
Abstract
We report the synthesis of spherical-shaped rare-earth (Eu3+ and Tb3+) ions doped CaMoO4 nanoparticles in double solvents (IPA and H2O) with the help of autoclave. The X-ray diffraction patterns well match with the standard values and confirm the crystallization in a tetragonal phase with an I41/a (88) space group. The luminescence spectra exhibit the strong red and green emissions from Eu3+ and Tb3+ ions doped samples, respectively. The X-ray photoelectron spectroscopy results show the oxidation states of all the elements present in the sample. The temperature-dependent luminescence spectra reveal the stability of Eu3+ and Tb3+ ions doped samples. The red- and green-emitting Eu3+ and Tb3+ ions doped CaMoO4 samples were used for detection and enhancement of latent fingerprints which are the common evidences found at crime scenes. The enhanced latent fingerprints obtained on different surfaces have high contrast with low background interference. The minute details of the fingerprint which are useful for individualization are clearly observed with the help of these nanopowders.
Similar content being viewed by others

Introduction
Nanoscience and nanotechnology evoke the research interest in synthesizing materials owing to their huge potential for applications in wide range of fields1. The self-assembled hierarchical structures having particular shapes and novel properties are important in the area of material synthesis and its application2, 3. They can bridge the nanoscale and microscale world and provide promising functions. Such features urge and initiate scientists in synthesizing inorganic hierarchical structures assembled by nanoparticles. This area of research has also got much attention and has an impact on forensic science for detecting latent fingerprints on potentially touched items4, 5. Latent fingerprints are not visible to the naked eye and need enhancement for identification and visualization. Among many identification techniques used for fingerprint detection, powdering is one technique which has been used since the early days of detection6. As we know, there are three main kinds of powders used for fingerprint detection i.e., standard, metallic and luminescent powders. The standard powder consists of adhesive polymer with a stain for contrast, and the metallic powder corresponds to powders of metals like Ag, Au and Pb7. These traditional powders have some difficulties and are unsafe to the human health and environment. The unique way to subdue such constrains is the use of rare-earth ions doped luminescent materials.
Rare-earth ions doped materials have characteristic properties and are applied in various fields ranging from electronics to biology and so on8,9,10. However, different types of host materials were studied and oxide-based materials were of significant interest because of their lower toxicity and robustness11. Metal molybdates of general formula AMoO4 (A = Ca, Sr, Ba) are one of the oxide-based host materials which give excellent luminescent properties due to the property of energy transfer from host lattice to the dopant ions12, 13. Among these metal molybdates, CaMoO4 is one of the host materials which belong to the scheelite family having tetragonal structure with a space group of I41/a. This material is highly transparent and allows a large range of light to pass through without weakening in luminescence. Additionally, these materials possess good chemical and physical properties in comparison to other oxide materials.
Many researchers have focused on and reported the synthesis of controlled CaMoO4 morphologies by following novel synthesis techniques. Different morphologies i.e., flower-like, doughnut-like, peanut-like, erythrocyte-like, walnut-like structures and so on were synthesized14,15,16,17. Recently, Liu et al. have demonstrated the controlled synthesis of CaMoO4 microspheres with PDDA18. The preparation of CaMoO4 takes a long synthesis time of 12 h and the particle size varies in the range of 1-2 μm. In this work, we demonstrate a relatively fast synthesis of spherical-shaped nanostructures formed with nano building blocks. The structural and optical properties were studied and the nanopowders were used to efficiently detect the latent fingerprints on different surfaces.
Results and Discussion
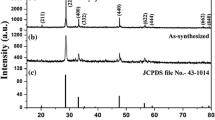
Figure 1(a) shows the XRD pattern of CaMoO4 sample along with the standard JCPDS #29-0351. The XRD data well matched with the standard values, the CaMoO4 sample was crystallized in tetragonal phase with a space group of I41/a (88) and no impurity peaks were found. To support the above mentioned statement, Rietveld refinement was performed on the XRD pattern using the FULLPROF software. The refined pattern with the observed (red), calculated (black), difference (blue) patterns and bragg-positions is shown in Fig. 1(b). The refinement parameters and the calculated and experimental cell parameters are tabulated as shown in Table 1. The lattice parameters obtained for CaMoO4 are a = b = 5.229 Å, c = 11.439 Å and V = 312.796 Å3. The CaMoO4 scheelite structure unit cell is shown in Fig. 1(c). The tetragonal structure with body centered inversion symmetry has Ca and Mo sites with S4 point symmetry. The crystal structure has CaO8 polyhedra and MoO4 tetrahedra units and the CaO8 unit shares four of its edges with four other polyhedrons through oxygen atoms. The MoO4 tetrahedra and CaO8 polyhedra are also connected through oxygen atoms and each oxygen atom is linked with one Mo atom and two Ca atoms19. The CaO8 units have two different Ca-O bond lengths (2.429 and 2.442 Å) and the MoO4 units have one Mo-O bond length (1.810 Å). The individual CaO8 and MoO4 units are shown in Fig. 1(c). Figure 1(d) shows the FTIR spectrum of CaMoO4 sample in the wavenumber range of 4000-450 cm−1. The spectrum consists of the bands at 3403 and 1583 cm−1 and the characteristic band of molybdate below 1000 cm−1. The bands at 3403 and 1583 cm−1 are due to the water molecules absorbed on to the sample and correspond to the H-O-H stretching and H-O-H bending vibrations, respectively20. The band centered at 770 cm−1 corresponds to the anti-symmetric stretching vibrations (ν3) of MoO4 tetragonal clusters21.

(a) XRD pattern, (b) XRD refinement, (c) crystal structure and (d) FTIR spectrum of CaMoO4.
The CaMoO4 spherical nanoparticles were synthesized by mixing appropriate amounts of calcium nitrate tetrahydrate, ammonium heptamolybdate tetrahydrate, sodium hydroxide and EDTA in IPA and DI water mixture as mentioned in the experimental section. The possible reaction mechanism for the formation of CaMoO4 is as follows:
When the solution-II was mixed with the solution-I, as described in the experimental section, the OH− anions from the NaOH solution react with the Ca2+ cations and form the Ca(OH)2. The formation of Ca(OH)2 is also confirmed by taking the XRD and it matches well with the JCPDS #72-0156 (Fig. S1). The heptamolybdate dissolved in DI water gives MoO4 2− monomeric oxyanion which is similar to the earlier reports22. The solution containing these monomeric oxyanions was added drop by drop into the solution containing Ca(OH)2, which further reacts and gives the final product CaMoO4 as shown in Eq. (3). The CaMoO4 synthesized in pure DI water rather gives nanoflake-like morphology as shown in Fig. S2. This is due to the ammonia-induced fast reaction between the calcium nitrate and ammonium heptamolybdate in DI water23. However, when IPA was added to the system, the reaction slows down and smaller particles are formed as shown in Fig. S3(a). Hence, it can be said that IPA plays a key role in obtaining the spherical-shaped CaMoO4 particles. These smaller particles aggregate and assemble into long spindle-shaped structures due to the presence of EDTA (inset of Fig. S3(a)) and are further formed into small clusters. These clusters grow further as the reaction time increases as shown in Fig. S3(b) and (c) and the Ostwald’s ripening also takes place, which results in the formation of bigger particles. Finally, the spherical-shaped CaMoO4 nanoparticles were obtained as shown in Fig. S3(d) due to the continuous assembly and Ostwald’s ripening processes.
Figure 2(a) shows the low-magnification FE-SEM image of the as-prepared CaMoO4 sample. From the FE-SEM image, it was observable that the formed particles are spherical in shape with sizes in the range of 300–500 nm. In Fig. 2(b), the high-magnification FE-SEM image revealed the rough surface on the spherical particles. Also, the spherical particles consist of smaller nanoparticles which are the building blocks. The chemical composition and atomic percentage distribution of elements were determined with the EDX spectrum, as shown in Fig. 2(c). The spectrum exhibited all the elements i.e., Ca, Mo and O and a small peak at approximately 2 keV which belongs to platinum (Pt) was observed. The presence is obvious due to the Pt coating done before measuring the sample. The atomic and weight percentage distribution of elements is tabulated and is shown in the inset of Fig. 2(c).

(a) Low-magnification FE-SEM image, (b) high-magnification FE-SEM image and (c) EDX spectrum (inset shows the elemental distribution table) of CaMoO4 sample.
Figure 3 shows the FE-SEM images, EDX patterns, XRD patterns and FTIR spectra of 1 mmol Eu3+ and 1 mmol Tb3+ ions doped CaMoO4 samples (hereafter referred as CaMoO4: 1Eu3+ and CaMoO4: 1Tb3+). The FE-SEM images in Fig. 3(a) and (b) displayed that the particles are spherical in shape and the doping of rare-earth elements does not have a great effect on the morphology of the CaMoO4 particles. As shown in Fig. 3(c) and (d), the EDX spectra revealed the elements present in Eu3+ and Tb3+ ions doped CaMoO4 samples. The spectra show all the elements i.e., Ca, Mo, O, Eu and Tb that are present in the samples. The insets show the table containing atomic and weight percentage distributions of elements. The XRD patterns of Eu3+ and Tb3+ ions doped CaMoO4 samples are shown in Fig. 3(e). The XRD patterns were well consistent with the standard JCPDS card value. Also, these XRD patterns well matched with that of the as-prepared CaMoO4 sample. With the doping of rare-earth elements, there are no significant changes in the XRD patterns, indicating that the Eu3+ and Tb3+ ions were completely doped into the Ca2+ sites. The difference in the ionic radii of Ca2+ and rare-earth ions of Eu3+ and Tb3+ was also very small (i.e., less than 30%) and very dilute concentrations were used in this experiment. Similarly, the FTIR spectra were also recorded for Eu3+ and Tb3+ ions doped CaMoO4 samples and they also matched well with that of the as-prepared CaMoO4 sample. The bending and stretching vibrations of H-O-H and H-O-H due to water molecules and the characteristic bands of molybdate were seen in the FTIR spectra.

(a,b) FE-SEM images, (c,d) EDX spectra, (e) XRD patterns and (f) FTIR spectra of the Eu3+ and Tb3+ ions doped CaMoO4 samples.
The oxidation states of the elements present in the molybdate samples were determined by a very sensitive technique i.e., XPS. The samples for XPS measurement were prepared by the following procedure; firstly, the powder samples were dispersed in DI water, drop-casted onto the cleaned silica substrate, dried and then used for measurements. The survey scan XPS spectra of the undoped and Eu3+ and Tb3+ ions doped CaMoO4 samples are shown in Fig. 4(a). The survey scan spectrum of CaMoO4 shows the peaks related to Ca, Mo and O and the doped samples also show similar peaks along with the Eu and Tb peaks for respective samples. The carbon 1 s peak in the spectra was due to the presence of trace amount of hydrocarbons during the XPS measurement. The high-resolution Ca 2p scan for CaMoO4 samples is shown in Fig. 4(b). As shown in Fig. 4(b), the two bands at binding energy (BE) values of 345.71 and 349.26 eV correspond to the Ca 2p3/2 and Ca 2p1/2, respectively. The full width at half maximum (FWHM) values for the two peaks were 1.41 and 1.41 eV and the area ratio, i.e., area of 2p3/2/area of 2p1/2 (ACa) was 2.00 for CaMoO4 sample. When the Eu3+ and Tb3+ ions were doped, the BE values of 2p3/2 and 2p1/2 did not show significant changes (Fig. 4(b)), and the ACa values also remained the same i.e., 2.00. The FWHM values slightly decreased as shown in Table 2. From the above obtained results and BE values, it is believed that the calcium is stabilized in +2 oxidation state in the undoped and doped samples. Figure 4(c) shows the high-resolution Mo 3d scan for all the CaMoO4 samples. The Mo 3d scan of CaMoO4 revealed two peaks at BE values of 231.24 and 234.37 eV corresponding to the 3d5/2 and 3d3/2. The AMo (area of 3d5/2/area of 3d3/2) value was found to be 1.29 and the FWHM values were found to be 1.24 and 1.24 eV, respectively. For Eu3+ and Tb3+ ions doped samples, the BE values of 3d5/2 and 3d3/2 did not show significant changes and the AMo values also remained almost the same. The FWHM value of the Eu3+ ions doped sample decreased and for the Tb3+ ions doped sample, the FWHM value remained the same, which can be seen in Table 2. The results indicate that the molybdenum is stabilized in +6 oxidation state in the undoped and doped samples. Figure 4(d) shows the high-resolution O 1 s spectra of the undoped and doped samples. The O 1 s spectrum of CaMoO4 exhibited a peak around 529 eV which is slightly asymmetric and two symmetric Gaussian peaks were fitted and labeled as O1 and O2. The O1 peak is attributed to the lattice oxygen of the CaMoO4 host lattice. The O2 peak is attributed to the chemisorbed oxygen or hydroxyl group as mentioned in earlier reports24. In spite of that, the O2 peak at the higher BE is normally due to the oxygen vacancies present in the sample, which was in consistent with previous reports25. The AO (O2/O1) values of the undoped and Eu3+ and Tb3+ ions doped samples were found to be 0.109, 0.176, and 0.124, respectively. The AO value of the undoped sample is smaller than those of the doped samples, suggesting the enrichment of oxygen vacancies. This result is reasonable as the defect and oxygen vacancy concentration increases with the doping of trivalent ions into the divalent calcium sites. Figure 4(e) shows the high-resolution Eu 3d and Tb 3d spectra of the Eu3+ and Tb3+ ions doped CaMoO4 samples, respectively. The Eu 3d spectrum exhibited a peak around BE value of 1134 eV which corresponds to the 3d5/2. This is consistent with the reported values and indicates that the europium ions exist in +3 oxidation state26. Similarly, the Tb 3d spectrum showed two peaks around BE values of 1240 and 1276 eV which are in accord with the reported values, indicating that the terbium ions exist in +3 oxidation state27, 28.

(a) Survey scan spectra, and high-resolution scan of (b) Ca 2p, (c) Mo 3d and (d) O 1 s for the undoped and doped CaMoO4 samples. High-resolution scan of (e) Eu 3d and (f) Tb 3d.
Figure 5(a) shows the photoluminescence (PL) excitation (PLE) spectrum of the Eu3+ ions doped CaMoO4 sample. The PLE spectrum was obtained by monitoring at an emission wavelength of 614 nm. The excitation spectrum revealed a broad band in the ultraviolet (UV) region and small f-f intraconfigurational transitions of Eu3+ ions in the longer wavelength region. The broad band centered at 267 nm is due to the charge transfer transition between the O2− and Mo6+ and so it is named as charge transfer band (CTB). The presence of intense CTB also indicates that there will be an energy transfer process from MoO4 2− moieties to Eu3+ ions. The weak bands observed at 394, 464 and 534 nm are assigned to the 7F0 → 5L6, 7F0 → 5D2 and 7F0 → 5D1 electronic transitions of Eu3+ ions, respectively. Figure 5(b) shows the PL emission spectrum of the Eu3+ ions doped CaMoO4 sample. The emission spectrum was obtained in the wavelength range of 500–725 nm by monitoring at an excitation wavelength of 267 nm. The PL emission spectrum showed an intense band centered at 614 nm which is due to the hypersensitive electric dipole 5D0 → 7F2 transition. The other bands centered at 591, 652 and 700 nm are due to the transition from the high energy 5D0 level to the ground states 7F1, 7F3 and 7F4, respectively. The asymmetric ratio, i.e., ratio of intensities of 5D0 → 7 F2/5D0 → 7F1 transitions, is higher than 1. This indicates that the Eu3+ ions occupy the non-inversion symmetry site and exhibit red emission. In general, the Ca2+ ions in the host lattice occupy inversion symmetry site, so the Eu3+ ions doped into the Ca2+ sites should give the asymmetric ratio less than 1. The opposite was observed due to the polarization effect of MoO4 2− moieties present in the host lattice and it also affects the luminescence of the sample. The MoO4 2− moieties of the host lattice are useful to get strong emission from activator ions by transferring the absorbed excitation energy non-radiatively to the Eu3+ ions12. The emission colors from the powder sample and the powder dispersed in DI water were snapped under normal and UV lights and are shown in the inset of Fig. 5(a). The PL emission intensity increased with the increase in the dopant concentration and reached its maximum at 1 mmol Eu3+ ion concentration, and then was quenched at higher concentrations. The concentration quenching effect occurs due to the decrease in Eu-Eu distance, which causes the non-radiative energy transfer between the activator ions. The variation in the emission intensity with activator ion concentration is shown in Fig. 5(c).

(a) PLE spectrum (inset shows the photographs of the powder sample and the powder dispersed in DI water under normal and UV lights), (b) PL emission spectrum and (c) PL emission intensity versus Eu3+ ion concentration for the Eu3+ ions doped CaMoO4 sample.
Figure 6 shows the PLE and PL emission properties of the Tb3+ ions doped CaMoO4 sample. As shown in Fig. 6(a), the PLE spectrum in the wavelength range of 200–550 nm was observed under the emission wavelength of 544 nm. The excitation spectrum consisted of CTB in the UV region which is due to the charge transfer between the O2− and Mo6+ ions and f-f transitions of Tb3+ ions in the longer wavelength region. The PL emission spectrum in the wavelength range of 450–675 nm was observed under the UV excitation of 267 nm (Fig. 6(b)). The emission spectrum shows the bands due to the transition from the higher energy 5D4 state to the ground state 7Fj (j = 2, 3, 4, 5 and 6) levels. Among all the emission bands, the PL emission intensity of the 5D4 → 7F5 electronic transition is high, which gives the characteristic green emission. The emission colors from the powder sample and the powder dispersed in DI water were snapped under normal and UV lights and are shown in the inset of Fig. 6(a). The PL emission intensity increased with the increase of dopant concentration and reached its maximum for 1 mmol of Tb3+ ion concentration. The emission intensity decreased further with the increase in the concentration of activator ions, which is due to the concentration quenching effect. The quenching in emission intensity is due to the shrinkage of Tb-Tb distance which causes the increase in the energy transfer to non-radiative sinks. The change in the emission intensity with respect to the activator ion concentration is shown in Fig. 6(c).

(a) PLE spectrum (inset shows the photographs of the powder sample and the powder dispersed in DI water under normal and UV lights), (b) PL emission spectrum and (c) PL emission intensity versus Tb3+ ion concentration for the Tb3+ ions doped CaMoO4 sample.
The luminescence decay curves of the Eu3+ and Tb3+ ions doped CaMoO4 samples were recorded as shown in Fig. 7(a). The decay curves were measured at an excitation wavelength of 267 nm and emission wavelengths of 614 and 544 nm for Eu3+ and Tb3+ ions doped CaMoO4 samples, respectively. The decay curves were well fitted to the double exponential decay function as shown below:
where I(t) and Io are the intensities at time t and 0. τ1 and τ2 are lifetime values and A and B are constants. The existence of double exponential behaviour is due to the presence of energy transfer from host lattice to activator ions and/or due to the presence of oxygen vacancies29, 30. The presence of oxygen vacancies in the Eu3+ and Tb3+ ions doped CaMoO4 samples was proved from the XPS results. The thermal stability is also an important parameter to be considered when it comes to the rare-earth doped phosphor materials. The temperature-dependent PL emission spectra of the Eu3+ and Tb3+ ions doped CaMoO4 samples in the wavelength rage of 400–700 nm are shown in Fig. 7(c) and (d). The emission intensity decreased gradually with the increase of temperature from 30 to 210 °C (step size of 20 °C). Generally, when the temperature is increased, the phonon manifold occurs in the high vibration levels and then is quenched to the ground states non-radiatively via the crossover between the ground and excited states. Hence, a drop in the emission intensity was observed with the increase of temperature. Additionally, the temperature at which the emission intensity drops to 50% of its original value called as thermal quenching temperature (T0.5) was also observed. The normalized intensity versus temperature was plotted as shown in Fig. S3. The T0.5 values for the Eu3+ and Tb3+ ions doped CaMoO4 samples were found to be 146 and 99 °C, respectively. The activation energy (Ea) values for the Eu3+ and Tb3+ ions doped CaMoO4 phosphor powders were obtained from the plots drawn between ln ((Io/I)−1) versus 1/KBT31. The plots were then fitted linearly as shown in the inset of Fig. 7(c) and (d). The slope of the straight line fitting gives the Ea values and the values were found to be 0.222 and 0.308 eV for the Eu3+ and Tb3+ ions doped CaMoO4 phosphor powders, respectively.

(a) Luminescence decay curves and (b,c) temperature-dependent PL emission spectra of the Eu3+ and Tb3+ ions doped CaMoO4 samples. Inset of (b,c) shows the linear fitting results for ln((Io/I)−1) versus 1/KBT.
The luminescence properties of CaMoO4 nanomaterials doped with Eu3+ and Tb3+ ions have been studied for being used in latent fingerprint detection. The size of these nanomaterials was useful for effective detection of minute details in the fingerprints. The strong visible light emissions (red and green) from these nanomaterials under UV (254 nm) light were also useful for latent fingerprint detection with low background interference and high contrast. Figure 8 shows the fingerprint images developed by Eu3+ and Tb3+ ions doped CaMoO4 phosphors on different substrates i.e., slide glass (Fig. 8(a) and (b)), compact disc (CD) (Fig. 8(c) and (d)) and stainless steel cup (Fig. 8(e) and (f)). The clear and bright images under the UV light indicate that the latent fingerprints on different kinds of substrates can be smoothly detected using these phosphor materials.

Digital photographs of the visualized CaMoO4:Eu3+ and CaMoO4:Tb3+ spheres stained latent fingerprint images on different substrates (a,b) slide glass, (b,c) CD and (e,f) stainless steel cup.
The latent fingerprint is one of the most common evidences found in any crime scenes and it is the one which presents the problem of invisibility. Therefore, application of chemical, physical or optical treatments is required to see the latent fingerprint32. Herein, we used the physical technique, i.e., powdering for the detection of latent fingerprints. To get into the details of the latent fingerprint, a specified substrate was selected, i.e., stainless steel cup. The detailed procedure of physical technique for developing the latent fingerprint was described in experimental section. The fresh fingerprint image and the powdered image are shown in Fig. 9(a) and (b). The latent fingerprint image shown in Fig. 9(b) is noticeable due to the attraction and adhesion of particles to the moisture and oily components present in the latent fingerprint. The fluorescent images under UV light showing the red and green emission colors are shown in Fig. 9(c) and (d). The fingerprint of one person is different from others and it is unique entity for the identification of a person. The fingerprint contains many minute details like bifurcations, islands, enclosures, etc. which are called as Galton details. The number and position of these minute details vary from person to person and are very important for individualization. The minutiae found in green color latent fingerprint image are shown from Fig. 9(e)-(h). The island found in the fingerprint is shown in Fig. 9(e) and the bifurcations and sweat pores found are shown in Fig. 9(f) and (g). All these details were marked with yellow circles in the figures. The ending of ridges are shown in Fig. 9(h) and displaying these minute details evidently makes these nanomaterials a potential candidate for latent fingerprint detection. The high-magnification optical images from Fig. 9(e)-(h) also revealed that the powder was attached only to the ridges, and the grooves were empty and do not show any emission color. To further confirm this, we have taken the pixel profile for both red and green optical latent fingerprint images. The pixel profile was obtained for a small area which was highlighted with a solid line on the digital photographs of the visualized latent fingerprint images (Fig. 10(a) and (b)). The corresponding fluctuations in red and green pixel values and the difference between ridges and furrows of the highlighted papillary ridges are shown in Fig. 10(c) and (d). From these pixel profile data, it is further confirmed that the grooves are empty and the powder was attached only on the ridges of the fingerprint. To test the quality of the powder and its affinity characteristics, the fingerprints taken on CD and stainless steel cup are preserved in ambient atmosphere for 9 days. Later, the latent fingerprint fluorescent images were observed under UV light as shown in Fig. 11. The imagesin Fig. 11(a) and (b) are the latent fingerprints on the CD and the images in Fig. 11(c) and (d) are the latent fingerprints on the stainless steel cup. As mentioned earlier, the affinity of the powder is due to the presence of moisture and oily components in the latent fingerprint. Actually, the finger ridges are appended only to the eccrine sweat glands. These glands are found all over the body and functions as thermoregulators. The secretions from these glands have 99% of water and the remaining 1% constituents include inorganic salts and organic derivatives. The derivatives include amino acids, proteins, lactic acid, urea, sugars, uric acid, creatinine and choline33. After a period of time, the water evaporated from the surface of the fingerprint and the oily components remained. Of these oily components, the amino acids remained as solid materials on the surface of the fingerprint and there are reports saying that the detectable amount of amino acids is found in fingerprints. The oxide materials react with the traceable amount of amino acids present in the latent fingerprint mark and have strong affinity. Hence, the finger marks were clearly observed even after 9 days of aging in ambient atmosphere.

(a) Bare image, (b) powdered image, (c,d) fluorescent images and (e–f) high-magnification images of the latent fingerprints under UV light irradiation, which were obtained from the surface of stainless steel cup.

(a,b) Area highlighted with solid line on the digital photographs of the visualized latent fingerprint images to verify the affinity of the rare-earth ions doped CaMoO4 spheres and (c,d) corresponding fluctuations in red and green values and the difference between ridges and furrows over the highlighted papillary ridges.

Digital photographs for the visualization of latent fingerprints on (a,b) CD and (c,d) stainless steel cup, which were stained with CaMoO4:Eu3+ and CaMoO4:Tb3+ spheres after 9 days of aging in ambient atmosphere.
Conclusion
In summary, the synthesis of spherical-shaped CaMoO4 nanoparticles using the mixture of IPA and DI water was studied. The samples were crystallized in tetrahedron phase with a space group I41/a (88) and the XRD patterns of the Eu3+ and Tb3+ ions doped samples well matched with that of the as-prepared sample along with the standard JCPDS card value. The luminescent properties of the Eu3+ and Tb3+ ions doped samples exhibited strong red and green visible emissions under UV irradiation. These materials were then used as an effective material for the detection and enhancement of latent fingerprints on different surfaces, including slide glass, compact disk and stainless steel cup. The fingerprint images were clear with high contrast and low background interference, and even showed the minute details which help for individualization. The affinity of the powders to the fingerprints was good, which results in an evident fingerprint ever after 9 days of aging in ambient atmosphere. The above mentioned results manifest the characteristics of CaMoO4 phosphor materials and the versatile nature as fluorescent powders for latent fingerprint detection.
Experimental Procedure
Materials
Calcium nitrate tetrahydrate (Ca(NO3)2∙4H2O), ammonium heptamolybdate tetrahydrate ((NH4)6Mo7O24∙4H2O), sodium hydroxide (NaOH), europium nitrate pentahydrate (Eu(NO3)3∙5H2O), terbium nitrate hexahydrate (Tb(NO3)3∙6H2O) and ethylene diamine tetraacetic acid (EDTA) were employed. All the chemicals are of analytical grade, purchased from Sigma-Aldrich Co. and are used as received without any further purification. Isopropyl alcohol (IPA) was purchased from ChemiTop Co., South Korea and the de-ionized (DI) water was obtained from a Milli-Q synthesis system (resistivity of 18.2 MΩ-cm).
Synthesis
For preparing the CaMoO4 spherical particles, three solutions, namely, solution-I, -II and -III were prepared as follows. Solution-I was prepared by mixing 35 mmol of calcium nitrate tetrahydrate in 70 ml of IPA, and 0.2 M 10 ml sodium hydroxide solution was prepared and labeled as solution-II. Solution-III was prepared by mixing 5 mmol of ammonium heptamolybdate tetrahydrate in 20 ml of DI. All the solutions were stirred to form transparent solutions. After few minutes, the solution-II was added dropwise to the solution-I. Later, the solution-III was added to the above mixture under continuous stirring. Finally, 0.1 g of EDTA was added and the solution was transferred to the teflon-lined stainless steel autoclave and heated to 160 °C for 5 h. The autoclave was further cooled down to room temperature, and the powder was collected, washed with DI water and dried at 70 °C. The europium and terbium doped samples were prepared by following the similar experimental procedure except adding stoichiometric amounts of europium nitrate pentahydrate/terbium nitrate hexahydrate to the solution-I.
Characterization
The prepared powders were characterized by using the following instruments or techniques. Field-emission scanning electron microscope (FE-SEM: LEO SUPRA 55, Carl Zeiss) equipped with energy dispersive X-ray spectrometer (EDX), X-ray diffractometer (XRD: M18XHF-SRA, Mac Science), Fourier transform infrared spectrometer (FTIR: Spectrum 100, PerkinElmer), X-ray photoelectron spectrometer (XPS: Thermo Electron Multilab2000) and spectroflourometer (FluroMate FS-2, Scinco, South Korea) equipped with a temperature controlled heating holder (25–250 °C) were employed.
Latent fingerprint detection
Substrates like slide glass, compact disk and stainless steel cup were selected for detecting the latent fingerprints. Fresh fingerprints were obtained by adopting the following procedure; the fingers were cleaned with soap and water and dried by exposing to air. Later, the finger was mildly rubbed over the forehead and pressed against different substrates as mentioned above. For studying the aging effect on fingerprints, the latent fingerprints on preferred substrates were preserved for 9 days in ambient atmosphere. Subsequently, the prepared fluorescent nanomaterials were brushed on to the surface of the substrates carefully. The excess powder was removed from the substrates carefully by smooth and mild motion for getting the latent fingerprint images. The latent fingerprints on different substrates under 254 nm UV light illumination were photographed with a Nikon D300 digital camera equipped with a Nikon AF-S VR 55 mm f/2.8 G IF-ED Macro lens without any filters.
Statement of authors and informed consent
The authors confirmed that all experiments (taking fingerprints of a volunteer/individual) were performed in accordance with relevant guidelines and regulations. An explicit informed consent was obtained from the anonymous volunteer providing the fingerprints. The individual explicitly allowed the authors to use the data in the present publication.
References
Balzani, V. Nanoscience and Nanotechnology: A Personal View of a Chemist. Small 1, 278–283 (2005).
Du, X. & He, J. Spherical silica micro/nanomaterials with hierarchical structures: Synthesis and applications. Nanoscale 3, 3984–4002 (2011).
Nie, Z., Petukhova, A. & Kumacheva, E. Properties and emerging applications of self-assembled structures made from inorganic nanoparticles. Nat Nano 5, 15–25 (2010).
Wang, Y. F., Yang, R. Q., Wang, Y. J., Shi, Z. X. & Liu, J. J. Application of CdSe nanoparticle suspension for developing latent fingermarks on the sticky side of adhesives. Forensic Sci. Int. 185, 96–99 (2009).
Liu, L., Gill, S. K., Gao, Y., Hope-Weeks, L. J. & Cheng, K. H. Exploration of the use of novel SiO2 nanocomposites doped with fluorescent Eu3+/sensitizer complex for latent fingerprint detection. Forensic Sci. Int. 176, 163–172 (2008).
Lennard, C. Fingerprint detection: current capabilities. Aust. J. Forensic Sci. 39, 55–71 (2007).
James, J. D., Pounds, C. A. & Wilshire, B. Production and Characterisation of Flake Metal Powders for Fingerprint Detection. Powder Metall. 34, 39–44 (1991).
Kenyon, A. J. Recent developments in rare-earth doped materials for optoelectronics. Prog. Quantum Electron. 26, 225–284 (2002).
Perera, T. S. H. et al. Rare Earth Doped Apatite Nanomaterials for Biological Application. J. Nanomater. 2015, 6 (2015).
Bouzigues, C., Gacoin, T. & Alexandrou, A. Biological Applications of Rare-Earth Based Nanoparticles. ACS Nano 5, 8488–8505 (2011).
Malhotra, B. D., Maumita, D. & Pratima, R. S. Opportunities in nano-structured metal oxides based biosensors. J. Phys: Conf. Ser. 358, 012007 (2012).
Gupta, S. K. et al. Energy transfer dynamics and luminescence properties of Eu3+ in CaMoO4 and SrMoO4. Dalton Trans. 44, 18957–18969 (2015).
Zhang, L. Y. et al. Morphology and luminescent properties of SrMoO4:Eu3+, Dy3+. J. Mater. Sci. - Mater. Electron. 27, 5164–5174 (2016).
Liu, X. et al. Synthesis and photoluminescence of novel 3D flower-like CaMoO4 architectures hierarchically self-assembled with tetragonal bipyramid nanocrystals. Opt. Mater. 43, 10–17 (2015).
Lv, L., Liu, X., Xu, C. & Wang, X. Synthesis and Optimum Luminescence of Doughnut-Shape Undoped and Doped CaMoO4. J. Nanosci. Nanotechnol. 14, 3521–3526 (2014).
Luo, Y.-S. et al. Controllable synthesis and luminescent properties of novel erythrocyte-like CaMoO4 hierarchical nanostructures via a simple surfactant-free hydrothermal route. Dalton Trans. 39, 2226–2231 (2010).
Wang, X.-F. et al. Hydrothermal synthesis and luminescence properties of 3D walnut-like CaMoO4:Eu3+ red phosphors. J. Alloys Compd. 599, 102–107 (2014).
Liu, X. et al. Controllable synthesis of uniform CaMoO4:Eu3+, M+ (M = Li, Na, K) microspheres and optimum luminescence properties. RSC Adv. 5, 9441–9454 (2015).
Parchur, A. K. & Ningthoujam, R. S. Preparation and structure refinement of Eu3+ doped CaMoO4 nanoparticles. Dalton Trans. 40, 7590–7594 (2011).
Gayatri Sharma, K. & Rajmuhon Singh, N. Synthesis and luminescence properties of CaMO4:Dy3+ (M = W, Mo) nanoparticles prepared via an ethylene glycol route. New J. Chem. 37, 2784–2791 (2013).
Krishna Bharat, L., Lee, S. H. & Yu, J. S. Synthesis, structural and optical properties of BaMoO4:Eu3+ shuttle like phosphors. Mater. Res. Bull. 53, 49–53 (2014).
Raju, G. S. R., Pavitra, E., Ko, Y. H. & Yu, J. S. A facile and efficient strategy for the preparation of stable CaMoO4 spherulites using ammonium molybdate as a molybdenum source and their excitation induced tunable luminescent properties for optical applications. J. Mater. Chem. 22, 15562–15569 (2012).
Yin, J., Gao, F., Wei, C. & Lu, Q. Water Amount Dependence on Morphologies and Properties of ZnO nanostructures in Double-solvent System. Sci. Rep. 4, 3736 (2014).
Al-Gaashani, R., Radiman, S., Daud, A. R., Tabet, N. & Al-Douri, Y. XPS and optical studies of different morphologies of ZnO nanostructures prepared by microwave methods. Ceram. Int. 39, 2283–2292 (2013).
Wan, B. et al. Glassy State Lead Tellurite Nanobelts: Synthesis and Properties. Nanoscale Res. Lett. 5, 1344 (2010).
Kumar, S., Prakash, R., Choudhary, R. J. & Phase, D. M. Structural, XPS and magnetic studies of pulsed laser deposited Fe doped Eu2O3 thin film. Mater. Res. Bull. 70, 392–396 (2015).
Lin, L., Sun, X., Jiang, Y. & He, Y. Sol-hydrothermal synthesis and optical properties of Eu3+, Tb3+-codoped one-dimensional strontium germanate full color nano-phosphors. Nanoscale 5, 12518–12531 (2013).
Tshabalala, K. G. et al. Enhanced green emission from UV down-converting Ce3+–Tb3+ co-activated ZnAl2O4 phosphor. J. Vac. Sci. Technol., B 30, 031401 (2012).
Gupta, S. K. et al. Multifunctional pure and Eu3+ doped β-Ag2MoO4: photoluminescence, energy transfer dynamics and defect induced properties. Dalton Trans. 44, 19097–19110 (2015).
Parchur, A. K. et al. Luminescence properties of Eu3+ doped CaMoO4 nanoparticles. Dalton Trans. 40, 7595–7601 (2011).
Hussain, S. K. & Yu, J. S. Broad red-emission of Sr3Y2Ge3O12:Eu2+ garnet phosphors under blue excitation for warm WLED applications. RSC Adv. 7, 13281–13288 (2017).
Gaensslen, H. C. L. R. R. R. E. Advances in Fingerprint Technology. second edn, (CRC Press, 1994).
Sodhi, G. S. & Kaur, J. Physical developer method for detection of latent fingerprints: A review. Egypt. J. Forensic Sci. 6, 44–47 (2016).
Acknowledgements
This work was supported by the National Research Foundation of Korea (NRF) Grant funded by the Korea Government (MSIP) (No. 2015R1A5A1037656 and No. 2017R1A2B4011998).
Author information
Authors and Affiliations
Contributions
L.K.B., G.S.R.R. and J.S.Y. conceived, designed and directed the project. L.K.B. synthesized and characterized the phosphor powders. G.S.R.R. directed the fabrication process and calculations. L.K.B., G.S.R.R. and J.S.Y. co-wrote the manuscript. All authors discussed the results and commented on the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Bharat, L.K., Raju, G.S.R. & Yu, J.S. Red and green colors emitting spherical-shaped calcium molybdate nanophosphors for enhanced latent fingerprint detection. Sci Rep 7, 11571 (2017). https://doi.org/10.1038/s41598-017-11692-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-11692-1
- Springer Nature Limited
This article is cited by
-
AI-authenticated latent fingerprint developed using ultra-bright nitrogen-doped carbon dots-intercalated Zn(OH)2 nanosheets
Chemical Papers (2024)
-
In-sensor reservoir computing system for latent fingerprint recognition with deep ultraviolet photo-synapses and memristor array
Nature Communications (2022)
-
Multicolour tuning of different RE ion doped CaMoO4 nanoparticles by single wavelength excitation
Journal of Materials Science: Materials in Electronics (2020)
-
Enhanced fluorescent intensity of magnetic-fluorescent bifunctional PLGA microspheres based on Janus electrospraying for bioapplication
Scientific Reports (2018)