Structure 602, 49–58 (2002).
Steiner, T. The hydrogen bond in the solid state. Angew. Chem. Int. Edit. 41, 48–76 (2002).
Lin, Q. F. et al. Intermolecular hydrogen bonding-assisted high contrast fluorescent switch in the solid state. Dyes and Pigments 114, 33–39 (2015).
Zhao, G.-J. & Han, K.-L. Hydrogen Bonding in the Electronic Excited State. Accounts. Chem. Res. 45, 404–413 (2012).
Han, Y.-F. et al. An AAA-DDD Triply Hydrogen-Bonded Complex Easily Accessible for Supramolecular Polymers. Chem-Eur J 20, 16980–16986 (2014).
Perrin, C. L. & Nielson, J. B. “Strong” hydrogen bonds in chemistry and biology. Annu. Rev. Phys. Chem. 48, 511–544 (1997).
Pang, X. F. Properties of proton transfer in hydrogen-bonded systems and its experimental evidences and applications in biology. Prog. Biophys. Mol. Bio. 112, 1–32 (2013).
Nakamura, R., Hamada, N., Abe, K. & Yoshizawa, M. Ultrafast Hydrogen-Bonding Dynamics in the Electronic Excited State of Photoactive Yellow Protein Revealed by Femtosecond Stimulated Raman Spectroscopy. J. Phys. Chem. B. 116, 14768–14775 (2012).
Zhao, G. J. & Han, K. L. Hydrogen Bonding in the Electronic Excited State. Accounts. Chem. Res. 45, 404–413 (2012).
Wu, Y. D., Houk, K. N., Valentine, J. S. & Nam, W. Is Intramolecular Hydrogen-Bonding Important for Bleomycin Reactivity-a Molecular Mechanics Study. Inorg. Chem. 31, 718 (1992).
Zhao, J. Z., Ji, S. M., Chen, Y. H., Guo, H. M. & Yang, P. Excited state intramolecular proton transfer (ESIPT): from principal photophysics to the development of new chromophores and applications in fluorescent molecular probes and luminescent materials. Phys. Chem. Chem. Phys. 14, 8803–8817 (2012).
Kwon, J. E. & Park, S. Y. Advanced Organic Optoelectronic Materials: Harnessing Excited-State Intramolecular Proton Transfer (ESIPT) Process. Advanced Materials 23, 3615–3642 (2011).
Goodman, J. & Brus, L. E. Proton-Transfer and Tautomerism in an Excited-State of Methyl Salicylate. J. Am. Chem. Soc. 100, 7472–7474 (1978).
Zhao, G. J., Liu, J. Y., Zhou, L. C. & Han, K. L. Site-selective photoinduced electron transfer from alcoholic solvents to the chromophore facilitated by hydrogen bonding: A new fluorescence quenching mechanism. J. Phys. Chem. B. 111, 8940–8945 (2007).
Chai, S. et al. Reconsideration of the excited-state double proton transfer (ESDPT) in 2-aminopyridine/acid systems: role of the intermolecular hydrogen bonding in excited states. Phys. Chem. Chem. Phys. 11, 4385–4390 (2009).
Zhang, Y. J., Sun, M. T. & Li, Y. Q. How was the proton transfer process in bis-3, 6-(2-benzoxazolyl)-pyrocatechol, single or double proton transfer? Sci. Rep. 6, 25568–25574 (2016).
Li, Y. Q., Yang, Y. F. & Ding, Y. The new competitive mechanism of hydrogen bonding interactions and transition process for the hydroxyphenyl imidazo [1, 2-a] pyridine in mixed liquid solution. Sci. Rep. 7, 1574–1588 (2017).
Kobayashi, K., Iguchi, M., Imakubo, T., Iwata, K. & Hamaguchi, H. ESIPT-induced photocyclization of N,N ‘-diphenyl-1,5-dihydroxy-9,10-anthraquinone diimine. Chem. Commun. 7, 763–764 (1998).
Manna, A., Sayed, M., Kumar, A. & Pal, H. Atypical Energetic and Kinetic Course of Excited-State Intramolecular Proton Transfer (ESIPT) in Room-Temperature Protic Ionic Liquids. J. Phys. Chem. B. 118, 2487–2498 (2014).
Kobayashi, K., Iguchi, M., Imakubo, T., Iwata, K. & Hamaguchi, H. Photocyclization of N,N′-diphenyl-1-hydroxy-9,10-anthraquinone diimines via excited-state intramolecular proton transfer (ESIPT). Journal of the Chemical Society-Perkin Transactions 2, 1993–1998 (1998).
Han, D. Y. et al. ESIPT-based anthraquinonylcalix 4 crown chemosensor for In3 + . Tetrahedron Lett 51, 1947–1951 (2010).
Lee, S., Lee, J. & Pang, Y. Excited state intramolecular proton transfer of 1,2-dihydroxyanthraquinone by femtosecond transient absorption spectroscopy. Current Applied Physics 15, 1492–1499 (2015).
Kaur, N. & Kumar, S. Aminoanthraquinone-based chemosensors: colorimetric molecular logic mimicking molecular trafficking and a set-reset memorized device. Dalton Transactions 41, 5217–5224 (2012).
Ranyuk, E. et al. Diaminoanthraquinone-Linked Polyazamacrocycles: Efficient and Simple Colorimetric Sensor for Lead Ion in Aqueous Solution. Org. Lett. 11, 987–990 (2009).
Areti, S., Khedkar, J. K., Chilukula, R. & Rao, C. P. Thiourea linked peracetylated glucopyranosyl-anthraquinone conjugate as reversible ON-OFF receptor for fluoride in acetonitrile. Tetrahedron Lett 54, 5629–5634 (2013).
Hussain, H. et al. A fruitful decade from 2005 to 2014 for anthraquinone patents. Expert. Opin. Ther. Pat. 25, 1053–1064 (2015).
Markovic, V. et al. Synthesis, cytotoxic activity and DNA-interaction studies of novel anthraquinone-thiosemicarbazones with tautomerizable methylene group. European Journal of Medicinal Chemistry 64, 228–238 (2013).
Ryu, J., Kim, H. W., Kim, M. S. & Joo, T. Ultrafast Excited State Intramolecular Proton Transfer Dynamics of 1-Hydroxyanthraquinone in Solution. B. Korean. Chem. Soc. 34, 465–469 (2013).
Cho, D. W., Kim, S. H., Yoon, M. & Jeoung, S. C. Transient Raman spectroscopic studies on the excited-state intramolecular reverse proton transfer in 1-hydroxyanthraquinone. Chem. Phys. Lett. 391, 314–320 (2004).
Choi, J. R., Jeoung, S. C. & Cho, D. W. Time-resolved anisotropy study on the excited-state intramolecular proton transfer of 1-hydroxyanthraquinone. B. Korean. Chem. Soc. 24, 1675–1679 (2003).
Marzocchi, M. P., Mantini, A. R., Casu, M. & Smulevich, G. Intramolecular hydrogen bonding and excited state proton transfer in hydroxyanthraquinones as studied by electronic spectra, resonance Raman scattering, and transform analysis. J. Chem. Phys. 108, 534–549 (1998).
Mech, J., Grela, M. A. & Szacitowski, K. Ground and excited state properties of alizarin and its isomers. Dyes and Pigments 103, 202–213 (2014).
Cho, D. W., Song, K. D., Park, S. K., Jeon, K. S. & Yoon, M. Excited-state intramolecular proton transfer of 1,5-and 1,8-dihydroxyanthraquinones chemically adsorpted onto SiO2, SiO2-Al2O3, and Al2O3 matrices. B. Korean. Chem. Soc. 28, 647–651 (2007).
Nagaoka, S.-i, Endo, H., Ohara, K. & Nagashima, U. Correlation between Excited-State Intramolecular Proton-Transfer and Singlet-Oxygen Quenching Activities in 1-(Acylamino)anthraquinones. J. Phys. Chem. B. 119, 2525–2532 (2015).
Smith, T. P. et al. Spectroscopic Studies of Excited-State Intramolecular Proton-Transfer in 1-(Acylamino)Anthraquinones. Journal of Physical Chemistry 95, 10465–10475 (1991).
Schmidtke, S. J., Underwood, D. F. & Blank, D. A. Following the solvent directly during ultrafast excited state proton transfer. J. Am. Chem. Soc. 126, 8620–8621 (2004).
Schmidtke, S. J., Underwood, D. F. & Blank, D. A. Probing excited-state dynamics and intramolecular proton transfer in 1-acylaminoanthraquinones via the intermolecular solvent response. J. Phys. Chem. A. 109, 7033–7045 (2005).
Mohammed, O. F., Xiao, D., Batista, V. S. & Nibbering, E. T. J. Excited-State Intramolecular Hydrogen Transfer (ESIHT) of 1,8-Dihydroxy-9,10-anthraquinone (DHAQ) Characterized by Ultrafast Electronic and Vibrational Spectroscopy and Computational Modeling. J. Phys. Chem. A. 118, 3090–3099 (2014).
Muller, C., Schroeder, J. & Troe, J. Intramolecular hydrogen bonding in 1,8-dihydroxyanthraquinone, 1-aminoanthraquinone, and 9-hydroxyphenalenone studied by picosecond time-resolved fluorescence spectroscopy in a supersonic jet. J. Phys. Chem. B. 110, 19820–19832 (2006).
Arzhantsev, S. Y., Takeuchi, S. & Tahara, T. Ultrafast excited-state proton transfer dynamics of 1,8-dihydroxyanthraquinone (chrysazin) studied by femtosecond time-resolved fluorescence spectroscopy. Chem. Phys. Lett. 330, 83–90 (2000).
Smulevich, G., Foggi, P., Feis, A. & Marzocchi, M. P. Fluorescence Excitation and Emission-Spectra of 1,8-Dihydroxyanthraqquinone-D0 and 1,8-Dihydroxyanthraquinone-D2 in N-Octane at 10 K. J. Chem. Phys. 87, 5664–5669 (1987).
Nagaoka, S. & Nagashima, U. Effects of node of wave function upon excited-state intramolecular proton transfer of hydroxyanthraquinones and aminoanthraquinones. Chem. Phys. 206, 353–362 (1996).
Moosavi-Tekyeh, Z. & Tayyari, S. F. Theoretical and spectroscopic studies on molecular structure and hydrogen bonding of 2-trifluoroacetylphenol. Spectrochim. Acta. A. 135, 820–827 (2015).
Nagaoka, S.-i, Uno, H. & Huppert, D. Ultrafast Excited-State Intramolecular Proton Transfer of Aloesaponarin I. J. Phys. Chem. B. 117, 4347–4353 (2013).
Paul, B. K. & Guchhait, N. A computational insight into the photophysics of a potent UV absorber Tinuvin P: Critical evaluation of the role of charge transfer interaction and topological properties of the intramolecular hydrogen bonding. Computational and Theoretical Chemistry 966, 250–258 (2011).
Furche, F. & Ahlrichs, R. Adiabatic time-dependent density functional methods for excited state properties. J. Chem. Phys. 117, 7433–7447 (2002).
Whitten, J. L. Coulombic Potential-Energy Integrals and Approximations. J. Chem. Phys. 58, 4496–4501 (1973).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Physical Review Letters 77, 3865–3868 (1996).
Becke, A. D. Density-Functional Exchange-Energy Approximation with Correct Asymptotic-Behavior. Phys. Rev. A. 38, 3098–3100 (1988).
Raghavachari, K. Perspective on “Density functional thermochemistry. III. The role of exact exchange” - Becke AD (1993) J Chem Phys 98:5648-52. Theor. Chem. Acc. 103, 361–363 (2000).
Miehlich, B., Savin, A., Stoll, H. & Preuss, H. Results Obtained with the Correlation-Energy Density Functionals of Becke and Lee, Yang and Parr. Chem. Phys. Lett. 157, 200–206 (1989).
Lee, C. T., Yang, W. T. & Parr, R. G. Development of the Colle-Salvetti Correlation-Energy Formula into a Functional of the Electron-Density. Phys. Rev. B. 37, 785–789 (1988).
Krishnan, R., Binkley, J. S., Seeger, R. & Pople, J. A. Self-Consistent Molecular-Orbital Methods 20. Basis Set for Correlated Wave-Functions. J. Chem. Phys. 72, 650–654 (1980).
Clark, T., Chandrasekhar, J., Spitznagel, G. W. & Schleyer, P. V. Efficient Diffuse Function-Augmented Basis Sets for Anion Calculations. Iii. The 3–21+ G Basis Set for First-Row Elements, Li-F. Journal of Computational Chemistry 4, 294–301 (1983).
Frisch, M. J. et al. Gaussian 09, revision D.01, Gaussian, Inc., Wallingford, CT (2010).
Andrienko, G. A. Chemcraft 1.8. website: http://www.chemcraftprog.com (Date of access: 08/08/2016) (2016).
Cossi, M., Rega, N., Scalmani, G. & Barone, V. Energies, structures, and electronic properties of molecules in solution with the C-PCM solvation model. Journal of Computational Chemistry 24, 669–681 (2003).
Barone, V. & Cossi, M. Quantum calculation of molecular energies and energy gradients in solution by a conductor solvent model. J. Phys. Chem. A. 102, 1995–2001 (1998).
Bader, R. F. W. A bond path: A universal indicator of bonded interactions. J. Phys. Chem. A. 102, 7314–7323 (1998).
Bader, R. F. W., Carroll, M. T., Cheeseman, J. R. & Chang, C. Properties of Atoms in Molecules - Atomic Volumes. J. Am. Chem. Soc. 109, 7968–7979 (1987).
Bader, R. F. W. Atoms in Molecules. Accounts. Chem. Res. 18, 9–15 (1985).
Nakanishi, W., Hayashi, S. & Narahara, K. Atoms-in-Molecules Dual Parameter Analysis of Weak to Strong Interactions: Behaviors of Electronic Energy Densities versus Laplacian of Electron Densities at Bond Critical Points. J. Phys. Chem. A. 112, 13593–13599 (2008).
Bader, R. F. W. Atoms in Molecules: A Quantum Theory (Oxford Univ. Press, 1990).
Gatti, C. In The Quantum Theory of Atoms in Molecules (eds Matta, C. F. & Boyd, R. J.) Ch. 7, 165–206 (Wiley-VCH, 2007).
Lu, T. & Chen, F. W. Multiwfn: A multifunctional wavefunction analyzer. Journal of Computational Chemistry 33, 580–592 (2012).
Espinosa, E., Molins, E. & Lecomte, C. Hydrogen bond strengths revealed by topological analyses of experimentally observed electron densities. Chem. Phys. Lett. 285, 170–173 (1998).
Grimme, S., Ehrlich, S. & Goerigk, L. Effect of the Damping Function in Dispersion Corrected Density Functional Theory. Journal of Computational Chemistry 32, 1456–1465 (2011).
Yu, X. F., Yamazaki, S. & Taketsugu, T. Concerted or Stepwise Mechanism? CASPT2 and LC-TDDFT Study of the Excited-State Double Proton Transfer in the 7-Azaindole Dimer. Journal of Chemical Theory and Computation 7, 1006–1015 (2011).
Ma, Y. Z., Yang, Y. F., Lan, R. F. & Li, Y. Q. Effect of Different Substituted Groups on Excited-State Intramolecular Proton Transfer of 1-(Acylamino)-anthraquinons. J. Phys. Chem. C. 121, 14779–14786 (2017).
Labhart, H. Z. Q. Beschreibung Des Einflusses Von Substituenten Auf Das Absorptionsspektrum Ebener Molekeln - Anwendung Auf Anthrachinon. Helv. Chim. Acta. 40, 1410–1420 (1957).
Zhao, G.-J. & Han, K.-L. Time-dependent density functional theory study on hydrogen-bonded intramolecular charge-transfer excited state of 4-dimethylamino-benzonitrile in methanol. Journal of Computational Chemistry 29, 2010–2017 (2008).
Zhao, G.-J. & Han, K.-L. Ultrafast hydrogen bond strengthening of the photoexcited fluorenone in alcohols for facilitating the fluorescence Quenching. J. Phys. Chem. A. 111, 9218–9223 (2007).
Zhao, G.-J., Liu, J.-Y., Zhou, L.-C. & Han, K.-L. Site-selective photoinduced electron transfer from alcoholic solvents to the chromophore facilitated by hydrogen bonding: A new fluorescence quenching mechanism. J. Phys. Chem. B. 111, 8940–8945 (2007).
Mishra, P., Verma, K., Bawari, D. & Viswanathan, K. S. Does borazine-water behave like benzene-water? A matrix isolation infrared and ab initio study. J. Chem. Phys. 144, 234307–234315 (2016).
Stasyuk, A. J., Cyranski, M. K., Gryko, D. T. & Sola, M. Acidic C-H Bond as a Proton Donor in Excited State Intramolecular Proton Transfer Reactions. Journal of Chemical Theory and Computation 11, 1046–1054 (2015).
Svechkarev, D. A., Doroshenko, A. O. & Kolodezny, D. Y. 1,4-bis-(3-hydroxy-4-oxo-4H-chromen-2-yl)-benzene (bis-flavonol): synthesis, spectral properties and principle possibility of the excited state double proton transfer reaction. Central European Journal of Chemistry 10, 205–215 (2012).
Acknowledgements
This work was supported by the National Natural Science Foundation of China (NSFC Nos 21422309 and 21573229). G.J.Z. also thanks the financial support from the Frontier Science Project of the Knowledge Innovation Program of Chinese Academy of Sciences (CAS) and the Project for Excellent Member of CAS Youth Innovation Promotion Association.
Author information
Authors and Affiliations
Contributions
G.Z. proposed this project and D.Z. performed the theoretical calculations and wrote the manuscript. M.Z. and G.Z. revised the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zheng, D., Zhang, M. & Zhao, G. Combined TDDFT and AIM Insights into Photoinduced Excited State Intramolecular Proton Transfer (ESIPT) Mechanism in Hydroxyl- and Amino-Anthraquinone Solution. Sci Rep 7, 13766 (2017). https://doi.org/10.1038/s41598-017-14094-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-14094-5
- Springer Nature Limited
This article is cited by
-
Molecular structure, FT-IR and NMR analyses of dihydrogen-bonded B3N3H6···HM complexes: a DFT and MP2 approach
Chemical Papers (2020)
-
Amino proton donors in excited-state intramolecular proton-transfer reactions
Nature Reviews Chemistry (2018)