
Abstract
A total of 807 entire VP1 sequences of Coxsackievirus A6 (CV-A6) from mainland of China from 1992 to 2015, including 520 in this study and 287 from the GenBank database, were analysed to provide a basic framework of molecular epidemiological characteristics of CV-A6 in China. Sixty-five VP1 sequences including 46 representative CV-A6 isolates from 807 Chinese strains and 19 international strains from GenBank were used for describing the genotypes and sub-genotypes. The results revealed that CV-A6 strains can be categorised into 4 genotypes designated as A, B, C, and D according to previous data and can be further subdivided into B1–B2, C1–C2, and D1–D3 sub-genotypes. D3 is the predominant sub-genotype that circulated in recent years in mainland of China and represents 734 of 807 Chinese isolates. Sixty-six strains belong to D2, whereas B1 and C1 comprise a single strain each, and five AFP strains formed B2. Sub-genotype D3 first circulated in 2008 and has become the predominant sub-genotype since 2009 and then reached a peak in 2013, while D2 was mostly undetectable in the past years. These data revealed different transmission stages of CV-A6 in mainland of China and that sub-genotype D3 may have stronger transmission ability.
Similar content being viewed by others

Introduction
Human enterovirus (EV), which belongs to the genus Enterovirus, family Picornaviridae, is a small non-enveloped, positive sense, single-stranded RNA virus1. EV infections are associated with a wide spectrum of illnesses, including mild diseases like a febrile illness and sometimes severe neurological disorders such as aseptic meningitis, myocarditis, acute flaccid paralysis, and encephalitis. Human EVs are classified into 4 species: EV-A, EV-B, EV-C, and EV-D; the EV-A species now consists of 25 serotypes: Coxsackievirus group A (CV-A; serotypes 2–8, 10, 12, 14, and 16); newly identified EVs (serotypes EV-A71, A76, A89–92, A114, A119–121); simian enteroviruses SV19, SV43, and SV46; and baboon enterovirus A132,3,4,5,6.
Hand, foot, and mouth disease (HFMD) is a common epidemic disease that affects young children and causes a series of symptoms such as a fever, sore throat, general rash on the hands and feet, and exanthema in the oral cavity and on the tongue. In rare cases, however, patients may also develop neurological complications such as neurogenic pulmonary oedema, aseptic meningitis, and acute flaccid paralysis; thus, this virus has become a significant public health threat worldwide7,8,9. More than 90% of HFMD cases are caused by EV-A, especially by EV-A71 and CV-A16. EV-A71 is more often associated with neurological diseases and sometimes death as compared to the other EV-A serotypes, whereas CV-A16-associated HFMD is usually mild10, 11. Nonetheless, in recent years, CV-A6-associated HFMD outbreaks have taken place worldwide, first in Finland in 2008, and then this disease circulated in France, Spain, and other European countries from 2009 to 2011. CV-A6 was the predominant pathogen for the subsequent HFMD outbreaks in these countries12,13,14. From 2014 to 2015, CV-A6-associated HFMD outbreaks occurred again in France15. Outbreaks in North America from 2011 to 2013 have also been reported16. In Asia, CV-A6 was responsible for HFMD outbreaks in Singapore in 2009, Japan in 2011, Thailand in 2012, and mainland of China in 201317,18,19,20,21. Since 2013, repeated larger-scale HFMD outbreaks caused by CV-A6 have occurred in China, and CV-A6 has become an important virus on the HFMD pathogen spectrum, even replaced EV-A71 and CV-A16 as the leading HFMD pathogen in many Provinces of China in 2013 and 201522, 23.
The clinical features of CV-A6-associated HFMD are slightly different from those caused by other EVs and represent so-called atypical HFMD, including spread of lesions beyond the typical sites of HFMD: to the trunk, neck, legs, and perioral area, and onychomadesis which causes nail loss24, 25. Severe cases caused by CV-A6 were also reported in China, characterised by high fever with shorter duration and twitching, as compared with EV-A71-associated severe cases26, and sometimes cause brainstem encephalitis and aseptic meningitis27. In addition to the largest number of HFMD cases, CV-A6 causes a large number of severe HFMD cases; therefore, CV-A6-related HFMD has been gradually becoming an important part of HFMD surveillance. Furthermore, CV-A6 is an important pathogen of herpangina14, but herpangina is not a disease that needs to be reported in the disease surveillance reporting system of China. Thus, the burden of the disease caused by CV-A6 infection may be strongly underestimated.
The VP1 coding region contains many important neutralisation epitopes and was demonstrated to help identify EV serotypes28. A phylogenetic dendrogram based on the entire VP1 capsid sequences of EV has been used for discrimination of genotypes; this approach is effective during temporal and geographical analysis of different outbreaks29. Studies have proven the correlation between the genotype identity and molecular epidemiological characteristics of EV-A71 and CV-A1629, 30. Nonetheless, there has been a lack of systematic research on classification of genotypes of CV-A6 strains based on the entire VP1 region; therefore, comprehensive application of bioinformatics methods is performed in this study to expound the genetic evolution of CV-A6 circulating in China and to improve the understanding of its genetic characteristics and genotypic and transmission patterns. The results of this study will provide important basic data for the prevention and control of enteroviral diseases in China and has crucial public health significance and practical utility for the development of effective HFMD prevention and control measures.
Results
Geographic and annual distribution of Chinese CV-A6 isolates
A total of 520 Chinese CV-A6 sequences collected in this study and 287 Chinese CV-A6 sequences downloaded from the GenBank database were used here and are summarised in Supplementary Materials (Supplementary Table S1). These 807 strains were isolated from 21 Provinces (or municipalities and autonomous regions), which represent 7 geographic regions of China including East China (Shandong, Jiangsu, Zhejiang, Fujian, and Shanghai), South China (Guangdong), Central China (Hunan, Henan, and Jiangxi), North China (Tianjin, Hebei, and Shanxi), Northwest China (Xinjiang, Shaanxi, and Gansu), Southwest China (Sichuan, Yunnan, Guizhou, and Chongqing), and Northeast China (Liaoning and Jilin), during the period 1992–2015 (Supplementary Table S1, Fig. 1). The strains isolated since 2008 are all HFMD-related strains (except that a Shandong strain 08216/SD/CHN/2008 was isolated from a patient with aseptic meningitis). Only 2 CV-A6 isolates were obtained between 1992 and 1996 and 9 isolates were reported between 2004 and 2007; strains isolated between 2008 and 2012 accounted for 18.8% (152/807) of all isolates, and strains isolated between 2013 and 2015 accounted for 79.8% (644/807; Supplementary Table S2).

Geographic and annual distribution of 807 Chinese CV-A6 strains included in this study.
Molecular phylogeny of modern CV-A6
First of all, a database of the CV-A6 entire VP1 sequences (915 bp) was built. We filtered the entire VP1 sequences of 65 CV-A6 strains by constructing a phylogenetic tree based on all the CVA-6 sequences released before October 10th, 2016, from the GenBank database plus 520 strains in this study, and excluded the laboratory adaptive strains, clones, strains with high passage numbers, and probably incorrect strains, meanwhile consulting other research on CV-A6, to ensure the representativeness of the chosen strains. The database covers CV-A6 strains isolated between 1949 (the prototype Gdula strain) and 2015 from 7 countries including 46 from mainland of China (between 1992 and 2015) (Table 1).
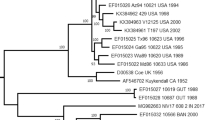
A phylogenetic dendrogram was constructed with the 65 CV-A6 VP1 sequences using the neighbour-joining method in MEGA 5.0 (Fig. 2). The bootstrap test was performed with 1000 replications. According to the rule that a difference of at least 15% in the entire VP1 nucleotide sequences should be used for distinguishing genotypes30, the VP1 tree clearly indicated that all the CV-A6 strains are segregated into 4 distinct genotypes which were designated as A, B, C, and D, referring to the other studies21, 22. The Gdula prototype strain isolated in the United States in 1949 formed a single lineage and differed from other stains by 19.1–21.2% and can be classified as the sole member of genotype A. Genotype B consists of 4 strains isolated in mainland of China between 1992 and 2007. Genotype C, represented by 2 strains, includes a virus from Shandong province of China in 1996 and India in 2008.

Phylogenetic trees based on the complete VP1 nucleotide sequences of CV-A6 of 65 representative strains isolated between 1949 (the prototype Gdula strain) and 2015. Numbers at the nodes indicate bootstrap support for each node (percentage of 1000 bootstrap replicates). A difference of at least 15% in the entire VP1 region of CV-A6 strains was used to distinguish genotypes. CV-A6 strains are labelled using the following format: ‘isolate name’/‘country of origin’/‘year of isolation’ (details are listed in Table 1). Different genotypes and sub-genotypes are shown to the right of the tree and the scale at the bottom indicates measurement of relative phylogenetic distance. Abbreviations of countries/Province: CHN, China; JPN, Japan; IND, India; TW, Taiwan, China; FIN, Finland; FRA, France; ESP, Spain.
Genotype D is represented by the rest of 58 strains isolated from 1999 to 2015 in mainland of China, Japan, Finland, Spain, and France. The group mean distances among the 4 genotypes range from 17.7% to 21.2%.
Genotypes B, C, and D can be further subdivided into 2, 2, and 3 sub-genotypes, respectively, with a difference of more than 8% in the VP1 region (Fig. 2). Cluster B1 contains a single strain isolated from Shandong Province, China, in 1992, and Cluster B2 consists of 3 strains isolated from patients with acute flaccid paralysis (AFP) from Guangdong Province, China, from 2004 to 2007; the mean distance between B1 and B2 sub-genotypes is 9.4%. Furthermore, the 2 strains of genotype C represent 2 sub-genotypes (C1 and C2) independently with a 10.1% VP1 sequence divergence. As to genotype D, Cluster D1 was composed of strains isolated in Japan in 1999 and in France and Spain from 2008 to 2010. Cluster D2 was composed of strains isolated in Japan and mainland of China from 2006 to 2011. Cluster D3 was composed of strains isolated in Finland, Spain, France, Japan, and China from 2008 to 2015. Isolates within sub-genotype D1 differed from one another by 0.4–6.8% and from strains in sub-genotype D2 and D3 by 8.3–11.7% and 4.1–10.9%, respectively. Viruses in sub-genotype D2 differ from one another by 1.1–6.4% and from strains in sub-genotype D3 by 6.4–12.0%, whereas strains in sub-genotype D3 differ from one another by 0.0–6.7%.
Except for the prototype Gdula strain (genotype A) and Indian strain N-313 (sub-genotype C2), all the other international CV-A6 strains belong to genotype D. On the basis of the above analysis, the outbreaks of CV-A6 that happened in Europe from 2008 to 2010 can be attributed to 2 major sub-genotypes, D1 and D3; however, sub-genotype D3 was responsible for most of the outbreaks worldwide after 2010. These data suggested that the sub-genotype D3 viruses probably have stronger transmissibility, infectivity, and virulence and may be the main reason for the persistent international circulation of CV-A6.
The pattern of circulation of CV-A6 in mainland of China: the switch from the co-circulation of sub-genotypes D2 and D3 to the predominance of D3
Phylogenetic trees were constructed using 807 Chinese entire CV-A6 VP1 sequences plus the selected 53 international CV-A6 strains, and the prototype CV-A2 strain (Fleetwood) served as an outgroup (Fig. 3).

Phylogenetic analyses based on 807 entire VP1 sequences of CV-A6 in China. The phylogenetic tree shows the evolutionary tendency of sub-genotype D2 and D3 strains in China since 2008; sub-genotype D3 was the predominant one. The branches of sequences from mainland of China are highlighted in different colours according to years from 2008 to 2015, the other international strains (including Taiwan strains) and strains detected before 2008 are uncoloured. The phylogenetic tree indicates that evolutionary branch D3a was responsible for the recent outbreaks since 2013. A ladder-like topology revealed that adaptation of CV-A6 occurred during the epidemic history. A prototype CV-A 2 strain (Fleetwood) served as an outgroup.
According to our previous research, all the CV-A6 strains isolated since 2008 circulating in mainland of China clustered with isolates of sub-genotypes D2 and D3, and most of them belong to D321. Strains 92022/SD/CHN/1992 and 96188/SD/CHN/1996 were the sole members of sub-genotypes B1 and C1, respectively. Five Guangdong AFP isolates (strains AFP024/GD/CHN/2004, AFP560/GD/CHN/2004, AFP262/GD/CHN/2005, AFP265/GD/CHN/2005, and AFP051/GD/CHN/2007) formed sub-genotype B2. Furthermore, sub-genotype D1 is composed of the strains free from Chinese sequences; therefore, it can be concluded that the molecular epidemiology of CV-A6 in China since 2008 reflects the pattern of endemic circulation of sub-genotype D2 and D3 viruses.
Sub-genotype D2 (66/807) consists mostly of the strains isolated from 2008 to 2011 and clustered with the Japanese strains isolated during the same period. This observation indicated that Chinese sub-genotype D2 strains co-evolved and co-circulated with those from neighbouring countries. Most of the sub-genotype D2 strains isolated between 2008 and 2011 were from South and East China especially from Guangdong Province, where they gave rise to the first reported CV-A6-associated HFMD circulation in mainland China31, 32. Nevertheless, most sub-genotype D2 strains isolated after 2011 were isolated outside South China, suggesting that the transmission route of sub-genotype D2 viruses in China was from south to north. In addition, 4 AFP strains (AFP569, AFP101, AFP147, and AFP149) isolated before 2008 clustered with D2, and originated from Guangdong Province as well. The above finding indicates that D2 genotype in China was probably the first circulating genotype in Guangdong for years, and then triggered the outbreaks in some southern cities, and then was transmitted nationally. The isolates from the European outbreaks did not appear in D2; this result further showed that D2 originated in Asian countries.
The sequences of D3 were analysed further. It was shown that 90.1% (734/807) of the Chinese isolates belong to sub-genotype D3 (except that 1 Guangdong strain was isolated in October 2008, and all the strains have been isolated since 2009), which revealed that sub-genotype D3 was the predominant sub-genotype circulating in mainland of China (Fig. 3b). Most of the Guangdong strains from the CV-A6 emergence period can be seen in this sub-genotype; this observation indicated that during the pre-outbreak period 2009–2012, D2 and D3 co-circulated, but D3 was the major sub-genotype.
Further research split sub-genotype D3 CV-A6 into 2 evolutionary branches, which were designated as D3a and D3b, respectively, the mean distance between these 2 branches reached 6.0% (Fig. 3). The stains that belonged to evolutionary branch D3a (700/734) have been isolated since 2009 (only 1 exception, from 2008), and evolutionary branch D3b (34/734) consists of the Chinese strains from 2011 to 2013 (with only 1 exception: a 2015 Yunnan CV-A6 strain, which independently formed a single lineage, suggesting that it might have been imported from another country) and clustered with the Finnish strains in 2008, the French strains in 2010, and the Japanese strains between 2010 to 2013. As we can see from evolutionary branch D3a of D3, most of the Chinese strains belong to this cluster, and almost all the strains detected since 2014 can be found only in D3a (except for a Yunnan CV-A6 strain in D3b and 2 Xinjiang strains in D2). This finding suggested that the D3a branch may be responsible for the recent virus circulation and may have high epidemic activity. Therefore, we can hypothesise that most descendants from evolutionary branch D3a were responsible mainly for the current large CV-A6-related HFMD outbreaks.
Annual and geographic distribution of D2 and D3 genotypes (D3a and D3b branches) circulating in mainland of China since 2008
In 2008, 80% (4/5) CV-A6 isolates from this period belonged to sub-genotype D2, 4 were isolated in Guandong and 1 in Shandong Province (Fig. 4). Because 4 Guangdong AFP isolates, which were detected before 2008, also belonged to sub-genotype D2, we can speculate that D2 may be the predominant sub-genotype of CV-A6 circulation in mainland of China before 2009. Nonetheless, since 2009, sub-genotype D2 viruses have been gradually decreasing in prevalence and a large number of strains from D3 have been detected. From 2009 to 2014, the percentages of isolates from sub-genotype D2 were 39.3% (11/28), 15.4% (4/25), 34.9% (15/43), 4.0% (2/50), 7.9% (24/305), and 1.5% (2/131), respectively, and no strains were detected in 2015. As to sub-genotype D3, strains from evolutionary branch D3b can be found only in 2011, 2012, and 2013 (except for 1 Yunnan strain from 2015), which suggests that evolutionary branch D3b circulated within a limited period. On the contrary, since 2009, strains from D3a have started to boom; in the CV-A6 outbreak year 2013, 87.5% (267/305) CV-A6 strains belonged to evolutionary branch D3a, 98.5% (129/131) in 2014, and 99.5% (207/208) in 2015. These data indicate that viruses from D3a were responsible for the recent CV-A6 outbreaks.

Yearly proportional distribution of HFMD-associated sub-genotypes D2 and D3 (evolutionary branches D3a and D3b) from 2008 to 2015 in mainland of China.
For the geographic distribution of HFMD-related sub-genotypes that circulated in mainland of China, the D3a evolutionary branch from the D3 sub-genotype has been predominant in all 7 regions of China since 2008 (Fig. 5). Sub-genotype D2 can be also seen in all 7 regions but only constituted a small proportion. This finding indicated again that since 2008, D2 and D3 sub-genotypes have co-circulated in mainland of China but D3, especially evolutionary branch D3a, has been the major one. As to evolutionary branch D3b, the strains from this branch can be detected only in 4 regions: East China, South China, North China, and Northwest China, and mostly (82.4%, 28/34) in Southern and East China, suggesting that D3b might be responsible for the small-scale transmission and probably was imported from the neighbouring countries.

The geographic distribution of HFMD-associated CV-A6 strains in 7 representative regions of China, the region and the number of the sequences are coloured according to the legend (Taiwan sequences are not included). The free map source was acquired from d-maps.com (URL: http://d-maps.com/carte.php?num_car=15272&lang=en; the content is properly attributed in the appropriate figure legend according to the attribution guidelines; the website is protected by Copyright France, Registration Number: 58KU297). The map was then generated and modified using Microsoft Excel 2010 software.
Discussion
The HFMD epidemics have become a serious public health threat in the Asia-Pacific region especially in China in the past decade33, 34. Systematic surveillance of HFMD established in mainland of China in 2008 has been contributing a lot to HFMD research. Nonetheless, studies have focused mostly on the predominant serotypes EV-A71 and CV-A16, whereas information about other EVs is still limited because other EV types usually have only caused sporadic cases. In 2013 and 2015, a series of CV-A6-associated HFMD outbreaks were reported in many provinces of China, and CV-A6 even surpassed EV-A71 and CV-A16 to become the predominant pathogen21,22,23. Since then, more and more researchers have given attention to the CV-A6 serotype. The reasons for the repeated larger-scale HFMD outbreaks and a large number of severe cases caused by CV-A6 remain unclear.
Although molecular epidemiologic analyses of Chinese CV-A6 have been conducted in limited geographic areas and periods21, 22, those studies may have some limitations. Firstly, previous research always have more or less following problems on the division of genotypes and sub-genotypes: (1) The division of genotypes was based on only partial VP1 sequences of CV-A6; (2) The division of genotypes was not following the consensus principle (a difference of at least 15% in the entire VP1 nucleotide sequences should be used for distinguishing genotypes, 8% for sub-genotypes), but according to the shape of the clusters in the phylogenetic dengrogram, which was too subjective. Secondly, currently, there has no published works of Chinese CV-A6 based on a relatively complete time span which includes the CV-A6 from the two large-scale outbreaks (2013 and 2015). Thirdly, because of the vast territory of China, the terrain and climate are different in different regions, which may lead to diverse spread and duplicate abilities of CV-A6. Therefore, an overall research based on the nationwide CV-A6 sequences is very important. The previous evolutionary analysis of CV-A6 of small-scale outbreaks may be quite different from national-based outbreaks. Therefore, a comprehensive study of CV-A6 molecular epidemiology covering most of Chinese areas, on a long-term timescale, and a phylogenetic analysis based on adequate entire VP1 nucleotide sequences of CV-A6 was still lacking. A unified classification of CV-A6 genotypes and sub-genotypes was still unclear.
However, in this study, we have precisely and comprehensively segregated the genotypes and sub-genotypes of worldwide CV-A6 based on the entire VP1 nucleotide sequences (915 bp), and then roughly revealed the circulation pattern of CV-A6 worldwide. Moreover, we accomplished a comprehensive molecular epidemiological analysis of CV-A6 from all seven regions in mainland of China, and analysed based on a long time span of 2008–2015, trying to find the explanations for the recent outbreaks.
The phylogenetic analysis indicated that the CV-A6 strains were segregated into 4 genotypes (A, B, C, and D). Still, like EV-A71 and CV-A1630, 35, the prototype strain of CV-A6 (strain Gdula/USA/1949) is the sole member of genotype A, and genotypes B, C, and D can be further subdivided into sub-genotypes B1–B2, C1–C2, and D1–D3, respectively. Most of the international strains cluster with sub-genotypes D3, suggesting that sub-genotype D3 may be responsible most of the circulation of CV-A6 worldwide.
According to our phylogenetic analysis of Chinese CV-A6 based on the entire VP1 sequences, the sub-genotype switch within genotype D occurred in the past decade. The Chinese CV-A6 strains that were associated with HFMD in the past 10 years are all clustered within genotype D. The most recent descendants of sub-genotype D3 viruses (especially evolutionary branch D3a) are the majority of CV-A6 strains detected during the HFMD outbreaks in China since 2013. In addition, the strains isolated from HFMD outbreaks in 2014 to 2015 can only be seen in evolutionary branch D3a (except for a Yunnan strain and 2 Xinjiang strains). These findings suggest that the evolution of sub-genotype D3 CV-A6 viruses is responsible for the CV-A6-related HFMD, and particularly the evolutionary branch D3a CV-A6 has caused large-scale outbreaks since 2013. The concept of evolutionary branch D3a has never been put forward in the previous studies, and it may have important public health significance in China.
To gain a better understanding of the molecular epidemiology of CV-A6 in mainland of China, the circulation of CV-A6 in China could be divided into three evolutionary stages based on our results above. The first stage should be called the sporadic stage that occurred before 2009, in which sub-genotype D2 might be the predominant sub-genotype of CV-A6. From 2009 to 2012, CV-A6 started to circulate from Guangdong Province and then gradually became one of the primary HFMD pathogens in different regions in China: this is the second stage, which could be regarded as the pre-large-scale outbreak stage. The isolates from this phase, which underwent sub-genotype transition co-circulated in both sub-genotype D2 and D3, and mostly in the D3a evolutionary branch. Evolutionary branch D3b was probably responsible for the small-scale transmission in Shenzhen and Shanghai city. At the third stage (since 2013, we can call it the large-scale outbreak stage), many outbreaks have occurred one after another in China, and the vast majority of the strains detected during this outbreak stage belong to the D3a branch of sub-genotype D3. In addition, there were very few isolates detected at this stage belonging to sub-genotype D2; after 2013, the strains that did not belong to branch D3a were nearly undetectable (only 1 Yunnan strain belonged to D3b and 2 Xinjiang strains belonged to D2). In a word, the decreasing prevalence of strains from the D2 genotype combined with the booming viruses from the D3 sub-genotype summarise the circulation pattern of CV-A6 in mainland of China.
In general, after the emergence and continued evolution, whether a genotype (or sub-genotype) will persistently circulate for a relatively long period like EV-A71 and CV-A16 did29, 33, 36, and whether genotype D3 CV-A6 will still remain the predominant pathogen that causes HFMD in the future in China, continuous surveillance for the coming years should be enhanced.
Large-scale HFMD outbreaks caused by CV-A6 that occurred in China suggest that CV-A6 has been becoming more aggressive in terms of virulence and transmissibility. Nevertheless, the relation between clinical features and molecular characteristics of CV-A6 is still unclear. Other studies have indicated that recombination events that happened in the 2C region of CV-A6 might be relevant to the more generalised rash22, 37; however, considerable studies need to be conducted to demonstrate the molecular evidence, which is our urgent and crucial point for the further research on CV-A6. The 520 CV-A6 entire VP1 sequences from 18 provinces (or municipalities and autonomous regions) involved in this study were obtained through HFMD case routine surveillance in mainland of China, which greatly enriched the GenBank database.
For prevention and control of HFMD, the vaccine strategy may be the most effective way, and should protect the population from infective viruses. Recently, more and more studies on EV-A71 vaccines modified them stepwise, and EV-A71 vaccines went through a clinical trial and have been put to use in mainland of China since 201638,39,40. Nonetheless, because CV-A6 is emerging as the predominant causative pathogen of HFMD with a huge number of severe cases in mainland of China, there is an urgent need to develop an effective vaccine against CV-A6 infection. Several studies on the CV-A6 vaccine based on a mouse model have yielded some results41, 42, but further experiments and development are required.
The National Notifiable Disease Reporting System (NNDRS) of HFMD organized in 2008 in mainland of China has been requiring the specific serotypes information of HFMD cases for EV-A71 and CV-A16, but not for CV-A6. Therefore, the molecular epidemiology of EV-A71 and CV-A16 nationwide has been maturely studied, but the molecular epidemiology of CV-A6 nationwide has been finitely focused on. However, in recent years, CV-A6 has become an important virus in the HFMD pathogen spectrum in China, research vacancies of Chinese CV-A6 nationwide may have a negative impact on China’s public health. Therefore, being the only laboratory in mainland of China that owns CV-A6 isolates from all regions of the nation, this may be the first study or even the only study in recent years that carried out a systematic research on the molecular epidemiology of CV-A6 in mainland of China.
In conclusion, the comparatively thorough molecular epidemiological study of Chinese CV-A6 indicates that CV-A6 has experienced a sub-genotype switch from the co-circulation of sub-genotypes D2 and D3 to the predominance of sub-genotype D3 along with the CV-A6-associated HFMD outbreaks in 2013. Most descendants of sub-genotype D3 are mostly CV-A6 strains isolated during the outbreaks since 2013. The growing virulence and transmissibility and increasing numbers of severe HFMD cases of CV-A6 infection call for further research to overcome the inherent problems of CV-A6.
Methods
Ethics Statement
This study did not involve human participants or human experimentation, the only human materials used were stool samples, throat swab samples, and vesicles collected from HFMD patients for public health purposes at the urging of the Ministry of Health, P. R. of China, and written informed consent for the use of their clinical samples was obtained from the parents of the children whose samples were analysed. This study was approved by the second session of the Ethics Review Committee of the National Institute for Viral Disease Control and Prevention (NIVDC), Chinese Center for Disease Control and Prevention, all experimental protocols were approved by NIVDC, and the methods were carried out in accordance with the approved guidelines.
Virus isolation
The CV-A6 strains used in this study were isolated between 2011 and 2015 from stool, throat swabs, or vesicles from HFMD patients from Yunnan, Zhejiang, Gansu, Guangdong, Guizhou, Hebei, Henan, Hunan, Jiangxi, Jilin, Liaoning, Shaanxi, Shandong, Shanxi, Tianjin, Chongqing, Xinjiang and Sichuan provinces (or municipalities and autonomous regions), which represent different geographic regions of China. Clinical samples were processed according to standard protocols43, and were first confirmed as CV-A6 positive by a commercial real-time PCR assay (Shuoshi Biotech, Jiangsu, China). A total of 520 CV-A6-positive samples were selected according to different years and geographic locations. The representative CV-A6-positive samples were inoculated into human rhabdomyosarcoma (RD) and human laryngeal epidermoid carcinoma (HEp-2) cell lines for virus propagation and purification. These 2 cell lines were obtained from the WHO Global Poliovirus Specialized Laboratory, USA, and were originally purchased from the American Type Culture Collection.
Determination of the entire VP1 nucleotide sequence of CV-A6
Viral RNA was extracted using a QIAamp Viral RNA Mini Kit (Qiagen, Valencia, CA, USA). Reverse transcription polymerase chain reaction (RT-PCR) was performed to amplify the entire VP1 capsid region (915 nucleotides) using PrimeScript One Step RT-PCR Kit Ver. 2 (TaKaRa, Dalian, China) with the primers designed in this study (CVA6-VP1-F: 5′-CTTCGTAGTGCCACCAGATA-3′ and CVA6-VP1-R: 5′-GTGGCGAGATGTCGGTTTA-3′). The PCR products were purified using the QIAquick PCR Purification Kit (Qiagen, Germany), and then amplicons were bi-directionally sequenced using ABI 3130 Genetic Analyser (Applied Biosystems, USA)44.
Phylogenetic analysis and genotyping
The entire VP1 sequence of CV-A6 from the GenBank database was first filtered to exclude the laboratory adaptive strains, clones, strains with high passage numbers, and the possible incorrect strains, and we eventually retrieved 340 entire CV-A6 VP1 sequences from the GenBank database before October 10th, 2016. In combination with the 520 entire CV-A6 VP1 nucleotide sequences determined in this study, a total number of 860 strains formed the dataset used in this study, containing 65 strains used for differentiating genotypes (including 46 Chinese strains and 19 international strains) (Table 1), plus other 36 international strains and other 747 Chinese strains (807 Chinese strains and 53 international strains in total) (Supplementary Table S1).
Sequence alignment was conducted by means of the ClustalW tool in MEGA 5.0, the bootstrap test was performed with 1,000 replications. Phylogenetic dendrograms based on complete VP1 coding sequence were constructed by the neighbour-joining method using the Kimura 2-parameter model45. Bootstrap values greater than 80% were considered statistically significant for grouping. Genotypes were described in a way similar to that originally used to describe EV-A71 and CV-A16: by the group mean distance computing method in MEGA45. A difference of at least 15% in the VP1 region was used to distinguish genotypes30.
Nucleotide sequence accession numbers
The entire VP1 nucleotide sequences of the representative CV-A6 strains which represent each sub-genotype and cluster from different years and regions included in this study were deposited in the GenBank database under the accession numbers KY211689–KY211741 and KY424356–KY424426.
References
Picornaviridae. Virus Taxonomy: Classification and Nomenclature of Viruses: Ninth Report of the International Committee on Taxonomy of Viruses. (Elsevier, San Diego, 2011).
Oberste, M. S., Penaranda, S., Maher, K. & Pallansch, M. A. Complete genome sequences of all members of the species Human enterovirus A. J Gen Virol 85, 1597–1607 (2004).
Oberste, M. S. et al. Enteroviruses 76, 89, 90 and 91 represent a novel group within the species Human enterovirus A. J Gen Virol 86, 445–451 (2005).
Ayukekbong, J. et al. Shift of Enterovirus species among children in Cameroon - Identification of a new enterovirus, EV-A119. J Clin Virol 58, 227–232, doi:10.1016/j.jcv.2013.07.005 (2013).
Razafindratsimandresy, R., Joffret, M. L., Delpeyroux, F. & Heraud, J. M. First full genome sequence of a human enterovirus a120, isolated in madagascar. Genome announcements 2, doi:10.1128/genomeA.00568-14 (2014).
Fan, Q. et al. A Novel Recombinant Enterovirus Type EV-A89 with Low Epidemic Strength in Xinjiang, China. Scientific reports 5, 18558, doi:10.1038/srep18558 (2015).
Zhang, Y. et al. An outbreak of hand, foot, and mouth disease associated with subgenotype C4 of human enterovirus 71 in Shandong, China. J Clin Virol 44, 262–267, doi:10.1016/j.jcv.2009.02.002 (2009).
Chang, L. Y. et al. Neurodevelopment and cognition in children after enterovirus 71 infection. N Engl J Med 356, 1226–1234, doi:10.1056/NEJMoa065954 (2007).
McMinn, P. C. An overview of the evolution of enterovirus 71 and its clinical and public health significance. FEMS Microbiol Rev 26, 91–107 (2002).
Chang et al. Comparison of enterovirus 71 and coxsackie-virus A16 clinical illnesses during the Taiwan enterovirus epidemic, 1998. Pediatr Infect Dis J 18, 1092–1096 (1999).
Zhang, Y. & Xu, W. B. Molecular epidemiology of enteroviruses associated with hand, foot, and mouth disease in the mainland of China. Biomedical and environmental sciences: BES 26, 875–876, doi:10.3967/bes2013.015 (2013).
Osterback et al. Coxsackievirus A6 and hand, foot, and mouth disease, Finland. Emerg Infect Dis 15, 1485–1488 (2009).
Montes et al. Hand, foot, and mouth disease outbreak and coxsackievirus A6, northern Spain, 2011. Emerg Infect Dis 19, 676–678, doi:10.3201/eid1904.121589 (2013).
Mirand et al. Outbreak of hand, foot and mouth disease/herpangina associated with coxsackievirus A6 and A10 infections in 2010, France: a large citywide, prospective observational study. Clin Microbiol Infect 18, E110–118, doi:10.1111/j.1469-0691.2012.03789.x (2012).
Mirand et al. Ambulatory Pediatric Surveillance of Hand, Foot and Mouth Disease as Signal of an Outbreak of Coxsackievirus A6 Infections, France, 2014–2015. Emerg Infect Dis 22, 1884–1893, doi:10.3201/eid2211.160590 (2016).
Flett et al. Hand, foot, and mouth disease caused by coxsackievirus a6. Emerg Infect Dis 18, 1702–1704, doi:10.3201/eid1810.120813 (2012).
Wu et al. The largest outbreak of hand; foot and mouth disease in Singapore in 2008: the role of enterovirus 71 and coxsackievirus A strains. Int J Infect Dis 14, e1076–1081, doi:S1201-9712(10)02473-2 (2010).
Fujimoto et al. Hand, foot, and mouth disease caused by coxsackievirus A6, Japan, 2011. Emerg Infect Dis 18, 337–339, doi:10.3201/eid1802.111147 (2012).
Puenpa et al. Hand, foot, and mouth disease caused by coxsackievirus A6, Thailand, 2012. Emerg Infect Dis 19, 641–643, doi:10.3201/eid1904.121666 (2013).
Han et al. Hand, foot, and mouth disease outbreak caused by coxsackievirus A6, China, 2013. J Infect 69, 303–305, doi:10.1016/j.jinf.2014.03.015 (2014).
Tan et al. Molecular epidemiology of coxsackievirus A6 associated with outbreaks of hand, foot, and mouth disease in Tianjin, China, in 2013. Arch Virol 160, 1097–1104, doi:10.1007/s00705-015-2340-3 (2015).
Feng et al. A novel recombinant lineage’s contribution to the outbreak of coxsackievirus A6-associated hand, foot and mouth disease in Shanghai, China, 2012-2013. Scientific reports 5, 11700, doi:10.1038/srep11700 (2015).
Zeng et al. The Epidemiological Study of Coxsackievirus A6 revealing Hand, Foot and Mouth Disease Epidemic patterns in Guangdong, China. Scientific reports 5, 10550, doi:10.1038/srep10550 (2015).
Chong et al. An atypical dermatologic presentation of a child with hand, foot and mouth disease caused by coxsackievirus A6. Pediatr Infect Dis J 33, 889, doi:10.1097/INF.0000000000000310 (2014).
Feder et al. Atypical hand, foot, and mouth disease: a vesiculobullous eruption caused by Coxsackie virus A6. The Lancet. Infectious diseases 14, 83–86, doi:10.1016/S1473-3099(13)70264-0 (2014).
Li et al. Characterization of Coxsackievirus A6- and Enterovirus 71-Associated Hand Foot and Mouth Disease in Beijing, China, from 2013 to 2015. Frontiers in microbiology 7, 391, doi:10.3389/fmicb.2016.00391 (2016).
Yang et al. Severe hand, foot, and mouth disease and coxsackievirus A6-Shenzhen, China. Clin Infect Dis 59, 1504–1505, doi:10.1093/cid/ciu624 (2014).
Oberste, M. S., Maher, K., Kilpatrick, D. R. & Pallansch, M. A. Molecular evolution of the human enteroviruses: correlation of serotype with VP1 sequence and application to picornavirus classification. J Virol 73, 1941–1948 (1999).
Zhang et al. Molecular evidence of persistent epidemic and evolution of subgenotype B1 coxsackievirus A16-associated hand, foot, and mouth disease in China. J Clin Microbiol 48, 619–622, doi:10.1128/JCM.02338-09 (2010).
Brown, B. A., Oberste, M. S., Alexander, J. P. Jr., Kennett, M. L. & Pallansch, M. A. Molecular epidemiology and evolution of enterovirus 71 strains isolated from 1970 to 1998. J Virol 73, 9969–9975 (1999).
Di et al. Circulation of Coxsackievirus A6 in hand-foot-mouth disease in Guangzhou, 2010-2012. Virol J 11, 157, doi:10.1186/1743-422X-11-157 (2014).
He et al. Emergence, circulation, and spatiotemporal phylogenetic analysis of coxsackievirus a6- and coxsackievirus a10-associated hand, foot, and mouth disease infections from 2008 to 2012 in Shenzhen, China. J Clin Microbiol 51, 3560–3566, doi:10.1128/JCM.01231-13 (2013).
Zhang, Y. et al. Emergence and transmission pathways of rapidly evolving evolutionary branch C4a strains of human enterovirus 71 in the Central Plain of China. PLoS One 6, e27895, doi:10.1371/journal.pone.0027895 (2011).
Xing, W. et al. Hand, foot, and mouth disease in China, 2008-12: an epidemiological study. Lancet Infect Dis 14, 308–318, doi:10.1016/S1473-3099(13)70342-6 (2014).
Perera et al. Molecular phylogeny of modern coxsackievirus A16. Arch Virol 152, 1201–1208 (2007).
Zhang, Y. et al. Complete genome analysis of the C4 subgenotype strains of enterovirus 71: predominant recombination C4 viruses persistently circulating in China for 14 years. PLoS One 8, e56341, doi:10.1371/journal.pone.0056341 (2013).
Gaunt et al. Genetic characterization of human coxsackievirus A6 variants associated with atypical hand, foot and mouth disease: a potential role of recombination in emergence and pathogenicity. J Gen Virol 96, 1067–1079, doi:10.1099/vir.0.000062 (2015).
Li, J. X. et al. Two-year efficacy and immunogenicity of Sinovac Enterovirus 71 vaccine against hand, foot and mouth disease in children. Expert Rev Vaccines 15, 129–137, doi:10.1586/14760584.2016.1096782 (2016).
Zhu, F. et al. Efficacy, safety, and immunogenicity of an enterovirus 71 vaccine in China. N Engl J Med 370, 818–828, doi:10.1056/NEJMoa1304923 (2014).
Li, R. et al. An inactivated enterovirus 71 vaccine in healthy children. N Engl J Med 370, 829–837, doi:10.1056/NEJMoa1303224 (2014).
Shen et al. Virus-like particle-based vaccine against coxsackievirus A6 protects mice against lethal infections. Vaccine 34, 4025–4031, doi:10.1016/j.vaccine.2016.06.028 (2016).
Yang et al. A neonatal mouse model for the evaluation of antibodies and vaccines against coxsackievirus A6. Antiviral Res 134, 50–57, doi:10.1016/j.antiviral.2016.08.025 (2016).
Xu, W. & Zhang, Y. Isolation and Characterization of Vaccine-Derived Polioviruses, Relevance for the Global Polio Eradication Initiative. Methods Mol Biol 1387, 213–226, doi:10.1007/978-1-4939-3292-4_10 (2016).
Zhang, Y. et al. Circulation of multiple serotypes of highly divergent enterovirus C in the Xinjiang Uighur Autonomous Region of China. Scientific reports 6, 33595, doi:10.1038/srep33595 (2016).
Tamura, K. et al. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 28, 2731–2739, doi:10.1093/molbev/msr121 (2011).
Acknowledgements
We would like to acknowledge the staff of Hand, Foot and Mouth Disease Surveillance program in the Yunnan, Zhejiang, Gansu, Guangdong, Guizhou, Hebei, Henan, Hunan, Jiangxi, Jilin, Liaoning, Shaanxi, Shandong, Shanxi, Tianjin, Chongqing, Xinjiang and Sichuan provinces (or municipalities and autonomous regions) Center for Disease Control and Prevention for clinical samples from the HFMD cases collected in this study. The study was supported by the National Natural Science Foundation of China (project nos. 81672070 and 81373049), National key research and development project (project no 2016YFC1200905), and the National Key Technology R&D Program of China (project no. 2013ZX10004-202).
Author information
Authors and Affiliations
Contributions
Y.Z. and W.X. conceived and designed the experiments. Y.S., T.J., Q.Y., X.G., Q.F., D.M., S.Z., W.X., Y.X., Y.S., X.H., Q.L., H.D., X.W., W.Y., S.W. and D.Y. performed the experiments. Y.S., T.J. and Y.Z. analysed the data. Y.S. and Y.Z. wrote the main manuscript and Y.S. prepared all the figures. All authors reviewed the manuscript.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Song, Y., Zhang, Y., Ji, T. et al. Persistent circulation of Coxsackievirus A6 of genotype D3 in mainland of China between 2008 and 2015. Sci Rep 7, 5491 (2017). https://doi.org/10.1038/s41598-017-05618-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-05618-0
- Springer Nature Limited
This article is cited by
-
Current status of hand-foot-and-mouth disease
Journal of Biomedical Science (2023)
-
Identification of a neutralizing linear epitope within the VP1 protein of coxsackievirus A10
Virology Journal (2022)
-
From Monovalent to Multivalent Vaccines, the Exploration for Potential Preventive Strategies Against Hand, Foot, and Mouth Disease (HFMD)
Virologica Sinica (2021)
-
Genetic Variation of Multiple Serotypes of Enteroviruses Associated with Hand, Foot and Mouth Disease in Southern China
Virologica Sinica (2021)
-
Pathogenic characteristics of hand, foot and mouth disease in Shaanxi Province, China, 2010–2016
Scientific Reports (2020)