
Abstract
In 2016 and 2017, monoclonal antibodies targeting PD-L1, including atezolizumab, durvalumab, and avelumab, were approved by the FDA for the treatment of multiple advanced cancers. And many other anti-PD-L1 antibodies are under clinical trials. Recently, the crystal structures of PD-L1 in complex with BMS-936559 and avelumab have been determined, revealing details of the antigen-antibody interactions. However, it is still unknown how atezolizumab and durvalumab specifically recognize PD-L1, although this is important for investigating novel binding sites on PD-L1 targeted by other therapeutic antibodies for the design and improvement of anti-PD-L1 agents. Here, we report the crystal structures of PD-L1 in complex with atezolizumab and durvalumab to elucidate the precise epitopes involved and the structural basis for PD-1/PD-L1 blockade by these antibodies. A comprehensive comparison of PD-L1 interactions with anti-PD-L1 antibodies provides a better understanding of the mechanism of PD-L1 blockade as well as new insights into the rational design of improved anti-PD-L1 therapeutics.
Similar content being viewed by others


Introduction
Programmed death 1 (PD-1) and its ligands PD-L1 and PD-L2 are key co-inhibitory molecules in the modulation of T-cell mediated immune responses1,2,3. PD-1 is a type I membrane protein with a single extracellular immunoglobulin superfamily (IgSF) V-set domain that is expressed on the surface of activated T cells in peripheral tissues4, 5. PD-L1 and PD-L2 are commonly expressed on dendritic cells and macrophages, and their ectodomains are composed of a membrane distal IgSF V-set and a membrane proximal IgSF C-set domains6, 7. Ligation of PD-1 with its two ligands initiates co-inhibitory signaling through the cytoplasmic domain of PD-1, containing an immunoreceptor tyrosine-based inhibitory motif and an immunoreceptor tyrosine-based switch motif, thus leading to activation of SHP phosphatases that downregulate TCR signaling by dephosphorylating effector molecules involved in the signaling8, 9. As a result, PD-1 signaling prevents excessive or harmful inflammation and maintains immune tolerance to self-antigens under normal conditions10.
PD-L1 is often overexpressed in different tumors, and its interaction with PD-1 on T cells enables cancer cells to evade T-cell-mediated immune responses11. Thus, blocking the PD-1/PD-L1 interaction can restore T-cell activation and antitumor responses12,13,14. The success of antibody-based PD-1/PD-L1 blockade therapies has provided a major breakthrough in the fight against human cancers, especially for solid tumors. The FDA approved the first anti-PD-L1 drug, atezolizumab (Tecentriq®), for the treatment of advanced urothelial carcinoma in May 2016 and metastatic non-small cell lung cancer (NSCLC) in October 201615,16,17,18. In 2017, avelumab (Bavencio®) and durvalumab (Imfinzi®) were also approved by the FDA for Merkel-cell carcinoma and advanced bladder cancer, respectively (Supplementary Table S1). Prior to the approval of the anti-PD-L1 antibodies, the anti-PD-1 drugs pembrolizumab (Keytruda®) and nivolumab (Opdivo®) have been widely used clinically and demonstrated unprecedented therapeutic benefits since 201419,20,21,22,23. In addition, other anti-PD-L1 antibodies including BMS-936559 are in multiple clinical trials for the treatment of NSCLC, renal cell cancer, head and neck cancer, gastric cancer, and other cancers24,25,26,27.
Combinations of anti-PD-1/PD-L1 antibodies with other therapies are being explored as potentially synergistic therapeutic strategies. Among these, the combination of anti-PD-1/PD-L1 and anti-CTLA-4 antibodies has been the most successful, probably because PD-1/PD-L1 and CTLA-4/B7 interactions play different roles in T-cell immunity6, 28,29,30. A clinical trial of a combination therapy involving durvalumab and an anti-CTLA-4 antibody, tremelimumab, showed antitumor activity in NSCLC regardless of PD-L1 status, suggesting that this combination has therapeutic potential for patients with PD-L1-negative tumors31. As PD-L1 is also expressed on activated T cells, the Fc domains of atezolizumab and durvalumab were engineered to eliminate antibody-dependent cellular cytotoxicity (ADCC) or complement-dependent cytotoxicity (CDC), thereby preventing the depletion of T cells expressing PD-L132, 33.
The structures of murine PD-1 in complex with human PD-L1, murine PD-1 in complex with murine PD-L2, and human PD-1 in complex with human PD-L1 have established the structural foundations of the interaction of PD-1 with its ligands, PD-L1 and PD-L234,35,36,37. Recently, the crystal structures of PD-L1 in complex with the Fab fragment of BMS-936559 and the single-chain Fv fragment (scFv) of avelumab have been determined, revealing details of the antigen-antibody interactions38, 39. However, the epitopes and the PD-L1 blocking mechanism of atezolizumab and durvalumab remain unclear. It is also unknown whether there are novel binding sites on PD-L1 for the design and improvement of anti-PD-L1 agents and whether atezolizumab and durvalumab utilize a similar or distinct competitive binding mode to PD-L1 compared with other anti-PD-L1 antibodies for the development of better combination therapies.
In the present study, we report the crystal structures of the N-terminal IgSF V-set domain of PD-L1 in complex with the Fab fragments of atezolizumab and durvalumab, thereby elucidating the structural basis for the blockade of the PD-1/PD-L1 interaction by these therapeutic antibodies. In addition, a comprehensive analysis of the PD-L1 interactions with the receptor PD-1 and anti-PD-L1 antibodies, including atezolizumab, durvalumab, BMS-963559, and avelumab, provides new insights into the development of future anti-PD-L1 therapeutics, including small-molecule modulators and next-generation therapeutic antibodies.
Results
Structure determination of PD-L1 in complex with atezolizumab and durvalumab
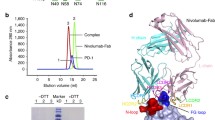
In this study, the IgSF V-set domain of PD-L1 was expressed as inclusion bodies in E. coli and refolded in vitro to obtain a soluble form. The Fab fragments of the anti-PD-L1 antibodies were produced by periplasmic expression in E. coli. Gel filtration analysis confirmed that that the 1:1 complexes of PD-L1 with each antibody exist as monomers in solution. The crystal structure of the IgSF V-set domain of human PD-L1 in complex with the atezolizumab Fab fragment was determined at a resolution of 3.10 Å with R/Rfree = 0.208/0.256 (Fig. 1a,c). PD-L1 and the atezolizumab Fab form a 1:1 complex in the crystal as in solution, and the crystallographic asymmetric unit contained five copies of the complex without any symmetric relationship among them (Supplementary Figure S1a). Superposition of the PD-L1 protein from the five copies in an asymmetric unit showed that the PD-L1 protein and the variable region of atezolizumab exhibited little structural deviation from each other, whereas the constant region swings almost 30° due to the intrinsic flexibility of the Fab elbow (Supplementary Figure S1b). Almost all residues in the complex, except for a few residues within the loops of the constant region of the atezolizumab Fab fragment, were well defined in the electron density map, clarifying the precise antigen-antibody interactions despite the relatively low resolution of the structural data (Supplementary Figure S2a). We also determined and refined the crystal structure of the IgSF V-set domain of human PD-L1 in complex with the durvalumab Fab fragment at a resolution of 2.65 Å with R/Rfree = 0.181/0.219 (Fig. 1b,d). The asymmetric unit contained only one PD-L1/durvalumab complex with 1:1 stoichiometry, and all residues in the complex were well defined by the electron density (Supplementary Figure S2b). Both atezolizumab and durvalumab use all three complementarity determining regions (CDRs) from the heavy chain (HCDR1, HCDR2, and HCDR3) and two from the light chain (LCDR1 and LCDR3) to form contacts with PD-L1 (Fig. 2). This is consistent with the general observation that the LCDR2 of antibodies is often not involved in antigen binding. The previously reported structures of PD-L1 in complex with BMS-963559 or avelumab demonstrated that these two antibodies similary involved only five of the six CDRs in the interaction with PD-L1, leaving LCDR2 without any binding to PD-L138, 39.

Crystal structures of PD-1 in complex with atezolizumab and durvalumab. (a) Ribbon representation of the complex structure of PD-L1/atezolizumab Fab fragment. (b) Ribbon representation of the complex structure of PD-L1/durvalumab Fab fragment. (c) Surface representation of the complex structure of PD-L1/atezolizumab Fab fragment. (d) Surface representation of the complex structure of PD-L1/durvalumab Fab fragment. PD-L1 and the antibody heavy and light chains are colored green, orange, and purple, respectively. In a–d, PD-L1 is in the same orientation. In c and d, the PD-1/PD-L1 complex (PDB code 4zqk) is superimposed onto the PD-L1 molecule in the PD-L1/anti-PD-L1 complexes with mixed ribbon/surface representation. PD-1 in the PD-1/PD-L1 complex are colored blue.

Sequence comparison of the antibodies against PD-L1. The CDRs are indicated with boxes and labeled. The residue numbers refer to those in atezolizumab. The identical and homologous residues are colored red and green, respectively.
Interactions between PD-L1 and atezolizumab
The interaction between atezolizumab and PD-L1 buries a total solvent accessible area of 2,106 Å2, which is larger than the PD-1/PD-L1 interface (1,970 Å2). Most of the buried solvent accessible area is contributed by the heavy chain (67%) (Fig. 3a). The atezolizumab epitope is formed on the surface of PD-L1 by the CC′FG antiparallel β-sheet and the BC, CC′, C′C″, and FG loops (Fig. 4a). In total, 23 residues of PD-L1 participate in the interaction with atezolizumab, forming nine hydrogen bonds, two salt bridges, and extensive van der Waals contacts (Supplementary Table S2). Of course, there should be water-mediated hydrogen bonds within the interface of the antigen-antibody pair, but these cannot be visualized due to the low resolution of 3.10 Å. The complex structure demonstrates that the interaction between PD-L1 and atezolizumab is mediated largely by residues within the central CC′FG β-sheet of PD-L1 and the heavy chain of atezolizumab. The side chain of PD-L1E58 makes two hydrogen bonds with the hydroxyl groups of heavyS52 and heavyS57. The side chain nitrogen atom of PD-L1Q66 and the backbone amide group of PD-L1V111 form hydrogen bonds with the backbone carbonyl group of heavyT58 and the side chain of heavyY54, respectively. PD-L1R113 and PD-L1R125 make salt-bridge interactions with the side chain of heavyD31 and hydrogen bonds with the backbone carbonyl group of heavyD31 and the hydroxyl group of heavyS30, simultaneously. The side chains of PD-L1I54, PD-L1Y56, PD-L1N63, PD-L1V111, PD-L1M115, PD-L1S117, PD-L1A121, and PD-L1Y123 in the CC′FG sheet of PD-L1 make van der Waals contacts with the residues within the HCDRs of atezolizumab, including heavyD31, heavyW33, heavyW50, heavyY54, heavyS57, and heavyW101, through alkyl-alkyl, alkyl-π, and T-shaped stacking interactions. In addition to the interaction mediated by residues of the central CC′FG sheet, residues in the BC, CC′, C′C″, and FG loops also contribute to the interaction with atezolizumab. The side chains of PD-L1E45 and PD-L1D49 in the BC loop make hydrogen bonds with the side chains of lightS30 and lightY93, respectively. A hydrophobic interaction is also formed between the BC loop and LCDR3, involving PD-L1A51 and PD-L1A52 in the BC loop and lightL92 and lightY93 in atezolizumab. These interactions draw the BC loop toward the antigen-antibody interface, thereby inducing a conformational change in the BC loop of PD-L1 that deviates from its conformation within PD-L1 in apo form or in complex with PD-1 and other anti-PD-L1 antibodies (Fig. 4b)34, 38, 39. PD-L1E60 and PD-L1D61 within the CC′ loop make van der Waals contacts with heavyY54, heavyG55, and heavyT74. PD-L1V68 and PD-L1H69 in the C′C″ loop also contact heavyW50, heavyY59, and lightH94 through alkyl-π and T-shaped stacking interactions. The residues in the FG loop, including PD-L1Y118 and PD-L1G119, produce hydrophobic interactions with the side chains of lightT31, lightA32, lightY91, lightL92, and heavyW33, as well as a hydrogen bond between the backbone amide group of PD-L1G119 and the side chain of heavyR99. Additionally, PD-L1A18 in the N-terminus of PD-L1 has a van der Waals interaction with heavyP102.

Interactions of atezolizumab and durvalumab with PD-L1. (a) Stereoview of the detailed PD-L1/atezolizumab Fab fragment interface. (b) Stereoview of the detailed PD-L1/durvalumab Fab fragment interface. In (a,b) the carbon atoms from PD-L1 and the antibody heavy and light chains are colored green, orange, and purple, respectively. Hydrogen bonds and salt bridges are indicated with dashed lines.

Intrinsic structural rigidity of PD-L1. (a) Canonical designation of the strands (red labels) and loops (black labels) within PD-L1. The N- and C-termini are labeled in blue. (b) Superposition of the PD-L1 protein extracted from the structures in complex with PD-L1 (blue), atezolizumab (red), durvalumab (cyan), BMS-963559 (yellow), and avelumab (green). The arrow indicates the conformational change of the BC loop in PD-L1 upon binding to atezolizumab. The bound PD-1 protein is shown as a gray surface model. The orientation of PD-L1 is the same as that in (a).
Interactions between PD-L1 and durvalumab
The interaction between durvalumab and PD-L1 buries a total solvent accessible area of 1,624 Å2, which is smaller than the PD-1/PD-L1 interface by 346 Å2. The binding interface is almost equally contributed by both the heavy and light chains (Fig. 3b). The durvalumab epitope is constituted by the C strand, F strand, G strand, CC′ loop, and N-terminal region of PD-L1 (Fig. 4a). In total, 16 residues of PD-L1 participate in the interaction with durvalumab through hydrogen bonds, salt bridges, and hydrophobic interactions (Supplementary Table S3). Fortunately, the resolution of the structure enabled the visualization of five water molecules mediating hydrogen bonds in the binding interface between PD-L1 and durvalumab. Most of the key interactions of PD-L1 with durvalumab are concentrated on the central CC′FG β-sheet within PD-L1. The side chain of PD-L1R113 makes a bidentate salt bridge with the side chain of heavyE57. PD-L1E58 also forms an ionic interaction with heavyK52. The side chain of PD-L1R125 makes two hydrogen bonds with the backbone carbonyl groups of heavyF103 and lightY92. Five water molecules mediating hydrogen bonds are involving three residues within the G strand, including PD-L1D122, PD-L1Y123, and PD-L1R125. A water molecule creates hydrogen bonds with the side chain of PD-L1D122 and the backbone carbonyl group of lightL95, enabling them to interact. The side chains of PD-L1Y123 and heavyE99 and the backbone carbonyl group of heavyF103 are also connected via water-mediated hydrogen bonds. Two water molecules are located between the backbone atoms of PD-L1Y123 and lightL95 and between PD-L1R125 and lightG93, mediating hydrogen bonds between them. The hydroxyl groups of lightS30 and lightS31 interact concurrently with the backbone carbonyl group of PD-L1R125 using a water molecule. In addition to the polar interactions, the side chains of PD-L1Y56, PD-L1V111, PD-L1M115, PD-L1A121, PD-L1Y123, PD-L1K124, and PD-L1T127 in the CC′FG β-sheet of PD-L1 make van der Waals contacts with the residues in the CDRs of durvalumab, including heavyW33, heavyS56, heavyY59, heavyF103, heavyE105, lightY33, lightS94, lightL95, and lightW97. Besides the main interaction by the residues within the CC′FG sheet, the CC′ loop and N-terminal region of PD-L1 also contribute to the interaction with durvalumab. The bulky side chain of heavyW102 in durvalumab makes van der Waals contacts with PD-L1E60 and PD-L1D61 within the CC′ loop of PD-L1. The N-terminal region of PD-L1 also participates in the interaction with durvalumab through an ionic interaction of PD-L1D26 with lightR28 and van der Waals contacts by PD-L1T20 and PD-L1V23.
Comparison of the interfaces between PD-L1 and PD-1 and anti-PD-L1 antibodies
A comprehensive comparison of the PD-L1 interactions with the receptor PD-1 and anti-PD-L1 antibodies, including atezolizumab, durvalumab, BMS-963559, and avelumab, can provide a better understanding of the mechanism of PD-L1 blockade by these therapeutic antibodies as well as new insights into the rational design of improved anti-PD-L1 therapeutics (Fig. 5)34, 37,38,39.

Comparison of the PD-L1 interactions with the receptor PD-1 and anti-PD-L1 antibodies. (a) Structure of PD-L1 (pale blue) in complex with PD-1 (orange) and the PD-1 binding site (orange) on the surface of the IgSF V-set domain of PD-L1. (b) Structure of PD-L1 in complex with atezolizumab Fab and its epitope on the surface of the IgSF V-set domain of PD-L1. (c) Structure of PD-L1 in complex with durvalumab Fab and its epitope on the surface of the IgSF V-set domain of PD-L1. (d) Structure of PD-L1 in complex with BMS-963559 Fab and its epitope on the surface of the IgSF V-set domain of PD-L1. (e) Structure of PD-L1 in complex with avelumab Fab and its epitope on the surface of the IgSF V-set domain of PD-L1. In (a–e) the IgSF V-set and IgSF C-set domains of PD-L1 are displayed in the same orientation, and the antibody heavy and light chains and their epitopes are colored red, yellow, and orange, respectively. The shared regions on the epitopes of the four antibodies and the PD-1 binding site are colored green.
The antibodies bind to PD-L1 from various directions and with different binding sites. Atezolizumab and BMS-963559 bind to the upper side close to the N-terminus of PD-L1, heavily tilted toward the face containing the central CC′FG β-sheet, implying the IgG form of these antibodies would take a narrow Y-shaped form when binding two neighboring PD-L1 molecules with two arms of IgG (Fig. 5b,d). In contrast, durvalumab and avelumab bind rather perpendicularly to PD-L1; therefore, the bivalent IgG of these antibodies would be T-shaped rather than Y-shaped when binding to two PD-L1 molecules at the same time (Fig. 5c,e). The dissociation constant (Kd) for the binding of avelumab-scFv to PD-L1 has been reported to be 42.1 pM by surface plasmon resonance (SPR) and other antibodies are also known to have Kd values less than 1 nM, whereas the binding affinity between PD-1 and PD-L1 was reported as 8.2 μM by SPR35, 39,40,41,42. The buried surface areas of PD-L1 in complex with PD-1, atezolizumab, durvalumab, BMS-963559, and avelumab are 1970, 2106, 1624, 1349, and 1865 Å2, respectively (Fig. 5). Simple comparison of these values cannot explain the large gap in the PD-L1 binding affinity between PD-1 and anti-PD-L1 antibodies. Considering the previous report that directed evolution on the binding surface of PD-1 by yeast-surface display generated a high-affinity PD-1 mutant (110 pM) that antagonizes PD-L1 competitively, the low affinity of PD-1 is probably due to the incomplete complementarity of the interface between PD-1 and PD-L143. The low affinity of the PD-1/PD-L1 pair should be more suitable for the transient interaction to modulate T-cell mediated immune responses in a timely manner through reversible interactions.
Although each antibody elicits a different epitope from the others, the central CC′FG β-sheet within PD-L1 provides key interactions for binding to all the antibodies (Fig. 5). The shared region on the epitopes of the four antibodies includes PD-L1Y56, PD-L1E58, PD-L1R113, PD-L1M115, and PD-L1Y123, which are located within the CC′FG β-sheet and provide pivotal interactions when PD-L1 binds to PD-1. Therefore, the overlap of the epitopes within the surface of the CC′FG sheet implies that the mechanism by which the anti-PD-L1 antibodies block the PD-1/PD-L1 interaction is by outcompeting PD-1 for binding to PD-L1. This is due to the much higher affinity of the antibodies and the increased avidity from the bivalency of IgG. In addition to the predominant interaction mediated by the residues of the central CC′FG β-sheet, the loops within PD-L1 are also involved in the interaction with each antibody, contributing to the stabilization of the antigen-antibody complexes. As described above, the BC, CC′, C′C″, and FG loops of PD-L1 make extensive interactions with atezolizumab, and the CC′ loop and N-terminal region are also involved in the interaction with durvalumab through a salt bridge and van der Waals contacts. The structure of PD-L1 in complex with BMS-963559 showed that the BC, C′C″, and FG loops provided key interactions, and the PD-L1-avelumab complex structure demonstrated that the CC′ loop contributed to a major interaction with avelumab through multiple hydrogen bonds provided by PD-L1D6138, 39. Taken together, the BC, CC′, C′C″, and FG loops as well as the CC′FG sheet could be vulnerable antigenic sites for anti-PD-L1 therapeutic antibodies. These loops may also be valuable hot-spots for the design of small-molecule modulators of the PD-1/PD-L1 interaction. As the surface of the central CC′FG β-sheet is flat, it is very difficult to design a small molecule with a high binding affinity by targeting only this region. Additional interactions with the loops would be critical to acquiring a high potency against PD-L1. Analysis of the diverse interactions of these loops with the antibodies should enable us to design promising small-molecule PD-1/PD-L1 blockers, which can overcome the drawbacks of antibody-based therapeutics.
Discussion
Cancer immunotherapy via antibody-based PD-1/PD-L1 blockade has provided a major breakthrough for the treatment of multiple advanced and metastatic cancers since the approval of the monoclonal antibodies targeting the PD-1/PD-L1 axis. Despite the great achievement of the therapeutic antibodies blocking the PD-1/PD-L1 interaction, they have specific shortcomings as therapeutics. Poor tissue/tumor penetrance of antibody drugs due to their large size can be problematic, especially when targeting PD-1/PD-L1 signaling as PD-1-expressing T cells are found infiltrated within the solid tissue of PD-L1-expressing tumors44, 45. To compensate in part for the possible suboptimal efficacy of therapeutic antibodies, the development of low-molecular weight protein drugs or small molecules modulating PD-1/PD-L1 signaling is urgently needed, and a combination therapy of small-molecule modulators and antibody drugs may be an excellent option for the treatment of cancers through complete PD-1/PD-L1 blockade in solid tumors. Directed evolution based on the crystal structure of PD-1/PD-L1 complex enabled to engineer the PD-1 ectodomain as a high-affinity (110 pM) competitive antagonist of PD-1 showing superior tumor penetration43. There are also several small-molecule immunomodulators targeting the PD-1/PD-L1 axis in preclinical or clinical investigations46. Structural studies on the PD-L1 interaction with therapeutic antibodies can provide insight into the design of small molecules targeting PD-L1, as their potency can be enhanced by mimicking the diverse interactions of these antibodies, including the involvement of the BC, CC′, C′C″, and FG loops for binding to atezolizumab; the CC′ loop and N-terminal region for durvalumab; the BC, C′C″, and FG loops for BMS-963559; and the CC′ loop for avelumab. We also believe that the accumulation of such structural studies will provide invaluable information for developing next-generation therapeutic antibodies, such as antibody drug conjugates (ADCs) and bi-specific antibodies, and for coping with any possible antigen mutational escape of PD-L1 in future.
PD-L2 also plays a role in maintaining peripheral tolerance in lung via interaction with PD-1 or other receptors such as RGMb47. PD-L2-deficient mice exhibit increased airway hyperactivity and lung inflammation48. While PD-1 inhibition by anti-PD-1 antibodies disrupts both PD-L1 and PD-L2 pathways, anti-PD-L1 antibodies, including atezolizumab, durvalumab, BMS-963559, and avelumab, are known to target only PD-L1 to inhibit the PD-1/PD-L1 interaction while preserving the PD-1/PD-L2 interaction, thereby avoiding the immune-related toxicity associated with PD-L2 blockade24. The complex structures of PD-L1 with these antibodies explain their lack of binding to PD-L2 (Fig. 6). The crystal structure of PD-1 in complex with PD-L2 showed that PD-L1 and PD-L2 have similar binding modes to PD-136. It has been reported that mutation of PD-L2W110 to alanine in PD-L2 reduces binding affinity to PD-1 to 40% of that of the wild type36. This is probably because PD-L2W110, which is located within the G strand of PD-L2, occupies a small hydrophobic pocket on the surface of PD-1, thereby contributing the binding energy for PD-1/PD-L2 interaction. The residue corresponding to PD-L2W110 is PD-L1A121 in PD-L1, and this difference in the side chain would lead to a 3-fold lower binding affinity of PD-L1 to PD-1 than that of PD-L2. The anti-PD-L1 antibodies contact PD-L1A121 with hydrophobic residues, including heavyW101 of atezolizumab, heavyY59 of durvalumab, heavyI54 of BMS-963559, and heavyI57 of avelumab (Fig. 6a–d). The substitution of PD-L1A121 with tryptophan should sterically collide with the residues of the anti-PD-L1 antibodies due to its bulky size, thereby leading to failure in binding to PD-L2. In addition, the structure-based sequence alignment of PD-L1 and PD-L2 shows the absence of the CC′ loop, C′ strand, C′C″ loop, and C″ strand in PD-L2 (Fig. 6e). As these regions in PD-L1 provide key interactions for the binding of the antibodies, the absence of them from PD-L2 should also negatively affect the binding of the antibodies to PD-L2.

Structural basis for the lack of the binding of anti-PD-L1 antibodies to PD-L2. (a) The partially transparent surface model of atezolizumab (green) when PD-L1 of PD-L1/atezolizumab is overlaid onto PD-L2 (red, PDB code 3bov). (b) The surface model of durvalumab (blue) when PD-L1 of PD-L1/durvalumab is overlaid onto PD-L2. (c) The surface model of BMS-963559 (gray) when PD-L1 of PD-L1/BMS-963559 is overlaid onto PD-L2. (d) The surface model of avelumab (yellow) when PD-L1 of PD-L1/avelumab is overlaid onto PD-L2. In (a–d) the residues of the antibodies, which collide with W110 of PD-L2, are shown in sticks and labeled. (e) Structure-based sequence alignment of PD-L1 and PL-L2. The strands in PD-L1 are denoted with arrows above the sequence. W110 of PD-L2 is indicated with an asterisk. The identical and homologous residues are colored red and green, respectively.
Protein glycosylation plays a critical role in many biological processes and cancer cells display numerous alterations in glycosylation patterns compared with normal cells, thereby contributing to altered cancer cell functions49, 50. As PD-L1 is overexpressed in cancer cells, possible alterations in the glycosylation patterns of PD-L1 would affect the binding of therapeutic antibodies to PD-L1. It has been reported recently that PD-L1 is exclusively N-glycosylated at PD-L1N35, PD-L1N192, PD-L1N200, and PD-L1N219 in cancer cells51. A structural analysis can estimate the influence of PD-L1 glycosylation on the interaction of the anti-PD-L1 antibodies, including atezolizumab, durvalumab, BMS-963559, and avelumab (Fig. 7). PD-L1N35 is located within the B strand, which is on the opposite side of the central CC′FG β-sheet, and PD-L1N192, PD-L1N200, and PD-L1N219 are residues of the IgSF C-set domain, which is not involved in the interaction with these antibodies. Therefore, the binding of these antibodies to PD-L1 should be independent of the glycosylation of PD-L1.

Glycosylation of PD-L1 and antibody binding. The four N-glycosylation sites (N35, N192, N200, and N219) of PD-L1 are shown in sticks and colored cyan. The IgSF V-set and IgSF C-set domains of PD-L1 are labeled. The bound antibodies, including atezolizumab, durvalumab, BMS-963559, and avelumab, are shown as green, blue, gray, and yellow surfaces, respectively.
The interaction between PD-1 and PD-L1 is associated with significant plasticity of PD-1. Through the conformational rearrangement within the CC′ loop of PD-1, four additional hydrogen bonds can be formed between PD-1 and PD-L1, thereby contributing to the binding energy of the ligand-receptor interaction34, 37. The complex formation-associated plasticity within PD-1 can also be seen in the interaction with the anti-PD-1 antibodies pembrolizumab and nivolumab38, 52,53,54. Binding of these antibodies induces drastic structural changes within the BC, C’D, FG, and N-terminal loops of PD-1, thereby stabilizing the antigen-antibody complexes. In contrast to PD-1, the structure of PD-L1 changes little upon binding to the receptor or to anti-PD-L1 antibodies34, 37,38,39. When binding to PD-1, no significant change within the backbone is induced and only minor adjustments in the arrangement of the side chains occur due to local steric constraints of the binding interface. The CC′, C′C″, and FG loops of PD-L1 are also involved in the interaction with the anti-PD-L1 antibodies including atezolizumab, durvalumab, BMS-963559, and avelumab. However, the binding of PD-L1 to the antibodies does not alter the conformations of these loops in PD-L1, implying that PD-L1 maintains these loops in the productive binding conformation prior to interacting with the receptor or antibodies. Only when binding to atezolizumab does the BC loop of PD-L1 move a little toward the binding interface to make additional interactions with atezolizumab. Compared to the high affinity of the anti-PD-L1 antibodies, the much weaker binding affinity of PD-1 to PD-L1 may be partly based on the intrinsic plasticity of PD-1.
In summary, we reported the crystal structures of the N-terminal IgSF V-set domain of PD-L1 in complex with the Fab fragments of atezolizumab and durvalumab, elucidating the precise epitopes involved and the structural basis for the blockade of the PD-1/PD-L1 interaction by these therapeutic antibodies. A comprehensive analysis of the PD-L1 interactions with the receptor PD-1 and anti-PD-L1 antibodies, including atezolizumab, durvalumab, BMS-963559, and avelumab, demonstrated that the overlap of the epitopes within the surface of CC′FG sheet implies the mechanism of the PD-1/PD-L1 blockade by the therapeutic antibodies. The epitopes and binding modes of the FDA-approved anti-PD-L1 antibodies can be references for the development of other antibodies in future and the BC, CC′, C′C″, and FG loops of PD-L1 should provide key interactions for the development of improved anti-PD-L1 therapeutics including next-generation therapeutic antibodies and small-molecule modulators.
Methods
Expression and purification of PD-L1
Genes encoding the IgSF V-set domain of human PD-L1 (aa 18-134) were subcloned into pET-21a (Novagen). The protein was expressed in E. coli BL21(DE3) as inclusion bodies. The cells were grown at 37 °C in LB medium supplemented with 50 μg mL−1 ampicillin until OD600 reached 0.6–1.0, and the protein expression was induced with 1 mM IPTG and incubated for 4 h at 37 °C. The cells were harvested by centrifugation, re-suspended in lysis buffer (20 mM Tris, pH 8.0, 200 mM NaCl) and lysed by sonication on ice. Inclusion bodies were recovered by centrifugation (25,000 × g for 0.5 h at 4 °C) and solubilized in 8 M urea, 20 mM Tris, pH 8.0, 200 mM NaCl by stirring overnight. After removing undissolved residue by centrifugation (25,000 × g for 0.5 h at 4 °C), solubilized fraction was applied to HisTrap HP column (GE Healthcare Life Sciences) and washed with five column volumes of wash buffer (8 M urea, 20 mM Tris, pH 8.0, 200 mM NaCl, 50 mM imidazole). The protein was then eluted with elution buffer (8 M urea, 20 mM Tris, pH 8.0, 200 mM NaCl, 400 mM imidazole). The eluted protein was refolded by dialysis 3 times against 20 mM Tris, pH 8.0, 200 mM NaCl and purified further by gel filtration chromatography using a HiLoad 16/60 Superdex 200 pg column (GE Healthcare Life Sciences). The protein purity was evaluated by reducing and nonreducing SDS-PAGE.
Expression and purification of Fab fragments
The DNA sequences for the Fab fragments of atezolizumab and durvalumab were synthesized after codon-optimization for expression in E. coli (Bioneer, Inc). The sequences for heavy chain and light chain were cloned into a modified pBAD vector, containing the STII signal sequence in each chain for periplasmic secretion and a C-terminal 6His-tag in heavy chain38. The plasmid pBAD-Fab was transformed into E. coli Top10F (Invitrogen). The cells were grown at 37 °C in LB medium supplemented with 50 μg mL−1 ampicillin. At an OD600 of 1.0, the protein expression was induced with 0.2% arabinose and cells were grown at 30 °C for 15 h. The cells were harvested by centrifugation, re-suspended in lysis buffer (20 mM Tris, pH 8.0, 200 mM NaCl) and lysed by sonication on ice. After removing cell debris by centrifugation (25,000 × g for 0.5 h at 4 °C), the supernatant containing soluble protein was applied to HisTrap HP column (GE Healthcare Life Sciences) and washed with five column volumes of wash buffer (20 mM Tris, pH 8.0, 300 mM NaCl, 50 mM imidazole). The protein was then eluted with elution buffer (20 mM Tris, pH 8.0, 300 mM NaCl, 400 mM imidazole). The eluted protein was concentrated for gel filtration chromatography using a HiLoad 16/60 Superdex 200 pg column (GE Healthcare Life Sciences). The column had previously been equilibrated with gel filtration buffer (20 mM Tris, pH 8.0, 300 mM NaCl). The elution profile of the protein showed a single major peak and the protein quality was evaluated by reducing and nonreducing SDS-PAGE.
Crystallization and structure determination
Details of the crystallization, X-ray data collection, structure determination, and refinement of the PD-L1/atezolizumab Fab and PD-L1/durvalumab Fab complexes are described in Supplementary Information. Data collection and refinement statistics are summarized in Table 1.
Data availability
The atomic coordinates and structure factors for the structures of PD-L1 in complex with the atezolizumab and durvalumab Fab have been deposited into Protein Data Bank (http://www.rcsb.org) under the accession codes 5X8 L and 5X8 M, respectively.
References
Freeman, G. J. et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J. Exp. Med. 192, 1027–1034 (2000).
Zou, W. et al. Inhibitory B7-family molecules in the tumour microenvironment. Nat. Rev. Immunol. 8, 467–477 (2008).
Chen, L. & Flies, D. B. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat. Immunol. Rev. 13, 227–242 (2013).
Greenwald, R. J. et al. The B7 family revisited. Annu. Rev. Immunol. 23, 515–548 (2005).
Lenschow, D. J., Walunas, T. L. & Bluestone, J. A. CD28/B7 system of T cell costimulation. Annu. Rev. Immunol. 14, 233–258 (1996).
Buchbinder, E. I. & Desai, A. CTLA-4 and PD-1 Pathways: Similarities, Differences, and Implications of Their Inhibition. Am. J. Clin. Oncol. 39, 98–106 (2016).
Latchman, Y. et al. PD-L2 is a second ligand for PD-1 and inhibits T cell activation. Nat. Immunol. 2, 261–268 (2001).
Chemnitz, J. M., Parry, R. V., Nichols, K. E., June, C. H. & Riley, J. L. SHP-1 and SHP-2 associate with immunoreceptor tyrosine-based switch motif of programmed death 1 upon primary human T cell stimulation, but only receptor ligation prevents T cell activation. J. Immunol. 173, 945–954 (2004).
Pardoll, D. M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 12, 252–264 (2012).
Collins, A. V. et al. The interaction properties of costimulatory molecules revisited. Immunity 17, 201–210 (2002).
Okazaki, T., Chikuma, S., Iwai, Y., Fagarasan, S. & Honjo, T. A rheostat for immune responses: the unique properties of PD-1 and their advantages for clinical application. Nat Immunol. 14, 1212–1218 (2013).
Couzin-Frankel, J. Breakthrough of the year 2013. Cancer immunotherapy. Science 342, 1432–1433 (2013).
Callahan, M. K., Postow, M. A. & Wolchok, J. D. Targeting T cell co-receptors for cancer therapy. Immunity 44, 1069–1078 (2016).
John, L. B. et al. Anti-PD-1 antibody therapy potently enhances the eradication of established tumors by gene-modified T cells. Clin. Cancer Res. 19, 5636–5646 (2013).
Powles, T. et al. MPDL3280A (anti-PD-L1) treatment leads to clinical activity in metastatic bladder cancer. Nature 515, 558–562 (2014).
Sidaway, P. Urological cancer: Atezolizumab: an alternative to cisplatin? Nat. Rev. Clin. Oncol. 14, 139 (2017).
Bellmunt, J., Powles, T. & Vogelzang, N. J. A review on the evolution of PD-1/PD-L1 immunotherapy for bladder cancer: The future is now. Cancer Treat. Rev. 54, 58–67 (2017).
Rittmeyerm, A. et al. Atezolizumab versus docetaxel in patients with previously treated non-small-cell lung cancer (OAK): a phase 3, open-label, multicentre randomised controlled trial. Lancet. 389, 255–265 (2017).
Sorscher, S. Pembrolizumab in Non-Small-Cell Lung Cancer. N. Engl. J. Med. 376, 996–997 (2017).
Hamid, O. et al. Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma. N. Engl. J. Med. 369, 134–44 (2013).
Garon, E. B. et al. Pembrolizumab for the treatment of non-small-cell lung cancer. N. Engl. J. Med. 372, 2018–2028 (2015).
Gunturi, A. & McDermott, D. F. Nivolumab for the treatment of cancer. Expert Opin. Invest. Drugs 24, 253–260 (2015).
Chen, L. & Han, X. Anti-PD-1/PD-L1 therapy of human cancer: past, present, and future. J. Clin. Invest. 125, 3384–3391 (2015).
Brahmer, J. R. et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N. Engl. J. Med. 366, 2455–2465 (2012).
Hamilton, G. & Rath, B. Avelumab: combining immune checkpoint inhibition and antibody-dependent cytotoxicity. Expert Opin. Biol. Ther. 17, 515–523 (2017).
Brower, V. Anti-PD-L1 inhibitor durvalumab in bladder cancer. Lancet Oncol. 17, e275 (2016).
Brahmer, J. R. Harnessing the immune system for the treatment of non-small-cell lung cancer. J. Clin. Oncol. 31, 1021–1028 (2013).
Das, R. et al. Combination therapy with anti-CTLA-4 and anti-PD-1 leads to distinct immunologic changes in vivo. J. Immunol. 194, 950–959 (2015).
Wolchok, J. D. et al. Nivolumab plus ipilimumab in advanced melanoma. N. Engl. J. Med. 369, 122–133 (2013).
Larkin, J., Hodi, F. S. & Wolchok, J. D. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N. Engl. J. Med. 373, 1270–1271 (2015).
Planchard, D. et al. A Phase III Study of Durvalumab (MEDI4736) With or Without Tremelimumab for Previously Treated Patients With Advanced NSCLC: Rationale and Protocol Design of the ARCTIC Study. Clin. Lung Cancer 17, 232–236 (2016).
Li, Y. et al. A Mini-Review for Cancer Immunotherapy: Molecular Understanding of PD-1/PD-L1 Pathway & Translational Blockade of Immune Checkpoints. Int. J. Mol. Sci. 17, 1151 (2016).
Wurz, G. T., Kao, C. J. & DeGregorio, M. W. Novel cancer antigens for personalized immunotherapies: latest evidence and clinical potential. Ther. Adv. Med. Oncol. 8, 4–31 (2016).
Zak, K. M. et al. Structure of the Complex of Human Programmed Death 1, PD-1, and Its Ligand PD-L1. Structure 23, 2341–2348 (2015).
Cheng, X. et al. Structure and interactions of the human programmed cell death 1 receptor. J. Biol. Chem. 288, 11771–11785 (2013).
Lázár-Molnár, E. et al. Crystal structure of the complex between programmed death-1 (PD-1) and its ligand PD-L2. Proc. Natl. Acad. Sci. USA 105, 10483–10488 (2008).
Lin, D. Y. et al. The PD-1/PD-L1 complex resembles the antigen-binding Fv domains of antibodies and T cell receptors. Proc. Natl. Acad. Sci. USA 105, 3011–3016 (2008).
Lee, J. Y. et al. Structural basis of checkpoint blockade by monoclonal antibodies in cancer immunotherapy. Nat. Commun. 7, 13354 (2016).
Liu, K. et al. Structural basis of anti-PD-L1 monoclonal antibody avelumab for tumor therapy. Cell. Res. 27, 151–153 (2016).
Bharathi, V. et al. Anti-pd-l1 antibodies and diagnostic uses thereof. WO 2016007235 A1 (2016).
John, P. C. et al. Cancer immunotherapy by disrupting pd-1/pd-l1 signaling. WO 2013173223 A1 (2013).
Lesniak, W. G. et al. PD-L1 detection in tumors using [64Cu]atezolizumab with PET. Bioconjug. Chem. 27, 2103–2110 (2016).
Maute, R. L. et al. Engineering high-affinity PD-1 variants for optimized immunotherapy and immuno-PET imaging. Proc. Natl. Acad. Sci. USA 112, 6506–6514 (2015).
Iwai, Y. et al. Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade. Proc. Natl. Acad. Sci. USA 99, 12293–12297 (2002).
Lee, C. M. & Tannock, I. F. The distribution of the therapeutic monoclonal antibodies cetuximab and trastuzumab within solid tumors. BMC Cancer 10, 255 (2010).
Zhan, M. M. et al. From monoclonal antibodies to small molecules: the development of inhibitors targeting the PD-1/PD-L1 pathway. Drug Discov. Today 21, 1027–1036 (2016).
Xiao, Y. et al. RGMb is a novel binding partner for PD-L2 and its engagement with PD-L2 promotes respiratory tolerance. J. Exp. Med. 211, 943–959 (2014).
Akbari, O. et al. PD-L1 and PD-L2 modulate airway inflammation and iNKT-cell-dependent airway hyperreactivity in opposing directions. Mucosal Immunol 3, 81–91 (2010).
Hakomori, S. & Kannagi, R. Glycosphingolipids as tumor-associated and differentiation Markers. J. Natl. Cancer Inst. 71, 231–251 (1983).
Pinho, S. S. & Reis, C. A. Glycosylation in cancer: mechanisms and clinical implications. Nat. Rev. Cancer 15, 540–555 (2015).
Li, C. W. et al. Glycosylation and stabilization of programmed death ligand-1 suppresses T-cell activity. Nat. Commun. 7, 12632 (2016).
Na, Z. et al. Structural basis for blocking PD-1-mediated immune suppression by therapeutic antibody pembrolizumab. Cell. Res. 27, 147–150 (2016).
Horita, S. et al. High-resolution crystal structure of the therapeutic antibody pembrolizumab bound to the human PD-1. Sci. Rep. 6, 35297 (2016).
Tan, S. et al. An unexpected N-terminal loop in PD-1 dominates binding by nivolumab. Nat. Commun. 8, 14369 (2017).
Acknowledgements
We are grateful to the staffs of beamline 7A and 5C at Pohang Accelerator Laboratory for help with the X-ray diffraction experiments. This work was supported by grants from the National Research Foundation of Korea (NRF-2015R1D1A1A01057706 and NRF-2015M3A9B5030302).
Author information
Authors and Affiliations
Contributions
H.T.L., J.Y.L., H.L., and Y.-S.H. designed the experiments. H.T.L., J.Y.L., H.L., S.H.L., Y.J.M., H.J.P., S.E.R., and W.S. performed the experiments including expression, purification, and crystallization. H.T.L., J.Y.L., and H.L. collected the X-ray data, determined, and analyzed the structures under the guidance of Y.-S.H. Y.-S.H. wrote the paper. All authors discussed the results and commented on the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lee, H.T., Lee, J.Y., Lim, H. et al. Molecular mechanism of PD-1/PD-L1 blockade via anti-PD-L1 antibodies atezolizumab and durvalumab. Sci Rep 7, 5532 (2017). https://doi.org/10.1038/s41598-017-06002-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-06002-8
- Springer Nature Limited
This article is cited by
-
Cancer therapy with antibodies
Nature Reviews Cancer (2024)
-
Comprehensive assessment of TECENTRIQ® and OPDIVO®: analyzing immunotherapy indications withdrawn in triple-negative breast cancer and hepatocellular carcinoma
Cancer and Metastasis Reviews (2024)
-
Developing a dual VEGF/PDL1 inhibitor based on high-affinity scFv heterodimers as an anti-cancer therapeutic strategy
Scientific Reports (2023)
-
Evaluation of therapeutic PD-1 antibodies by an advanced single-molecule imaging system detecting human PD-1 microclusters
Nature Communications (2023)
-
The RESP AI model accelerates the identification of tight-binding antibodies
Nature Communications (2023)