
Abstract
Extensive phytochemical investigation on the whole herbs of Delphinium anthriscifolium var. majus led to the identification of fourteen diterpenoid alkaloids, including three new C20–diterpenoid alkaloids (anthriscifolsines A–C, 1–3), six new C19–diterpenoid alkaloids (anthriscifolrines A–F, 4–9), and five know compounds (10–14). Among them, anthriscifolsine A represents a novel C20–diterpenoid alkaloid characterized by a seco C–ring. The structures of the isolated compounds were elucidated by extensive spectroscopic methods, including HR-ESI–MS, X–ray, and 1D and 2D NMR experiments. Bioactivity of compounds 3–6 was evaluated for their cytotoxicity against the MCF–7, HepG2 and H460 cancer cell lines.
Similar content being viewed by others


Introduction
Delphinium is a large genus comprising 350 species and distributed in the temperate regions of the Northern Hemisphere, of which 173 are found in mainland China1. In our continuous phytochemical studies on the pharmacologically interesting plants of the genera Aconitum and Delphinium, we obtained a series of structurally and chemotaxonomically diverse diterpenoid alkaloids2,3,4,5. Delphinium anthriscifolium var. majus is an herbaceous plant, belonging to the Sect. Anthriscifolium of the genus Delphinium, and widely distributed in Guizhou, Sichuan, Hubei and Shanxi provinces in China6. Our earlier chemical investigation of this plant led to the discovery of two new C18–diterpenoid alkaloids2. Further studies on the whole extract of this plant resulted in the isolation and structural determination of three new C20–diterpenoid alkaloids, anthriscifolsines A–C (1–3), six new C19–diterpenoid alkaloids, anthriscifolrines A–F (4–9) (Fig. 1), and five know alkaloids nudicaulamine (10)7, anthriscifolmine C (11)8, anthriscifolmine D (12)9, anthriscifolmine I (13)10, and hetisine 13–O–acetate (14)11. Anthriscifolsine A represents a new type of C20–diterpenoid alkaloid, featuring a seco C–ring through an unprecedented C11–C12 bond cleavage of hetisine-type skeleton, whose stereostructure has been unambiguous established by an X–ray crystallographic analysis. Cytotoxicity of diterpenoid alkaloids against MCF–7, HepG2 and H460 cancer cell lines was also evaluated by the MTT method. Herein, we report the isolation, structural elucidation and bioactivity of these diterpenoid alkaloids.

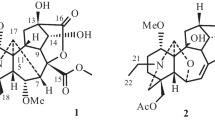
Structures of compounds 1–14.
Results
Anthriscifolsine A (1) was obtained as needles via crystallization from MeOH. Its molecular formula, C29H31NO7, was deduced from the HR–ESI–MS (m/z 506.2182 [M + H]+, calcd for C29H32NO7, 506.2179) and 13C NMR spectroscopic data. The 1H NMR data (Table 1) displayed characteristic resonances of two methyl [δ H 1.10, 1.97 (each 3 H, s)], an acetyl [δ H 2.29 (3 H, s)], a benzoyl [δ H 7.32 (2 H, t, J = 7.8 Hz), 7.51 (1 H, t, J = 7.8 Hz), 7.89 (2 H, d, J = 7.8 Hz)], and an aldehyde [δ H 9.84 (1 H, br.s)] groups. The 13C NMR and DEPT spectra of 1 exhibited the presence of two methyls (δ C 18.8, 24.6), three methylenes (δ C 33.2, 33.5, 36.3), eight sp3 methines (δ C 54.9, 58.5, 59.1, 61.1, 63.9, 67.3, 74.3, 88.4), and three sp3quaternary carbons (δ C 44.4, 47.1, 47.6), one trisubstituted double bond (δ C 124.6, 158.5), one aldehyde (δ C 201.0), one keto carbonyl (δ C 196.1). In addition, an acetyl group [δ C 21.6 (q), 169.8 (s)], a benzoyl group [δ C 129.8 (s), 129.7 × 2 (d), 128.4 × 2 (d), 133.2 (d), 165.5 (s)] were presented in the structure according to the NMR spectra. These characteristic spectroscopic data suggested that 1 was a typical skeleton of C20–diterpenoid alkaloid diester9. The proton and corresponding carbon resonances in the 2D NMR spectra of 1 were assigned by the HMQC experiment. The existence of three oxygenated carbons deduced from its 13C NMR spectrum suggests that 1 has a hydroxyl group, in addition to two ester groups. The absence of a typical C–19 methylene signals in its NMR spectra suggested that a hydroxyl group might be located at C–19, which was confirmed by the HMBC correlations (Fig. 2) from H–3, H3–18 and H–5 to C–199. The acetoxy group could be assigned to C–2 and the benzoyl group at C–3 respectively, on the basis of the HMBC correlations from H–2 (δ H 5.59, m) to the carbonyl carbon of the acetyl group at δ C 169.8 and H–3 (δ H 5.18, d, J = 4.8 Hz) to the carbonyl carbon at δ C 165.5 of the benzoyl group. Compound 1 has the same macular formula and similar NMR spectraoscopic data with those of anthriscifolmine C (11)9, which also possesses an acetyl group at C–2 and a benzoyl group at C–3. However, compound 1 differs from anthriscifolmine C (11) mainly at C–11 where an aldehyde group and a trisubstituted double bond between C–12 and C–16 were deduced. Two methys group were shown to be attached at C–4 and C–16 according to the HMBC correlations from H3–18 to C–3, C–4, C–5 and C–19, and H3–17 to C–12, C–15 and C–16. The substitution pattern and the assigned planar structure of 1 were confirmed by complete 1H−1H COSY and HMBC spectroscopic analysis.

Key HMBC and 1H–1H COSY interactions of compounds 1–3.
The NOESY correlations of H–1β and H–3, H–3 and H–5, H–1α and H–11, H–1α and H–20, H–19 and H–20, proved that H–3 was β–oriented, H–11 and H–19 were in α–orientation (Fig. 3). The NOESY correlations indicated that H–2 was in an equatorial position, which indicated a β–orientation. Moreover, an X–ray diffraction experiment with a suitable crystal was conducted and the absolute configuration of 1 was established as H–2β, H–3β, H–11α, H–19α (19–s) (Fig. 4), consistent with the absolute configuration determined by NOESY correlations. Thus, the structure of 1 was assigned as shown in Fig. 1.

Key NOESY correlations of compounds 1–3.

ORTEP projection of compound 1 (crystallographic numbering).
A possible biogenetic pathway of anthriscifolsine A (1) was proposed as shown in Fig. 5. Aldehyde A could be generated from the known alkaloid anthriscifolmine C (11) through a critical retro–aldol process involving the cleavage of C11–C12 bond. The latter has been also isolated from this plant, which was obtained as needles crystal (MeOH), and the structure of which was unambiguously confirmed by an X–ray crystallographic analysis (Fig. 6). The unstable intermediate A then underwent proton shift and epimerization of the C9 stereochemistry, thus leading to anthriscifolsine A (1). Finally, the artificial possibility of anthriscifolsine A (1) had been explicitly excluded used UPLC–HRESI–MS method and the detailed experiments were added the Supporting Information.

Postulated biogenetic pathway of anthriscifolsine A (1).

ORTEP projection of compound 11 (crystallographic numbering).
Anthriscifolsine B (2) was obtained as a white amorphous powder. Its molecular formula C24H31NO7 was derived from a pseudomolecular ion at m/z 446.2196 [M + H]+ in its HR–ESI–MS. It exhibited characteristic NMR features of a hetisine–type C20–diterpenoid alkaloid bearing groups including two acetyl groups, and an exocyclic double bond (Table 1)12. Two acetyl groups can be installed at C–2 and C–3, respectively, on the basis of the HMBC correlations from H–2 (δ H 5.35, m) to the carbonyl carbon of one acetyl group at δ C 170.2 and H–3 (δ H 4.93, d, J = 4.8 Hz) to the carbonyl carbon of another acetyl group at δ C 170.6. Along with the abovementioned signals, its 13C NMR spectrum displayed five oxygenated carbon signals, suggesting that this compound possessed three additional hydroxyl groups in addition to two ester groups. Two hydroxyl groups were assigned at C–11 and C–13 based on the HMBC correlations from H–11 to C–10, C–13 and C –16, and H–13 to C –11, and COSY correlations of H–11/H–12/H–13/H–14. The observation of HMBC crosspeak between C–9 (δ C 80.2) and H–1 (δ H 1.99), H–7(δ H 1.39), H–12 (δ H 2.51), and H–20 (δ H 3.71), facilitated the location of the third hydroxyl group at C–9.
The relative configuration of 2 was deduced from the vicinal coupling constants and a NOESY experiment (Fig. 3). The coupling constant (J = 4.8 Hz) of H–2 with H–3, indicated that H–2 was in an equatorial position, which indicated a β–orientation9. The large coupling constant of H–13 (J = 8.4 Hz) with H–14α revealed that the dihedral angle between these two H–atoms was ca. 0 °C, which implied that H–13 was in an α–orientation9. In the NOESY spectrum of 2, the cross–peak between H–1β and H–3, H–3 and H–5, H–1α and H–11, H–1α and H–20, proved that H–3 was β–oriented and H–11was in α–orientation. Therefore, the structure of anthriscifolsine B was determined as shown in Fig. 1, and the full assignment of its spectroscopic data was achieved based on the 1D– and 2D NMR analysis (Table 1, Fig. 2).
Anthriscifolsine C (3) was isolated as a white amorphous powder and its molecular formula was deduced to be C30H43NO7 by HR–ESI–MS at m/z 530.3118 [M + H]+. The 1H NMR and 13C NMR data (Table 1) of 3 indicated the presence of the signals of two 2–methylbutanoyloxy groups (MbO) at [(δ H 2.44 (1 H, m), 1.73 (2 H, m), 0.91(3 H, t, J = 7.2 Hz), 1.20 (3 H, d, J = 7.2 Hz) and δ C 177.3 (s), 41.9 (d), 26.8 (t), 11.9 (q), 16.8 (q)] and [(δ H 2.40 (1 H, m), 1.50 (2 H, m), 0.93 (3 H, t, J = 7.2 Hz), 1.16 (3 H, d, J = 7.2 Hz) and δ C 176.8 (s), 41.6 (d), 26.7 (t), 11.8 (q), 17.0 (q)]2. The remaining 20 carbons were assigned based on 1D– and 2D–NMR data and exhibited characteristic NMR features of a hetisine–type C20–diterpenoid alkaloid12 bearing five methylenes, nine methines (four oxygenated) and five quaternary carbons (one ester carbonyl), in addition to one methyl group that was attached to a quaternary carbon (Table 1). The presence of an exocyclic double bond was evidenced by singals in the 1H NMR spectrum (δ H 4.87, d, J = 1.8 Hz, 4.98, d, J = 1.8 Hz) and 13C NMR spectrum (δ C 111.2 and 148.5). The locations of two 2–methylbutanoyloxy groups at C–2 and C–15 were determined by the correlations in the HMBC experiment (Fig. 2). The 13C NMR spectrum showed a singlet at δ C 97.0, indicative of a carbinolamine carbon (C–6). Besides the two ester groups and the carbinolamine carbon, there were two OH groups in the molecule, which were placed at C–3 and C–11, respectively, according to the HMBC displayed in Fig. 2. The coupling constant (J = 5.4 Hz) of H–2 with H–3 indicated that H–2 was in an equatorial position, namely, a β–orientation.
The key NOE correlations of H–1β with H–3, H–3 with H–5, H–1α and H–11, H–1α with H–20, H–15 with H–7α, H–15 with H–14, indicated the orientation of H–3β, H–11α and H–15α. On the basis of the aforementioned evidence, the structure of 3 was determined, and the trivial name anthriscifolsine C was assigned to this compound.
HR–ESI–MS of anthriscifolrine A (4) gave a molecular ion at m/z 448.2757 [M + H]+ (calcd. for C25H38NO6, 448.2699), corresponding to the molecular formula C25H37NO6. Its NMR data indicated seven methylene (one oxymethylene), seven (two oxymethines), and five quaternary carbons (a carbonyl and two oxygen–bearing), in addition to a methylenedioxy group, an N–ethyl, and three methoxy substituents, suggesting that 4 was a typical lycoctonine C19–diterpenoid alkaloid13. The 2D NMR and NOESY experiments confirmed the NMR data and configuration assignments of 4. In particular, HMBC correlations of C–14 with H–9, H–12, H–13, and H–16 confirmed the 14–keto group, while HMBC correlations of the protons of the methylenedioxy with C–7 and C–8, OCH3–1 with C–1, OCH3–16 with C–16, OCH3–18 with C–18, confirmed the locations of the methylenedioxy and three methoxy groups. The α–oriented 1–OCH3 and β–oriented 16–OCH3 in compound 4 were deduced from the vicinal coupling constants (Table 2)and a NOESY experiment (Fig. 7). The structure of anthriscifolrine A was thus established.

Key 1H–1HCOSY, HMBC and NOSEY correlations of 4.
The molecular formula of anthriscifolrine B (5) was determined as C27H41NO8 by HR–ESI–MS at m/z 508.2952 [M + H]+ (calcd. for C27H42NO8, 508.2910). The NMR spectroscopic data of 5 were similar to those of 4, suggesting 5 was also a lycoctonine C19–diterpenoid alkaloid with an acetyl group, an N–ethyl group, three methoxyl groups, and a methylenedioxy group13. In the HMBC spectrum of 5, critical correlations for the protons of the methylenedioxy/C–7 and C–8, OCH3–1/C–1, OCH3–16/C–16, OCH3–18/C–18, H–14/OAc, suggested the location of three methoxyl groups at C–1, C–16 and C–18, the acetyl group at C–14, and the methylenedioxy group at C–7 and C–8. Its 13C NMR spectrum displayed seven oxygenated carbon signals, suggesting that it possessed an additional hydroxyl group in addition to three methoxy groups, an ester group, and a methylenedioxy group. The additional hydroxyl group in 5 was assigned to C–10 on the basis of the correlations of C–10 (δc 79.8) with H–1 (δ H 3.57), H–9 (δ H 2.42), H–13 (δ H 2.76) and H–17 (δ H 2.98) in the HMBC spectrum. The configurations of 1α–OCH3, 16β–OCH3, 14α–OAc and constant18β–OCH3 were deduced by the vicinal coupling constants (Table 2) and a NOESY experiment. Thus, the structure of anthriscifolrine B was confirmed by NMR experiments.
The molecular formula of anthriscifolrine C (6) was determined as C27H41NO9 (HR–ESI–MS). The 1H and 13C NMR data (Table 2) of 6 showed close structural similarity to compound 5, and the distinction between the two sets of spectra was demonstrated by the presence of an additional hydroxyl group signal in 6, which was validated by the additional 16 mass units in mass spectrometry. The proton signal of H–6 at [δ H 2.11 (1 H, m), 1.50 (1 H, m) and δ C 32.8 (t)] in compound 5 was shifted downfield to [δ H 5.52 (1 H, s), δ C 78.3(d)] in compound 6, suggesting that the hydroxyl group in 6 might be located at C–6, which was further confirmed by the HMBC correlations. The hydroxyl group at C–6 was determined to have a β–orientation based on the multiplicity of H–6 (singlet) in the 1H NMR spectrum2. Thus, the structure of anthriscifolrine C was determined as shown in Fig. 1.
Anthriscifolrine D (7), a white amorphous powder, C27H39NO9 (HR–ESI–MS), was also a lycoctonine–type C19–diterpenoid alkaloid. By comparison of the NMR data of 7 with those of 4, the main difference was the presence of a acetyl and a hydroxyl groups. In the HMBC experiment, long–range correlations were observed from H–6 (5.54) to the carbonyl carbon of the acetyl group at δ C 170.2, and H–1 (δ H 3.91), H–9 (δ H 3.49), H–13 (δ H 2.82) and H–17 (δ H 3.71) to C–10 (δc 79.8) supported the hydroxyl group at C–10. The structure of anthriscifolrine D (Fig. 1) was confirmed by the analysis of its 2D NMR data.
The molecular formula of anthriscifolrine E (8) was deduced to be C26H39NO8 from its HR–ESI–MS at m/z 494.2752 [M + H]+. From its NMR data (Table 3), an N–ethyl group, two methoxy groups, an acetoxy group, and a methylenedioxy group could be easily recognized. Compound 8 shared highly similar 1H– and 13C–NMR spectral patterns with those of 5. The only difference is that the absence of a methoxy group and the presence of a hydroxy group at C–18 in 8, which was further supported by comparison of the NMR data: the C–18 signal in 8 appeared at δ C 68.3 instead of at δ C 79.1 in 5. The structure of anthriscifolrine E was unquestionably confirmed by extensive analyses of its 1D and 2D NMR spectra.
Comparison of spectroscopic data of anthriscifolrine F (9) and E (8) indicated that an acetyl group in 8 was substituted by a methoxy group in 9. According to 2D NMR analysis, especially the HMBC correlation of OCH3/C–14, the OCH3 group was attributed to C–14 in 9. The corresponding structure of 9 was confirmed by DEPT, HMQC, 1H–1H COSY, and HMBC experiments. Thus, anthriscifolrine F was assigned as shown in Fig. 1.
To evaluate the biological activities of these compounds isolated from the whole plant of D.anthriscifolium var. majus, compounds 3–6 were tested for their in vitro cytotoxicity against the MCF–7, HepG2 and H460 cancer cell lines. Unfortunately, all of the compounds were inactive (IC50 > 50 μM, n = 3).
Discussion
Investigation on the whole plant of Delphinium anthriscifolium var. majus resulted in the isolation of nine new diterpenoid alkaloids named anthriscifolsines A–C (1–3) and anthriscifolrines A–F (4–9), together with five known alkaloids (10–14). Notably, anthriscifolsine A (1) is the first naturally occurring C20–diterpenoid alkaloid with a unique seco C–ring generated by an unprecedented C11–C12 bond cleavage, and its possible biogenetic pathway was proposed. Since the Sect. Anthriscifolium only comprises three species (D. anthriscifolium, D. anthriscifolium var. majus, and D. anthriscifolium var. savatieri), the present research would be particularly valuable in understanding their chemotaxonomical significance. The identification of various C19– and C20–diterpenoid alkaloids from D. anthriscifolium var. majus revealed its transitional position among the Delphinium plants.
Materials and Methods
General Experimental Procedures
Optical rotations were measured using a Perkin–Elmer 341 polarimeter. The IR spectra were obtained using a Thermo Fisher Nicolet 6700 spectrometer and KBr pellets in cm−1. The HR–ESI–MS data were measured using a Q–TOF micro mass spectrometer (Waters). The 1D and 2D NMR spectra were recorded using a Bruker AV 600 with TMS. Silica gel (Qingdao Haiyang Chemical Co., Ltd., 200–300 mesh) was used for column chromatography (CC). The TLC plates were precoated with silica gel GF254 (Qingdao Haiyang Chemical Co., Ltd., China), and it was visualized under a UV lamp at 254 nm or by spraying with Dragendorff’s reagent or iodine.
Plant Material
The whole herbs of D. anthriscifolium var. majus were collected in Longshanwa, Zhuxi county, Hubei province of China, in April 2015, and were identified (voucher specimen: L H. Shan & J X. Wang 801) by Prof. Qing–Er Yang of the Institute of Botany, Chinese Academy of Sciences.
Extraction and Isolation
Dried and powdered whole herbs of D. anthriscifolium (21.5 kg) were extracted with 95% EtOH four times at room temperature, with each soaking process lasting a week. After removal of the solvent by evaporation, the ethanol extract (2000 g) was recovered. The extract was suspended in H2O (3 L) and adjusted to pH 2 with HCl, and successively extracted with petroleum ether (4 × 1 L) and ethyl acetate (4 × 1 L). The pH of aqueous layer was adjusted to 10 with aqueous ammonia solution and extracted with CH2Cl2 (4 × 1 L). The CH2Cl2 extracts were concentrated to produce the crude alkaloid extract (28.5 g). Column chromatography of the crude alkaloid extract over silica gel, using a CH2Cl2:MeOH (80:1, v/v) mixture with increasing polarity afforded fractions A–D based on TLC analysis.
Fraction A (10.7 g) was submitted to silica gel CC eluting with petroleum ether/Me2CO/Et2N (50: 1: 0.1 to 20:1:0.1, v/v/v) to yield compounds 4 (14 mg), 10 (20 mg) respectively.
Fraction B (7.2 g) was submitted to silica gel CC eluting with petroleum ether/Me2CO/Et2N (15: 1: 0.1 to 6:1:0.1, v/v/v) to yield compounds 5 (20 mg), 6 (43 mg) and 12 (8 mg).
Fraction C (5.6 g) was subjected to silica gel CC, petroleum ether/Me2CO/Et2N (15: 1: 0.1 to 6:1:0.1, v/v/v) to yield compounds 1 (16 mg), 11 (14 mg), 7 (10 mg) and 3 (4.9 mg).
Fraction D (5.0 g) was subjected to silica gel CC, eluted with CH2Cl2:MeOH (30:1 to 10:1, v/v) to get four fractions (D1–D4), fraction D1 was subjected to Sephadex LH–20 column chromatography (MeOH) to yield compounds 13 (14 mg) and 14 (8 mg), fraction D2 was further purified using an RP–18 silica gel column with MeOH: H2O (10:90 to 30:70, v/v) as the mobile phase to yield compounds 2 (7.6 mg), 8 (20 mg), and 9 (11 mg).
Spectroscopic data of 1–9
Anthriscifolsine A (1): needle crystal (MeOH); \({[{\rm{\alpha }}]}_{{\rm{D}}}^{20}\) + 11.1 (c 0.38, CHCl3); IR (KBr) v max: 3465, 3070, 2928, 2880, 2854, 2747, 1744, 1716, 1667, 1630, 1450, 1381, 1340, 1276, 1233, 1121, 1062, 1038, 997, 965, 942, 909, 752, 715; 1H and 13C NMR data see Table 1; HR–ESI–MS at m/z 506.2182 [M + H]+ (calcd. for C29H32NO7, 506.2179).
Anthriscifolsine B (2): white, amorphous powder; \({[{\rm{\alpha }}]}_{{\rm{D}}}^{20}\) + 1.1 (c 0.38, CHCl3); IR (KBr) v max: 3408, 2925, 2853, 1741, 1655, 1371, 1251, 1063, 1042, 985, 769, 719; 1H and 13C NMR data see Table 1; HR–ESI–MS at m/z 446.2196 [M + H]+ (calcd. for C24H32NO7, 446.2179).
Anthriscifolsine C (3): white, amorphous powder; \({[{\rm{\alpha }}]}_{{\rm{D}}}^{20}\) −9.8 (c 0.25, CHCl3); IR (KBr) v max: 3443, 2967, 2935, 2878, 1727, 1656, 1462, 1383, 1265, 1237, 1187, 1152, 1077, 1135, 1009, 997, 906, 754, 715; 1H and 13C NMR data see Table 1; HR–ESI–MS at m/z 530.3118 [M + H]+ (calcd. for C30H44NO7, 530.3118).
Anthriscifolrine A (4): white, amorphous powder; \({[{\rm{\alpha }}]}_{{\rm{D}}}^{20}\) −4.2 (c 0.50, CHCl3); IR (KBr) v max: 3423, 2953, 2925, 2854, 1752, 1648, 1463, 1377, 1094, 954, 734, 721; 1H and 13C NMR data see Table 2; HR–ESI–MS at m/z 448.2757 [M + H]+ (calcd. for C25H38NO6, 448.2699).
Anthriscifolrine B (5): white, amorphous powder; \({[{\rm{\alpha }}]}_{{\rm{D}}}^{20}\) −26.7 (c 0.30, CHCl3); IR (KBr) v max: 3472, 2958, 2923, 2875, 2823, 2755, 1740, 1718, 1456, 1369, 1248, 1206, 1092, 1075, 1054, 961, 934, 755, 730; 1H and 13C NMR data see Table 2; HR–ESI–MS at m/z 508.2952 [M + H]+ (calcd. for C27H42NO8, 508.2910).
Anthriscifolrine C (6): white, amorphous powder; \({[{\rm{\alpha }}]}_{{\rm{D}}}^{20}\) −2.3 (c 0.70, CHCl3); IR (KBr) v max: 3438, 2972, 2927, 2875, 2829, 2750, 1738, 1666, 1453, 1387, 1368, 1246, 1232, 1090, 1045, 961, 918, 756, 715; 1H and 13C NMR data see Table 2; HR–ESI–MS at m/z 524.2867 [M + H]+ (calcd. for C27H42NO9, 524.2860).
Anthriscifolrine D (7): white, amorphous powder; \({[{\rm{\alpha }}]}_{{\rm{D}}}^{20}\) −12.8 (c 0.50, CHCl3); IR (KBr) v max: 3461, 2962, 2924, 2873, 2854, 2827, 2752, 1757, 1742, 1647, 1456, 1368, 1245, 1227, 1103, 1088, 1043, 958, 926, 761, 742; 1H and 13C NMR data see Table 3; HR–ESI–MS at m/z 522.2702 [M + H]+ (calcd. for C27H40NO9, 522.2703).
Anthriscifolrine E (8): white, amorphous powder; \({[{\rm{\alpha }}]}_{{\rm{D}}}^{20}\) −10.7 (c 0.30, CHCl3); IR (KBr) v max: 3447, 2962, 2926, 2885, 2857, 2818, 2746, 1741, 1717, 1463, 1370, 1265, 1230, 1092, 1075, 1052, 953, 913, 756, 729, 710; 1H and 13C NMR data see Table 3; HR–ESI–MS at m/z 494.2752 [M + H]+ (calcd. for C26H40NO8, 494.2754).
Anthriscifolrine F (9): white, amorphous powder; \({[{\rm{\alpha }}]}_{{\rm{D}}}^{20}\) −15.0 (c 0.30, CHCl3); IR (KBr) v max: 3528, 3382, 2958, 2925, 2889, 2855, 2817, 1742, 1666, 1464, 1388, 1371, 1327, 1239, 1158, 1132, 1045, 1103, 1087, 1072, 1051, 1001, 970, 955, 921, 781, 757, 731; 1H and 13C NMR data see Table 3; HR–ESI–MS at m/z 466.2817 [M + H]+ (calcd. for C25H40NO7, 466.2805).
Crystal Data of 1: C29H31NO7, Mr 505.21, a = 12.9571(5) Å, b = 20.9703(17) Å, c = 21.7049(12) Å, V = 5897.5(6) Å3, space group P212121, Z = 8, D calc = 1.204 Mg/m3, λ = 0.71073 Å, μ(Moka) = 0.086 mm−1, F(000) = 2272.0, and T = 293.15 K; Data were collected using an orthorhombic of size 0.4 × 0.1 × 0.05 mm3 in the range −15 ≤ h ≤ 16, −24 ≤ k ≤ 26, −27 ≤ l ≤ 16. 21557 reflections measured, 11486 unique reflections R int = 0.0297. Refinement by full–matrix least–squares on F 2 converged to give final R indices R 1 = 0.0781, wR 2 = 0.1779 [I > 2σ(I)] and R 1 = 0.1399, wR 2 = 0.2106 (all data).
Data/restraints/parameters = 11486/1/725, goodness–of–fit on F 2 = 0.988, largest difference peak and hole are 0.25 and −0.19 e Å−3. Crystallographic data for 1 have been deposited with the Cambridge Crystallographic Data Center as supplementary publication number CCDC 1487703. These data can be obtained free of charge via www.ccdc.cam.ac.uk/deposit (or from the CCDC, 12 Union Road, Cambridge CB2 1EZ, UK; fax: + 44 1223336033; deposit@ccdc.cam.ac.uk).
Crystal Data of 11: C29H31NO7, Mr 505.21, a = 13.1758(7) Å, b = 18.3883(9) Å, c = 21.9022(13) Å, V = 5306.5(5) Å3, space group P212121, Z = 8, D calc = 1.263 Mg/m3, λ = 0.71073 Å, μ(Moka) = 0.090 mm−1, F(000) = 2136.0, and T = 293.15 K; Data were collected using an orthorhombic of size 0.4 × 0.08 × 0.08 mm3 in the range −16 ≤ h ≤ 15, −13 ≤ k ≤ 22, −23 ≤ l ≤ 27. 17628 reflections measured, 9715 unique reflections R int = 0.0250. Refinement by full–matrix least–squares on F 2 converged to give final R indices R 1 = 0.0551, wR 2 = 0.1345 [I > 2σ(I)] and R 1 = 0.07969, wR 2 = 0.1498 (all data).
Data/restraints/parameters = 9715/0/687, goodness–of–fit on F 2 = 0.999, largest difference peak and hole are 0.36 and −0.14 e Å−3. Crystallographic data for 11 have been deposited with the Cambridge Crystallographic Data Center as supplementary publication number CCDC 1487702. These data can be obtained free of charge via www.ccdc.cam.ac.uk/deposit (or from the CCDC, 12 Union Road, Cambridge CB2 1EZ, UK; fax: + 44 1223336033; deposit@ccdc.cam.ac.u k).
Cell Culture and Cytotoxicity Assay
The cytotoxicity of the compounds against cultured human tumor cell lines such as MCF–7, HepG2 and H460 cell lines was evaluated by the MTT method as described in our previous paper14. Cells treated with DMSO (0.1% v/v) were used as negative controls, whereas adriamycin (≥98%; Sigma Chemical Co., Ltd., Shanghai, China) was used as the positive control.
References
Wang, W. C. & Wamock, M. Flora of China, Vol. 6 (eds by Wu Z. Y., Raven P. and Hong D. Y.), 223–274 (Science Press, Beijing, 2001).
Shan, L. et al. Two New C18–Diterpenoid Alkaloids from Delphinium anthriscifolium. Nat prod commun 10, 2067–2068 (2015).
Zhang, J. F. et al. Iliensines A and B: Two new C 19–diterpenoid alkaloids from Delphinium iliense. Phytochem Lett 17, 299–303 (2016).
Chen, L. et al. Diterpenoid Alkaloids from Aconitum soongaricum var. pubescens. Nat prod commun 10, 2063–2065 (2015).
Chen, L. et al. A new C20–diterpenoid alkaloid from Aconitum soongaricum var. pubescens. Nat Prod Res 31, 523–528 (2016).
Wang, W. C. & Warnock, M. Flora of China, Vol. 6 (eds by Wu, Z. Y., Raven, P., Hong, D. Y.), 273 (Science Press, Beijing, 2001).
Kulanthaivel, P. & Benn, M. Diterpenoid alkaloids from Delphinium nudicaule Torr. and Gray. Heterocycles 23, 2515–2520 (1985).
Liu, X. Y., Chen, Q. H. & Wang, F. P. Three new C20–diterpenoid alkaloids from Delphinium anthriscifolium var. savatieri. Chinese Chem Lett 20, 698–701 (2009).
Liu, X. Y., Chen, Q. H. & Wang, F. P. New C20–Diterpenoid Alkaloids from Delphinium anthriscifolium var. savatieri. Helv Chim Acta 92, 745–752 (2009).
Liu, X. Y. et al. Two new C20–diterpenoid alkaloids from Delphinium anthriscifolium var. savatieri. Nat prod commun 5, 1005–1008 (2010).
Benn, M., Richardson, J. F. & Majak, W. Hetisine 13–O–acetate, a new diterpenoide alkaloid from Delphunium nuttallianum Pritz. Heterocycles 24, 1605–1607 (1986).
Wang, F. P. & Liang, X. T. In Cordell G.A. (Ed.), The Alkaloids: Chemistry and Biology, vol. 59, Elsevier Science, New York, 2002, pp. 23–27 (Chapter 1) (2002).
Song, L. et al. New C19–and C18–Diterpenoid Alkaloids from Delphinium anthriscifolium var. savatieri. Chem Pharm Bull 57, 158–161 (2009).
Huang, S. et al. Pyrrolizidine alkaloids from Liparis nervosa with inhibitory activities against LPS–induced NO production in RAW264. 7 macrophages. Phytochemistry 93, 154–161 (2013).
Acknowledgements
This research was supported by grants from National Natural Foundation of China (81402803), the Research Foundation for the Educational Commission of Sichuan Province (15TD0048), Applied Basic Research of Sichuan Province (2014JY0125) and the Fundamental Research Funds for the Central Universities (2682015ZT04).
Author information
Authors and Affiliations
Contributions
X.-L.Z. initiated and designed the project. S.H. contributed to study design, coordinated the project, and cytotoxicity assay. L.-H.S. and J.-F.Z. performed the extraction, isolation, structural identification of the compounds. G.F. contributed to structure determination, helped with data analysis. All authors reviewed the manuscript.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Shan, LH., Zhang, JF., Gao, F. et al. Diterpenoid Alkaloids from Delphinium anthriscifolium var. majus. Sci Rep 7, 6063 (2017). https://doi.org/10.1038/s41598-017-05372-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-05372-3
- Springer Nature Limited