
Abstract
Mitotic spindles, which consist of microtubules (MTs) and associated proteins, play critical roles in controlling cell division and maintaining tissue homeostasis. The orientation of the mitotic spindle is closely related with the duration of mitosis. However, the molecular mechanism in regulating the orientation of the mitotic spindles is largely undefined. In this study, we found that Palladin is a novel MT-associated protein and regulator of spindle orientation, which maintains proper spindle orientation by stabilizing astral MTs. Palladin depletion distorted spindle orientation, prolonged the metaphase, and impaired proliferation of HeLa cells. Results showed that Palladin depletion-induced spindle misorientation and astral MT instability could be rescued by constitutively active AKT1 or dominant negative GSK3β. Our findings revealed that Palladin regulates spindle orientation and mitotic progression mainly through the AKT1–GSK3β pathway.
Similar content being viewed by others

Introduction
Spindle orientation determines the axis of cell division. Thus, precise orientation and architectural integrity of the mitotic spindle is critical for mitosis1. Perturbation of mitosis due to spindle misorientation causes chromosome instability, which is an important aspect of tumorigenesis2. Proper spindle orientation depends mainly on astral microtubule (MT) stabilization, proper distribution of cell cortical complexes, and intact F-actin3,4. Various force generators and cytoskeleton-related kinases have been found to be involved in the regulation of spindle orientation2,5,6,7,8,9,10,11,12, but the molecular mechanism is largely unknown.
Palladin (PALLD, RIG-K) was originally cloned from the acute promyelocytic leukemia NB4 cell line induced by all-trans retinoic acid (ATRA) in our lab and was one of the up-regulated ATRA-inducing genes13. It has been characterized as a key actin-binding and microfilament-associated protein14. Furthermore, PALLD is responsible for cell morphology, mobilization, adhesion, invasion and metastasis of cancer cells15,16,17,18. However, the function of PALLD in regulating mitosis remains unknown.
In this study, we investigated the biological function of PALLD in spindle orientation and cell division. Our results showed that PALLD influenced cell proliferation mainly through regulating mitotic progression, especially at the metaphase. Furthermore, we identified that PALLD was a novel MT-associated protein involved in spindle orientation regulation. We also found that PALLD interacted with AKT1 via the third IgC domain to maintain AKT1-GSK3β activation and spindle orientation. These findings suggested that PALLD played important roles in spindle orientation maintenance and mitotic progression.
Results
PALLD regulates mitotic progression
To investigate the roles of PALLD in mitosis, we knocked it down in HeLa cells (KD cells) using shRNAs (Supplementary Fig. 1a). Analysis of growth curves showed that average total cell number at seventh day decreased significantly in the KD group compared with the control (scramble 5.46 × 105 ± 0.13 × 105 vs. KD 3.92 × 105 ± 0.12 × 105, P < 0.001; average inhibition rate, 28.21%) (Fig. 1a). This finding indicated that PALLD depletion impaired cell proliferation in HeLa cells. To study the underlying mechanism, we calculated their mitotic index (MI). Average MI increased from 2.88% ± 0.53% to 6.25% ± 0.87% upon PALLD knockdown (scramble vs. KD, P < 0.01) (Fig. 1b,c). Consistent with this result, post-synchronization cell cycle analysis revealed that the transition from G2/M to G1 phase was delayed in KD cells (P < 0.05) (Supplementary Fig. 1b,c). No obvious difference in the rate of apoptosis was observed (Supplementary Fig. 1d). We further monitored the mitosis of H2B–EGFP-expressing cells in the scramble and KD groups in real time using high-resolution time-lapse microscopy. We found that PALLD knockdown significantly prolonged cell division time at the metaphase (scramble 24.67 ± 4.42 min vs. KD 39.00 ± 12.98 min, P < 0.001; average duration of metaphase prolonged by 58.09%) (Fig. 1d,e and Supplementary Videos 1 and 2). These data indicated that PALLD could regulate mitotic progression and cell proliferation.

PALLD regulates mitotic progression. (a) Proliferation of scramble and KD cells were monitored from day 1 to day 7 (n = 3). (b and c) MI was calculated in non-synchronized HeLa cells (randomly counted the number of pSer10 H3-positive cells from 130–200 cells/slide, from three independent experiments). pSer10 H3 red, DAPI blue, scale bar 25 µm. (d and e) H2B–EGFP (green)-expressing scramble and KD cells were monitored in real time by time-lapse imaging, and the duration of mitotic phases was calculated (total randomly counted 15 division cells each group, from four independent experiments). H2B–EGFP signal and cytoplasm are green and red, respectively. Scale bar, 5 µm. ***P < 0.001; **P < 0.01 in t-test for each graph.
PALLD is a MT-associated protein
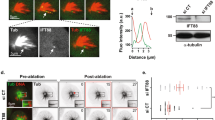
Next, we investigated how PALLD affects mitosis. PALLD was localized at mitotic spindles from the pro-metaphase to cytokinesis in HeLa cells (Fig. 2a). In addition, it was localized at the mitotic spindles in the metaphase of MDA-MB-231, a breast cancer cell line (Supplementary Fig. 2a). Therefore, the association of PALLD with mitotic spindles was common in different cell types. As a known microfilament-associated protein, PALLD was also localized at the cell cortex in HeLa cells (Supplementary Fig. 2b). PALLD2 and PALLD4 (two splice variants of PALLD) were constructed by one proline-rich region and immunoglobulin C-type domains (Fig. 2b). Endogenous PALLD (Fig. 2c,d) and overexpressed PALLD2 and PALLD4 could interact with α-tubulin via the third IgC domain (Supplementary Fig. 3a–c) in Hela cells. MT co-sedimentation assay showed that the GST-tagged third to fifth IgC domain of PALLD was able to co-pellet with polymerized MTs in vitro (Fig. 2e). Thus, our data demonstrated that PALLD is a novel MT-associated protein.

PALLD is associated with α-tubulin localizing at mitotic spindles. (a) Co-localization of PALLD and MTs in HeLa cells was displayed. PALLD green, α-tubulin red, DAPI blue, scale bar 10 µm. (b) Schematic representations of PALLD are shown, and the truncated PALLDs were constructed as indicated in Figure. PR, proline-rich region; IgC, immunoglobulin C2 type domain. (c and d) Detection of the interaction between endogenous PALLD and α-tubulin in HeLa cells. (e) In vitro MT co-sedimentation of GST-tagged third to fifth IgC domain was performed. The MT-associated protein MAPF was used as positive control. The original Western blot files are presented in Supplementary Figure 13.
PALLD sustains proper spindle orientation
Furthermore, we found evidence of various mitotic defects caused by PALLD knockdown, including spindle tilt, off-centered spindle, and multipolar spindle at the metaphase and misaligned chromosome at the pro-metaphase. However, the abnormalities were not increased by PALLD knockdown at the anaphase or cytokinesis such as lagging chromosomes or chromosomal bridges (Supplementary Fig. 4).We focused on studying spindle tilt among these mitotic defects because it showed the greatest change (scramble 19.33% ± 7.57% vs. KD 56.67% ± 4.16%, P < 0.01, n = 50; three independent experiments). Spindle angle was calculated in 50 metaphase cells in each group, and KD cells exhibited lower percentage of spindle angle within 10° (scramble 80.67% ± 7.57% vs. KD 43.33% ± 4.16%, P < 0.01) and larger spindle angle (median of scramble 4.48° vs. KD 10.74°, P < 0.001) (Fig. 3a–c ). Spindle misorientation caused by PALLD knockdown was completely rescued by shRNA-resistant PALLD2 or PALLD4 (Fig. 3c ). As U2OS (osteosarcoma cell line) is recognized as another classic model for mitosis research, we performed spindle orientation assay and confirmed that PALLD depletion also led to spindle misorientation in U2OS cells (Supplementary Fig. 5). Thus, our results demonstrated that PALLD plays an important role in sustaining proper spindle orientation.

PALLD sustains proper spindle orientation. (a) After staining with anti-γ-tubulin (red) and DAPI (blue), image stacks of mitotic HeLa cells were acquired at 0.5 µm interval along the z-axis. The spindle angle was calculated using an inverse trigonometric function. (b) Distribution of spindle angles in each group was assessed (n = 50, from three independent experiments). (c) Median of spindle angles was calculated in scramble and KD cells, scramble and KD cells expressing an empty vector and shRNA-resistant PALLD2 or PALLD4, and KD cells expressing PALLD2 or PALLD4 with deletion of the third IgC domain (n = 50). (d–f) The mean relative astral MT intensity was calculated as depicted in the schematic overview in (d) in both groups after DMSO or 1 µM Taxol treatment (n = 20) (e and f). α-Tubulin white, scale bar 5 µm. (g and h) Mitotic HeLa cells were stained with anti-EB1 (white) (g), and the percentage of cells with cortical anchoring of astral MTs (h) was counted (n = 50, from two independent experiments). ***P < 0.001; **P < 0.01; *P < 0.05; NS, P > 0.05. Mann–Whitney U test was used to compare the median of spindle angles, whereas t-test was used for other graphs.
As the instability of astral MTs could contribute to spindle misorientation, we compared the levels of astral MTs between the scramble and KD cells. Our data showed that PALLD depletion decreased astral MTs by 69.90% (scramble 36.45% ± 8.00% vs. KD 10.97% ± 2.96%, P < 0.001), and loss of astral MTs could be rescued by a MT-stabilizing drug Taxol (Fig. 3d–f ). At the same time, instability of astral MTs caused by PALLD depletion could be completely rescued by shRNA-resistant PALLD2 or PALLD4 (Supplementary Fig. 6a). Furthermore, we treated HeLa cells with Taxol (1 µM) or Nocodazole (10 ng/mL) to stabilize or depolymerize astral MTs, respectively. We found that the spindle tilt in KD cells could be rescued by Taxol, whereas spindle tilt in scramble cells was increased similar to that in KD cells by Nocodazole (Supplementary Fig. 6b). These results showed that PALLD could maintain proper spindle orientation by stabilizing astral MTs.
To visualize the MT plus ends, we performed MT plus end-binding protein EB1 staining in HeLa cells19,20,21. Our data showed that EB1 tracked MT plus end of spindle MTs and astral MTs connected to the cell cortex in scramble cells. By contrast, EB1 was present at the tips of spindle MTs in KD cells, and fewer cells showed EB1 signal near the cell cortex (scramble 75.00% ± 4.24% vs. KD 44.00% ± 2.83%, P < 0.05) (Fig. 3g,h). Similarly, dynein/dynactin complex mediated the connection between astral MTs and cell cortex22. Our data showed that significantly less dynein (scramble 62.67% ± 6.43% vs. KD 37.33% ± 7.57%, P < 0.05)/dynactin (P150Glued, scramble 67.33% ± 3.06% vs. KD 32.00% ± 5.29%, P < 0.001) complex was distributed on the cell cortex in PALLD KD group (Supplementary Fig. 7). In addition, PALLD knockdown did not affect centrosome nucleation and MT regrowth in mitotic HeLa cells (Supplementary Fig. 8). Taking this into account, our results indicated that PALLD knockdown led to loss of connection between astral MTs and cell cortex.
PALLD interacts with AKT1 via the third IgC domain to maintain AKT1-GSK3β activation and spindle orientation
AKT1 can regulate astral MT intensity in HeLa cells23 and spindle orientation in early Drosophila melanogaster embryos24. We confirmed that PALLD, including PALLD2 and PALLD4, was associated with AKT1 via the third IgC domain in HeLa cells (Fig. 4a,b and Supplementary Fig. 9). In addition, the third IgC domain in PALLD had a high-affinity binding with AKT1 to support their direct interaction (Supplementary Fig. 10). Without the third IgC domain, AKT1 binding to PALLD was abrogated and led to spindle misorientation in PALLD KD cells not being rescued (Fig. 3c). After cell synchronization to M phase, protein expression was detected by Western blot. The results showed that PALLD deletion caused down-regulation of pAKT T308 and S473 levels in mitotic HeLa cells (Fig. 4c), indicating that PALLD could regulate AKT1 activation at the metaphase. Next, our data exhibited that the AKT inhibitor MK2206 or LY294002 significantly increased spindle angle in scramble cells, but no obvious change was observed in KD cells (Supplementary Fig. 11). Furthermore, PALLD knockdown induced spindle misorientation, and astral MT instability could be rescued by constitutive overexpression of activated AKT1 (myristoylated AKT1, AKT1 CA). Meanwhile, control cells with dominant negative AKT1 (AKT1 T308A and S473A, AKT1 DN) overexpression showed similar phenotypes as KD cells (Fig. 4d and Supplementary Fig. 12a). Thus, these data demonstrate that PALLD sustained correct spindle orientation by maintaining AKT1 activity.

PALLD interacts with AKT1 to maintain AKT1-GSK3β activation and spindle orientation. (a and b) Results of co-immunoprecipitation between endogenous AKT1 and PALLD in mitotic HeLa cells. (c) After synchronization using double Thymidine, HeLa cells in the scramble and KD groups were collected, and proteins were extracted. Levels of proteins in the AKT1–GSK3β pathway were examined by Western blot. (d and e) Spindle angles of scramble and KD cells overexpressing AKT1 CA, AKT1 DN, GSK3β CA, or GSK3β DN were analyzed to determine the extent of rescue of spindle misorientation caused by PALLD knockdown (n = 50). (f) Molecular model illustrating the mechanism by which PALLD regulates spindle orientation and mitotic progress. ***P < 0.001; **P < 0.01; *P < 0.05; NS, P > 0.05. Mann–Whitney U test was used to compare median spindle angles across experimental conditions. The original Western blot files are presented in Supplementary Figure 13.
AKT1 mediates Ser9 phosphorylation of GSK3β, which inhibits GSK3β and promotes MT anchoring to the periphery in interphase via regulation of APC and CLASP2 phosphorylation25,26. Unexpectedly, PALLD depletion down-regulated the pGSK3β Ser9 level in mitotic HeLa cells (Fig. 4c). Random spindle angles or weakened astral MTs were induced by constitutively overexpressed GSK3β (GSK3β K85R, GSK3β CA) in scramble cells, and their phenotype was similar to that in KD cells. These PALLD knockdown-induced abnormal phenotypes could be rescued by overexpressing dominant negative GSK3β (GSK3β S9A, GSK3β DN) (Fig. 4e and Supplementary Fig. 12b). Thus, our data showed that PALLD maintains AKT1–GSK3β pathway activation to stabilize astral MTs and sustain proper spindle orientation (Fig. 4f).
Discussion
As an actin-binding and microfilament-associated protein, PALLD has been reported to regulate cell morphology, mobilization, adhesion, invasion, and metastasis of cancer cells27,28,29,30. Surprisingly, our data showed that PALLD also is a novel MT-associated protein and plays an important role in cell proliferation and mitosis. Knockdown PALLD in HeLa cells led to mitotic defects and metaphase extension. Consistently, MI increase and transition delay from M to G1 were observed in KD cells. Mitotic defects, such as multipolar spindle, could induce cell apoptosis19, but we did not find significant difference in cell apoptotic rate between the PALLD KD and control groups. The reason might be that the percentage of cells with multipolar spindle was low, and self-adjustment was not disturbed significantly. Therefore, cells without PALLD can overcome these defects by increasing division time.
In our study, PALLD knockdown-induced mitotic defects occurred mainly at metaphase, so PALLD depletion significantly prolonged the duration of metaphase. Among those mitotic defects, we focused on studying spindle tilt. Proper spindle orientation determines well-oriented cell division, which is critical for cell fate specification, tissue homeostasis, and tissue organization3,31. Spindle orientation is mainly determined by astral MT stability, F-actin integrity, and cell cortical complex distribution3. We found that PALLD knockdown induced astral MT instability. The evidence includes high-frequency spindle tilt and off-centered spindles occurred. In addition, stabilizing astral MTs by Taxol rescued spindle misorientation due to PALLD depletion. Astral MT stability depends on MT nucleation at the centrosome, MT dynamics, MT–cortex interactions, and MT behavior at the cortex4. We further found that PALLD depletion impaired the connection between astral MTs and the cell cortex. Therefore, we identified PALLD as a new regulator of spindle orientation.
Our results showed that the third IgC domain of PALLD was required for its interaction with MTs. It also mediated its interaction with F-actin14,32,33. The interaction between MT and microfilament cytoskeleton exists at the cell cortex and around mitotic spindles34,35. However, we do not know whether simultaneous or competitive binding to PALLD exists between MTs, F-actin, and other proteins. Therefore, we presumed that PALLD plays different roles through interacting the third IgC domain with different partners.
Furthermore, we found that AKT1 was associated with PALLD, and its activity was reduced after PALLD depletion. In turn, the inhibitory effect on GSK3β was lost in mitotic HeLa cells. AKT1 phosphorylates PALLD at Ser507 to regulate its actin-bundling function36,37. Meanwhile, our results showed that PALLD could also regulate AKT1 phosphorylation at Thr308 and Ser473 to maintain its activation in the metaphase. Several groups have reported that mitosis-specific AKT phosphorylation levels were up-regulated23,38. AKT is mainly activated by PI3K–PDK1, but many cell signaling molecules, such as Src, ERK, MEK, or PKC, also crosstalk with this central axis39. We presumed that PALLD might promote the interaction between AKT1 and its upstream molecules as a scaffold protein and might help anchor AKT1 to proper subcellular structures for its phosphorylation. AKT1 was reported to regulate spindle orientation via inhibiting GSK3β functions24,40. Inactivation of GSK3β leads to the accumulation of APC and CLASP2 at the plus end of MTs, helping them interact with microfilaments at the cell cortex and making them more stable25,26. Therefore, AKT1 and GSK3β also can function as astral MT stabilizers through promoting their cell cortical anchorage. In our studies, PALLD depletion-induced spindle misorientation can be rescued by AKT1 CA or GSK3β DN. Therefore, AKT1–GSK3β might be the major downstream pathway of PALLD for sustaining proper spindle orientation, but the detailed mechanisms of how PALLD regulated AKT1 activation need to be further investigated in future.
Interestingly, PALLD regulates spindle orientation acting as a novel MT-associated protein and modulator of the AKT1–GSK3β pathway, which was quite different from known regulators2,38,41,42,43,44,45,46,47. PALLD had no influence on MT nucleation or regrowth in HeLa cells although it bound to MTs. Whether PALLD could directly control MTs anchoring to the cell cortex still needs to be investigated. We speculated that PALLD is a linker of the AKT1–GSK3β pathway and MTs. It might be a new class of spindle orientation regulators, which anchor cytoskeleton-related kinase to the cytoskeleton.
In summary, our data demonstrated that PALLD, a novel MT-associated protein, played important roles in spindle orientation, mitotic progression, and cell proliferation. These activities were mainly mediated by the interaction between PALLD and AKT1 in mitotic cells, which maintained activation of the AKT1–GSK3β pathway and stabilized astral MTs to sustain proper spindle orientation.
Methods
Cell culture, cell cycle synchronization, and reagents
HeLa, MDA-MB-231, U2OS, and 293 T cells were supplied with DMEM (Gibco) containing 10% fetal bovine serum (HyClone), 100 U/mL penicillin, and 100 mg/ml streptomycin (Gibco) in a 5% CO2 humidified incubator at 37 °C. To synchronize cells in M phase, HeLa cells were blocked with 100 ng/mL Nocodazole (Sigma) for 16 h or treated with 2 mM Thymidine (Sigma) for 16 h and released to Thymidine-free medium for 10 h, followed by second exposure to 2 mM Thymidine for 16 h before being released for 9 h. The following reagents were also used: Taxol (1 µM, Selleck), LY294002 (50 µM, Selleck), and MK2206 (40 µM, Selleck).
Plasmid construction, transfection, and stable cell lines
The scramble (targeted sequence: AGCGTGTAGCTAGCAGAGG) and PALLD shRNA (targeted sequence: TGGAGATCTGACTGTTCAA) sequences were cloned into the BamH I–EcoR I sites of the pLVX-ShRNA2 vector. PALLD2 and PALLD4 were amplified from the cDNA library and cloned to the pPBCAG vector at Afl II–Pme I sites. Different truncated forms of PALLD representing different domains were amplified from plasmids pPBCAG-PALLD2 and pPBCAG-PALLD4 and then cloned into pPBCAG vector as shown above. ShRNA-resistant PALLD variants were constructed by site-directed mutagenesis. In addition, the third to fifth IgC domain and full length of PALLD4 were also cloned into BamH I–Xho I sites of the pGEX-4T2 vector for protein purification in vitro. All products were confirmed by DNA sequencing. HeLa cells and 293 T cells were transfected with Lipofectamine 2000 (Invitrogen) and the Calcium Phosphate Cell Transfection Kit (Beyotime), respectively. HeLa cells with stable knockdown of PALLD were made by transfecting with lentivirus-expressing PALLD ShRNA, which were selected by flow cytometry-based sorting. Knockdown efficiency was determined by Western blot.
Cell proliferation, apoptosis, and cell cycle analysis
To monitor the proliferation of HeLa cells, 1000 cells of each group were placed into a 96-well plate. After four days in culture, cells in each well were passaged at 1:10 ratio because of limited culture surface. Afterward, 1:10 volume of CCK-8 (Dojindo) was added to the medium and cultured for 2 h at 37 °C. The absorbance was tested at 450 nm, whereas the standard curve of HeLa cells was measured to convert absorbance value to cell counts.
For detecting the apoptotic rate of cells, cells were collected after treatment and washed twice in 1 × PBS. Following staining with Annexin-V FITC antibody (Biotool) for 10 min, cells were subjected to flow cytometry analysis.
For cell cycle analysis, cells were collected after synchronization and then fixed with 75% ethanol at −20 °C for 24 h. After washing with 1 × PBS once, 100 µg/mL RNase and 100 µg/mL PI were added in turn for 30 min each. The cells were then subjected to flow cytometry for cell cycle analysis.
Immunoblots
Cells were collected and extracted using lysis buffer (50 mM Tris-HCl (pH 7.4),150 mM NaCl, 1% Triton X-100, 1% sodium deoxycholate, 0.1% SDS) to which protease inhibitor cocktail (Roche) and PhosSTOP (Roche) tablets had been added. The lysates were centrifuged at 15,600 × g for 10 min, and the supernatants were collected for Western blot after quantification of protein concentration by Bradford assay (Beyotime). The following antibodies were applied: anti-Palladin (Proteintech), anti-α-tubulin (Sigma, Proteintech), anti-Akt (Cell Signaling), anti-phospho-Thr308-Akt (Cell Signaling), anti-phospho-Ser473-Akt (Cell Signaling), anti-GSK3β (Proteintech), anti-phospho-Ser9-GSK3β (Cell Signaling), anti-β-actin (Sigma), anti-FLAG tag (Abmart, Cell Signaling), and anti-HA tag (Cell Signaling).
Co-immunoprecipitations
For co-immunoprecipitation of endogenous proteins, HeLa cells were lysed with a buffer containing 50 mM Tris-HCl (pH 7.4), 150 mM NaCl, 1 mM EDTA, 1% Triton X-100, protease inhibitor cocktail (Roche), and PhosSTOP (Roche) tablets. Subsequently, the supernatants were collected after centrifugation at 15,600 × g for 10 min. After incubation with 1–2 µg primary antibody for 6 h at 4 °C, 20 µL protein A/G agaroses (Santa Cruz) were added and incubated for another 2 h. The immunoprecipitates were washed with 1 × PBS four times and eluted using 2 × SDS sample buffer (125 mM Tris-HCl, pH 6.8, with 4% SDS, 20% (v/v) glycerol, and 0.004% bromophenol blue) containing 5% DTT. For co-immunoprecipitation of overexpressed FLAG-tagged proteins, the supernatant extracts were incubated with 20 µL Anti-FLAG Affinity Gel for 3 h and then eluted in 100 µL 3 × FLAG peptide (150 ng/µL) after washing. The final products were then subjected to detection by Western blot.
Immunofluorescence staining
For detecting PALLD, cells were fixed with 3.7% formaldehyde at 37 °C for 10 min, followed by 0.5% Triton-X 100 in PBS at 37 °C for 10 min. To display α-tubulin, γ-tubulin, P150Glued, and dynein heavy chain staining, cells were incubated with methanol at −20 °C for 20 min. Cells were blocked with 3% BSA for 1 h and then incubated at 4 °C overnight with the following primary antibodies: Rabbit anti-Palladin (1:50, Proteintech), Mouse anti-α-tubulin (1:500, Sigma or Abcam), and Mouse anti-γ-tubulin (1:100, Abcam or Sigma, GTU88), Rabbit anti-P150Glued (1:50, Proteintech), and Rabbit anti-dynein heavy chain (1:50, Proteintech). The cells were then washed with 1 × PBS three times and incubated for 2 h with the following secondary antibodies: Alexa Fluor 488- or 594-goat anti-mouse or anti-rabbit IgG [H + L] antibodies (Invitrogen). Finally, DAPI (Invitrogen) staining was conducted for 10 min after washing.
Time-lapse images
Scramble or KD cells with H2B–EGFP expression were plated in 3 cm glass bottom dishes. After synchronization using double Thymidine, cells were cultured in Leibovitz’s L-15 Medium (Gibco) containing 10% FBS at 37 °C, and time-lapse images were obtained using a DeltaVision OMX microscope every 5 min for 6 h.
MT co-sedimentation assay
In vitro MT binding assay was performed using the Microtubule Binding Protein Spin-down Assay Kit (Cytoskeleton, BK029) following the manufacturer’s instructions. In test samples, 5 µg GST or GST-third to fifth IgC domian proteins with purity more than 95% was added. The results were analyzed with SDS-PAGE followed by Coomassie Blue staining.
Mitotic defect calculation
For detecting multipolar spindle, misaligned chromosome, off-centered spindle position, or spindle tilt, we randomly picked 50 metaphase cells in the scramble or KD group and then calculated the percentage of these mitotic defects. For calculating the frequency of lagging chromosomes and chromosomal bridge, 50 anaphase cells were randomly picked. All data were from three independent experiments.
Spindle orientation analysis
HeLa cells were plated on coverslips and synchronized at M phase using double Thymidine and then fixed and stained with γ-tubulin antibody and DAPI. Cells at the metaphase were selected for spindle orientation analysis by confocal imaging with 0.5 µm z-sections. Spindle angle calculation formula was α = arctan[Z/H], where the vertical (Z) and horizontal distances (H) between the two spindle poles were measured and spindle angle (α = arctan[Z/H]) was calculated using an inverse trigonometric function2,10.
MT regrowth assay
HeLa cells were placed in 4 °C for 2 h to depolymerize MTs completely and then to 37 °C and fixed at 0, 2, 5, and 8 min after recovery. Co-staining of α-tubulin antibody and DAPI was displayed, and the length of MTs was analyzed19.
Centrosome nucleation assay
After depolymerizing MTs completely at 4 °C for 2 h, HeLa cells were moved to 37 °C and fixed at 1.5 min after return. Centrosomes were marked by α-tubulin antibody, and their intensity was measured by ImageJ software48.
Protein binding kinetics and affinities
Binding of GST-AKT1 CA (85 kDa), α-tubulin (55 kDa), or GST (25 kDa, negative control) to GST-PALLD third IgC domain (40 kDa) was tested using an Octet RED96 instrument. All proteins were dissolved in the same buffer (20 mM NaH2PO4/Na2HPO4, 50 mM NaCl, pH = 7.6). Assays were performed at 30 °C in solid black 96-well plates, and the final volume for each well was 200 µL. GST-PALLD third IgC was labelled by biotin, and streptavidin-coated sensors were used. The final concentration of GST-AKT1 CA or α-tubulin was 200, 100, 50, 25, 12.5, and 0 nM. Also, a sensor with only PALLD third IgC biotin or testing proteins was displayed for correction of baseline drift. The running protocol was set as follows: 60 s biosensor baseline, 360 s loading biotin-labelled protein, 120 s protein loaded biosensor baseline, 600 s protein association, and 180 s protein dissociation. Octet data were processed by ForteBio Data Acquisition software v 8.1. K D values or other kinetic parameters were calculated from 1:1 global fitting for GST-PALLD third IgC domain49.
Statistical analysis
For spindle orientation analysis, Mann–Whitney U test was used to compare median spindle angles between scramble and knockdown groups due to non-normal distribution of data. For other data, Student’s t-test was applied to compare the differences between the two groups. A value of P < 0.05 was considered statistically significant.
Data availability
All relevant data supporting the findings of this study are available within the article and its Supplementary Information files, or from the corresponding author on request.
References
Garzon-Coral, C., Fantana, H. A. & Howard, J. A force-generating machinery maintains the spindle at the cell center during mitosis. Science 352, 1124–1127, https://doi.org/10.1126/science.aad9745 (2016).
Thoma, C. R. et al. VHL loss causes spindle misorientation and chromosome instability. Nature cell biology 11, 994–1001, https://doi.org/10.1038/ncb1912 (2009).
Lu, M. S. & Johnston, C. A. Molecular pathways regulating mitotic spindle orientation in animal cells. Development 140, 1843–1856, https://doi.org/10.1242/dev.087627 (2013).
di Pietro, F., Echard, A. & Morin, X. Regulation of mitotic spindle orientation: an integrated view. EMBO reports 17, 1106–1130, https://doi.org/10.15252/embr.201642292 (2016).
Godin, J. D. et al. Huntingtin is required for mitotic spindle orientation and mammalian neurogenesis. Neuron 67, 392–406, https://doi.org/10.1016/j.neuron.2010.06.027 (2010).
Kotak, S., Busso, C. & Gonczy, P. Cortical dynein is critical for proper spindle positioning in human cells. The Journal of cell biology 199, 97–110, https://doi.org/10.1083/jcb.201203166 (2012).
Mimori-Kiyosue, Y. & Tsukita, S. “Search-and-capture” of microtubules through plus-end-binding proteins (+TIPs). Journal of biochemistry 134, 321–326 (2003).
Matsumura, S. et al. ABL1 regulates spindle orientation in adherent cells and mammalian skin. Nat Commun 3, 626, https://doi.org/10.1038/ncomms1634 (2012).
Petridou, N. I. & Skourides, P. A. FAK transduces extracellular forces that orient the mitotic spindle and control tissue morphogenesis. Nat Commun 5, 5240, https://doi.org/10.1038/ncomms6240 (2014).
Hanafusa, H. et al. PLK1-dependent activation of LRRK1 regulates spindle orientation by phosphorylating CDK5RAP2. Nature cell biology 17, 1024–1035, https://doi.org/10.1038/ncb3204 (2015).
Kwon, M., Bagonis, M., Danuser, G. & Pellman, D. Direct Microtubule-Binding by Myosin-10 Orients Centrosomes toward Retraction Fibers and Subcortical Actin Clouds. Developmental cell 34, 323–337, https://doi.org/10.1016/j.devcel.2015.06.013 (2015).
Gallini, S. et al. NuMA Phosphorylation by Aurora-A Orchestrates Spindle Orientation. Current biology: CB 26, 458–469, https://doi.org/10.1016/j.cub.2015.12.051 (2016).
Liu, T. X. et al. Gene expression networks underlying retinoic acid-induced differentiation of acute promyelocytic leukemia cells. Blood 96, 1496–1504 (2000).
Dixon, R. D. et al. Palladin is an actin cross-linking protein that uses immunoglobulin-like domains to bind filamentous actin. The Journal of biological chemistry 283, 6222–6231, https://doi.org/10.1074/jbc.M707694200 (2008).
Yadav, R., Vattepu, R. & Beck, M. R. Phosphoinositide Binding Inhibits Actin Crosslinking and Polymerization by Palladin. J Mol Biol 428, 4031–4047, https://doi.org/10.1016/j.jmb.2016.07.018 (2016).
von Nandelstadh, P. et al. Actin-associated protein palladin promotes tumor cell invasion by linking extracellular matrix degradation to cell cytoskeleton. Mol Biol Cell 25, 2556–2570, https://doi.org/10.1091/mbc.E13-11-0667 (2014).
Grooman, B., Fujiwara, I., Otey, C. & Upadhyaya, A. Morphology and viscoelasticity of actin networks formed with the mutually interacting crosslinkers: palladin and alpha-actinin. PloS one 7, e42773, https://doi.org/10.1371/journal.pone.0042773 (2012).
Azatov, M., Goicoechea, S. M., Otey, C. A. & Upadhyaya, A. The actin crosslinking protein palladin modulates force generation and mechanosensitivity of tumor associated fibroblasts. Sci Rep 6, 28805, https://doi.org/10.1038/srep28805 (2016).
Zhu, M. et al. MISP is a novel Plk1 substrate required for proper spindle orientation and mitotic progression. The Journal of cell biology 200, 773–787, https://doi.org/10.1083/jcb.201207050 (2013).
Guesdon, A. et al. EB1 interacts with outwardly curved and straight regions of the microtubule lattice. Nature cell biology 18, 1102–1108, https://doi.org/10.1038/ncb3412 (2016).
Ran, J. et al. Phosphorylation of EB1 regulates the recruitment of CLIP-170 and p150glued to the plus ends of astral microtubules. Oncotarget. https://doi.org/10.18632/oncotarget.14222 (2016).
Yang, Y. et al. CYLD regulates spindle orientation by stabilizing astral microtubules and promoting dishevelled-NuMA-dynein/dynactin complex formation. Proceedings of the National Academy of Sciences of the United States of America 111, 2158–2163, https://doi.org/10.1073/pnas.1319341111 (2014).
Wakefield, J. G., Stephens, D. J. & Tavare, J. M. A role for glycogen synthase kinase-3 in mitotic spindle dynamics and chromosome alignment. Journal of cell science 116, 637–646 (2003).
Buttrick, G. J. et al. Akt regulates centrosome migration and spindle orientation in the early Drosophila melanogaster embryo. The Journal of cell biology 180, 537–548, https://doi.org/10.1083/jcb.200705085 (2008).
Akhmanova, A. et al. Clasps are CLIP-115 and -170 associating proteins involved in the regional regulation of microtubule dynamics in motile fibroblasts. Cell 104, 923–935 (2001).
Zumbrunn, J., Kinoshita, K., Hyman, A. A. & Nathke, I. S. Binding of the adenomatous polyposis coli protein to microtubules increases microtubule stability and is regulated by GSK3 beta phosphorylation. Current biology: CB 11, 44–49 (2001).
Goicoechea, S. M., Arneman, D. & Otey, C. A. The role of palladin in actin organization and cell motility. European journal of cell biology 87, 517–525, https://doi.org/10.1016/j.ejcb.2008.01.010 (2008).
Mykkanen, O. M. et al. Characterization of human palladin, a microfilament-associated protein. Molecular biology of the cell 12, 3060–3073 (2001).
Parast, M. M. & Otey, C. A. Characterization of palladin, a novel protein localized to stress fibers and cell adhesions. The Journal of cell biology 150, 643–656 (2000).
Goicoechea, S. M. et al. Palladin contributes to invasive motility in human breast cancer cells. Oncogene 28, 587–598, https://doi.org/10.1038/onc.2008.408 (2009).
Ahringer, J. Control of cell polarity and mitotic spindle positioning in animal cells. Current opinion in cell biology 15, 73–81 (2003).
Beck, M. R. et al. Structure and function of palladin’s actin binding domain. Journal of molecular biology 425, 3325–3337, https://doi.org/10.1016/j.jmb.2013.06.016 (2013).
Liang, W. et al. Expression, crystallization and preliminary X-ray studies of the immunoglobulin-like domain 3 of human palladin. Acta crystallographica. Section F, Structural biology and crystallization communications 62, 556–558, https://doi.org/10.1107/S1744309106016411 (2006).
Chan, Y. W. et al. Mitotic control of kinetochore-associated dynein and spindle orientation by human Spindly. The Journal of cell biology 185, 859–874, https://doi.org/10.1083/jcb.200812167 (2009).
Toyoshima, F. & Nishida, E. Integrin-mediated adhesion orients the spindle parallel to the substratum in an EB1- and myosin X-dependent manner. The EMBO journal 26, 1487–1498, https://doi.org/10.1038/sj.emboj.7601599 (2007).
Chin, Y. R. & Toker, A. The actin-bundling protein palladin is an Akt1-specific substrate that regulates breast cancer cell migration. Molecular cell 38, 333–344, https://doi.org/10.1016/j.molcel.2010.02.031 (2010).
Chin, Y. R. & Toker, A. Akt isoform-specific signaling in breast cancer: uncovering an anti-migratory role for palladin. Cell adhesion & migration 5, 211–214 (2011).
Toyoshima, F., Matsumura, S., Morimoto, H., Mitsushima, M. & Nishida, E. PtdIns(3,4,5)P3 regulates spindle orientation in adherent cells. Developmental cell 13, 796–811, https://doi.org/10.1016/j.devcel.2007.10.014 (2007).
Carracedo, A. & Pandolfi, P. P. The PTEN-PI3K pathway: of feedbacks and cross-talks. Oncogene 27, 5527–5541, https://doi.org/10.1038/onc.2008.247 (2008).
Goold, R. G., Owen, R. & Gordon-Weeks, P. R. Glycogen synthase kinase 3beta phosphorylation of microtubule-associated protein 1B regulates the stability of microtubules in growth cones. Journal of cell science 112(Pt 19), 3373–3384 (1999).
Bompard, G. et al. P21-activated kinase 4 (PAK4) is required for metaphase spindle positioning and anchoring. Oncogene 32, 910–919, https://doi.org/10.1038/onc.2012.98 (2013).
Hehnly, H. & Doxsey, S. Rab11 endosomes contribute to mitotic spindle organization and orientation. Developmental cell 28, 497–507, https://doi.org/10.1016/j.devcel.2014.01.014 (2014).
Machicoane, M. et al. SLK-dependent activation of ERMs controls LGN-NuMA localization and spindle orientation. The Journal of cell biology 205, 791–799, https://doi.org/10.1083/jcb.201401049 (2014).
Matsumura, S. et al. ABL1 regulates spindle orientation in adherent cells and mammalian skin. Nature communications 3, 626, https://doi.org/10.1038/ncomms1634 (2012).
Matsumura, S. & Toyoshima, F. ABL1 joins the cadre of spindle orientation machinery. Cell structure and function 37, 81–87 (2012).
Mitsushima, M., Toyoshima, F. & Nishida, E. Dual role of Cdc42 in spindle orientation control of adherent cells. Molecular and cellular biology 29, 2816–2827, https://doi.org/10.1128/MCB.01713-08 (2009).
Petridou, N. I. & Skourides, P. A. FAK transduces extracellular forces that orient the mitotic spindle and control tissue morphogenesis. Nature communications 5, 5240, https://doi.org/10.1038/ncomms6240 (2014).
Nakayama, Y. et al. c-Src but not Fyn promotes proper spindle orientation in early prometaphase. The Journal of biological chemistry 287, 24905–24915, https://doi.org/10.1074/jbc.M112.341578 (2012).
He, L. et al. Presenting native-like trimeric HIV-1 antigens with self-assembling nanoparticles. Nature communications 7, 12041, https://doi.org/10.1038/ncomms12041 (2016).
Acknowledgements
We thank X.-B. Yao, X.-L. Zhu and J. Zhu for critical comments on the work, H.-J. Yu and X.-M. Yan for experimental assistance, and all members of the Shanghai Institute of Hematology for their encouragement. X.-B. Yao and H.-J. Yu are from University of Science & Technology of China. X.-L. Zhu and X.-M. Yan are from Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences. This work was supported by the National Key Basic Research Program of China (2013CB966800), Ministry of Health Grant (201202003), Mega-projects of Scientific Research for the 12th Five-Year Plan (2013ZX09303302), National Natural Science Foundation of China (81123005), and Samuel Waxman Cancer Research Foundation.
Author information
Authors and Affiliations
Contributions
X.Z., Q.-H.H., T.Y., Z.C., and S.-J.C. designed the study. X.Z., X.-L.C., J.L., X.X., Y.-L.Z., Z.R., Y.-Y.X., and T.Y. performed the experiments. X.Z., Q.-H.H., and T.Y. analyzed the data. X.Z., Q.-H.H., T.Y., and S.-J.C. wrote the manuscript. X.Z., X.-L.C., and J.L. should be considered equal first authors.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhang, X., Chen, X., Liu, J. et al. Palladin is a novel microtubule-associated protein responsible for spindle orientation. Sci Rep 7, 11806 (2017). https://doi.org/10.1038/s41598-017-12051-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-12051-w
- Springer Nature Limited