
Abstract
Microbes in the sediments across a series of seamounts along the island arc of the Yap and Mariana trenches were investigated by metagenome. In this study, we reconstructed 107 metagenome-assembled genomes (MAGs), including 100 bacteria and 7 archaea. All the MAGs exhibited >75% completeness and <10% contamination, with 26 MAGs being classified as ‘nearly complete’ (completeness >90%), while 50 falling within 80–90% range and 31 between 75–80% complete. Phylogenomic analysis revealed that 86% (n = 92) of these MAGs represented new taxa at different taxonomical levels. The species composition of these MAGs was most consistent with the previous reports, with the most abundant phyla being Proteobacteria (n = 39), Methylomirabilota (n = 27), and Nitrospirota (n = 7). These draft genomes provided novel data on species diversity and function in the seamount microbial community, which will provide reference data for extensive comparative genomic studies across crucial phylogenetic groups worldwide.
Similar content being viewed by others
Background & Summary
Seamounts are defined as abrupt rising structure from the seafloor with height greater than 100 m below the sea surface1, and there are more than 170,000 seamounts distributed across the global seafloor2. Seamounts represent unique marine environments, their specific topographic characteristics and complex hydrodynamics directly or indirectly enrich the concentrations of inorganic nutrients and particle organic matter, and was proposed as ‘oasis’ harboring generally higher biomass than surrounding waters3. Hydrological dynamics produced by seamounts could cause significant disturbance to the surrounding water bodies, thus impacts on the metabolic functions, taxonomy diversity and population distributions of microbes4. Therefore, a comprehensive insight into the diversity and distribution patterns of microbial communities around the deep-sea seamounts is crucial.
Typical oligotrophic characteristics, complex hydrological characteristics and massive seamounts make the western Pacific Ocean become an ideal region to study the effect of seamount on microbes5. The Yap and Mariana trenches were formed by the collision of plates6. Yap-Mariana Junction cuts across the Mariana Ridge and Yap Ridge, and is located just to the west of the Mariana Trench. Formed by volcanic magmatic activity associated with plate subduction and compression, a series of seamounts are located on the island arc of the two trenches7. In recent years, the microbial diversity of seamounts has been largely investigated by 16 S rDNA amplicon sequencing7,8,9,10,11,12. However, amplicon analysis focusing on one or a few gene regions often fails to distinguish closely related species when assessing community diversity. Alternatively, metagenomics provides abundant gene information about microbes through high-throughput sequencing, and the assembly of these genes could identify a large number of uncultured microbes13. In this study, we further demonstrated the potential microbial diversity by retrieving and assembling their metagenomic sequences into near complete microbial genomes, because metagenome-assembled genomes (MAGs) can provide more accurate information about microbial species and their communities14,15.
We successfully reconstructed 107 MAGs by collecting sediment samples from various locations along the two Trenches. These locations included the summit, flank, and base of seamounts and the deepest point of the Challenger Deep of the Mariana Trench as a control (Table 1; Fig. 1a–c). All of these MAGs have a completeness of >75% with a contamination <10%. In other words, all of the 107 MAGs meet the medium quality of the MIMAG standards16. Of these MAGs, 26 (24%) were ‘near complete’ (completeness >90%), 50 (47%) were >80% completeness, and 31 (29%) were >75% completeness (Table S1). In addition, 60 (56%) MAGs had <5% contamination, and 2 (2%) MAGs had no contamination at all (Tables S1). A total of 40 (37%) MAGs had a N50 length greater than 10,000 bp (Table S1), indicating excellent assembly quality. The genome size that was calculated from MAG completeness using CheckM v1.2.217, ranged from 1.00 to 7.62 Mbp, with an average value of 2.35 Mbp (Table S1) Overviews of the MAGs were presented in Fig. 2. At the phylum level, Thermoplasmatota had the highest GC content (average 64.06%), in contrast, Bacteroidota had the lowest GC content (41.25%, Tables S1, S2; Fig. 2e). There was no significant correlation between genome size and N50 length (Fig. 2c). Of all the MAGs, there was no correlation between their completeness and contamination, despite the fact that MAGs with much lower completeness (completeness < 80%) usually had lower contamination (Table S1; Fig. 2d). According to the Genome Taxonomy Database (GTDB)18, these draft genomes were classified into 100 bacteria and 7 archaea (Fig. 1c). A total of 15 phyla were identified; the most abundant phyla were Proteobacteria (n = 39), Methylomirabilota (n = 27) and Nitrospirota (n = 7) (Figs. 2a, 3). Notably, 92 (86%) MAGs cannot be assigned to any named entry in GTDB, indicating that most of these MAGs represent novel taxa (Table S2). In sum, 2 class, 3 order, 21 families, 32 genera, and 34 species (57 bacteria and 6 archaea) were novel taxa (Table S2; Fig. 2b). The abundance of these MAGs varied among different samples; in general, B02 had more MAGs than others (Fig. 4). The repertoire of such microbial genomes from seamounts can further facilitate the understanding of the species diversity, structure and function of these microbial communities, which will provide reference data for extensive comparative genomic studies across crucial phylogenetic groups worldwide.

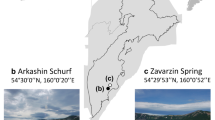
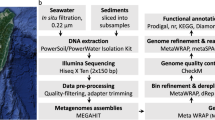
Sample collection and data analysis. (a) Geographical location of the sample sites. (b) The manned submersible Shenhaiyongshi for sample collection. (c) Schematic representation for the metagenomic analysis conducted. A bolded font represents the key processes, and directly below are the tools implemented.

Overview of the MAGs. (a) The distribution of all MAGs at the phylum level. (b) Potential taxonomic novelty of MAGs at different taxonomical levels. (c) The relationship between genomic size and N50 length among MAGs. (d) The relationship between the completeness and contamination of MAGs. (e) Boxplots compare the distribution of genomic size and GC content among MAGs at the phylum level.

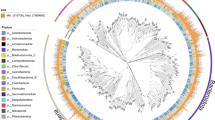
A phylogenetic tree of all species-level bacterial MAGs (n = 100) constructed from 120 conserved bacterial marker genes. The circle colors at the ends of the phylogenetic branches represent known species (green) and unknown species (orange) in GTDB. Different phyla of these MAGs were colored in the outermost ring.

Stacked bar plot of the relative distribution of the 107 MAGs at phylum level across different samples.
Methods
Sample collection and metagenomic sequencing
Sediment samples were collected from a series of seamounts along the Yap Island Arc (YIA: SY222 and SY223), Yap-Mariana Junction area (YMJ: SY203, SY206, SY207 and SY220), Mariana Island Arc (MIA: SY190, SY191, SY192, SY194, SY196 and SY212) and the Challenger Deep (CD: B02), using a pushcore, during cruise TS14 on R/V “Tan Suo Yi Hao” in September 2019 (Fig. 1). In situ hydrographic parameters (i.e., location, depth, temperature and salinity) were measured with the manned submersible, SHENHAI YONGSHI. Three stations (SY220, SY212 and SY223) contained samples of summit, flank and base of seamounts. The surface (0–4 cm) and subsurface (4–8 cm, SY206 and SY220-base) deposits were immediately stored at −80°C for further analysis. Before sediment characteristics analysis, samples were dried in an oven. The concentrations of total organic carbon (TOC), total nitrogen (TN) ammonia (NH4+) and nitrate (NO3−) were determined according to Wang et al.19. In short, TOC and TN contents were estimated by an element analyzer (Elementar vario Macro cube, Germany) on 5 g of dried sediments. NO3− and NH4+ were extracted with 2 M HCl and determined using a colorimetric auto-analyzer (AutoAnalyzer 3, SEAL Analytical, Germany).
Total genomic DNA were extracted from sediment samples using the MoBio PowerSoil DNA extraction kit following the manufacturer’s instructions. The quantity of extracted DNA was measured using the Qubit dsDNA assay kit in combination with a Qubit® 2.0 fluorometer (Life Technologies, USA) and verified by 1% agarose gel electrophoresis. The paired-end sequencing was performed on the Illumina NovoSeq 6000 platform (Illumina Inc., San Diego, CA, USA) at Novogene Co., Ltd. (www.novogene.com).
Quality control and assembly
The quality filtering of short reads was achieved by removing the adapters and barcodes, as well as reads containing poly-N or that were of low-quality from the raw data using the FASTX-Toolkit (http://hannonlab.cshl.edu/fastx_toolkit) and Fastqc softwares (https://github.com/s-andrews/FastQC). Then all of the quality-controlled reads were co-assembled with MEGAHIT v1.2.9 with parameters ‘--k-min 21 --k-max 144 --k-step 10’20. The quality of the assembly was assessed using QUAST v5.0.221.
Genome binning, refinement, and dereplication
Based on tetranucleotide frequencies, coverage, and GC content, genome bins were recovered using the MetaWRAP v1.3.2 pipeline (parameters: default)22, including MaxBin 2.023, metaBAT 2.024 and CONCOCT v1.0.025 metagenomic binning software. The binning results were refined using the MetaWRAP-Bin_refinement module (parameters: -c 50 -x 10). A lineage-specific work flow of CheckM was used to estimate the completeness and contamination of these genome bins. The refinement bins were dereplicated using dRep v2.6.226 (parameters: -comp 50 -con 10) at the 95% average nucleotide identity (ANI).
Taxonomic classification and Phylogenomic analysis of MAGs
The classification of 100 MAGs was performed by the classify_wf workflow of GTDB-TK v2.0.027 with GTDB release 207 (parameters: default). A phylogenetic tree of 100 species-level bacterial MAGs was constructed by 120 bacterial marker genes using the gtdbtk infer module in GTDB-TK (parameters: default). The tree was annotated and visualized by iTOL v528.
Data Records
The raw reads and MAGs of these metagenomic datasets have been deposited in the NCBI under BioProject ID PRJNA113162029. Sequence Read Archive (SRA) accession number SRP51791030. Additionally, the MAGs are available in the NCBI with the Sequence Read Archive (SRA) entries under accessions SRP517910 and the figshare31.
Technical Validation
To avoid contamination of samples, all sampling tools and containers have been sterilized before sampling. After the samples were obtained, they were immediately placed on −80 °C and kept away from light. DNA extraction was carried out in a specialized lab area, the entire sample processing was expedited and completed within 48 hours. We consistently used the PowerSoil DNA Isolation Kit for sediment samples from the same batch to ensure uniformity. To guarantee the integrity of the assembled contigs, different k-mer sizes were selectively used during the MEGAHIT assembly process (ranging from 21 to 141, step by 10). Following assembly, rigorous binning standards were applied, and the sequences obtained post-binning were re-assembled to ensure the highest possible quality of the resulting data. The completeness and contamination of the draft genomes were validated using CheckM.
Usage Notes
Investigating the microorganisms in seamount sediments is crucial for understanding microbial ecology and evolution. This study provides comprehensive metagenomic and microbial genomic datasets from the seamount sediments along the Yap and Mariana trenches. These datasets were acquired using a next-generation sequencing platform and a commonly used metagenomic analysis pipeline. Detailed information about the samples was provided in Table 1. Metagenome sequencing statistics for the MAGs results are listed in Tables S1 and S2.
Ethics approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Code availability
Custom scripts used to generate or process this dataset were deposited in the figshare (https://doi.org/10.6084/m9.figshare.26139184.v1). Software versions and non-default parameters used have been appropriately specified where required.
References
Staudigel, H., Koppers, A. A., Lavelle, J. W., Pitcher, T. J. & Shank, T. M. Defining the word “seamount”. Oceanography 23(1), 20–21 (2010).
Sonnekus, M. J., Bornman, T. G. & Campbell, E. E. Phytoplankton and nutrient dynamics of six south West Indian Ocean seamounts. Deep Sea Res., Part II 136, 59–72 (2017).
Morato, T., Hoyle, S. D., Allain, V. & Nicol, S. J. Seamounts are hotspots of pelagic biodiversity in the open ocean. Proc. Natl. Acad. Sci. USA 107, 9707–9711 (2010).
Liu, J. et al. Bacterial community structure and novel species of magnetotactic bacteria in sediments from a seamount in the Mariana volcanic arc. Sci. Rep. 7, 17964 (2017).
Ma, J. et al. Environmental characteristics in three seamount areas of the tropical western Pacific Ocean: focusing on nutrients. Mar. Pollut. Bull. 143, 163−–174 (2019).
Crawford, A. J., Beccaluva, L., Serri, G. & Dostal, J. Petrology, geochemistry and tectonic implications of volcanics dredged from the intersection of the Yap and Mariana trenches. Earth Planet. Sci. Lett. 80, 265–280 (1986).
Xu, K. Exploring seamount ecosystems and biodiversity in the tropical Western Pacific Ocean. J. Oceanol. Limnol. 39, 1585–1590 (2021).
Sunamura, M., Higashi, Y., Miyako, C., Ishibashi, J. & Maruyama, A. Two bacteria phylotypes are predominant in the Suiyo Seamount hydrothermal plume. Appl. Environ. Microbiol. 70(2), 1190–1198 (2004).
Sudek, L. A., Templeton, A. S., Tebo, B. M. & Staudigel, H. Microbial ecology of Fe (hydr)oxide mats and basaltic rock from Vailulu’u Seamount, American Samoa. Geomicrobiol. J. 26, 581–596 (2009).
Mottl, M. J., Komor, S. C., Fryer, P. & Moyer, C. L. Deep-slab fluids fuel extremophilic Archaea on a Mariana forearc serpentinite mud volcano: Ocean Drilling Program Leg 195. Geochem. Geophys. Geosyst. 4, 9009 (2003).
Davis, R. E. & Moyer, C. L. Extreme spatial and temporal variability of hydrothermal microbial mat communities along the Mariana Island Arc and southern Mariana back-arc system. J. Geophys. Res. 113(B8), 325–334 (2008).
Zhang, Y. & Jing, H. Deterministic process controlling the prokaryotic community assembly across seamounts along in the Yap and Mariana trenches. Ecol. Indic. 158, 111538 (2024).
Nishimura, Y. & Yoshizawa, S. The OceanDNA MAG catalog contains over 50,000 prokaryotic genomes originated from various marine environments. Sci. Data 9, 305 (2022).
Zhou, L., Huang, S. H., Gong, J. Y., Xu, P. & Huang, X. D. 500 metagenome-assembled microbial genomes from 30 subtropical estuaries in South China. Sci. Data 9, 301 (2022).
Haroon, M. F., Thompson, L. R., Parks, D. H., Hugenholtz, P. & Stingl, U. A catalogue of 136 microbial draft genomes from Red Sea metagenomes. Sci. Data 3, 160050 (2016).
Bowers, R. M. et al. Minimum information about a single amplified genome (MISAG) and a metagenome-assembled genome (MIMAG) of bacteria and archaea. Nat. Biotechnol. 35, 725–731 (2017).
Parks, D. H., Imelfort, M., Skennerton, C. T., Hugenholtz, P. & Tyson, G. W. CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 25, 1043–1055 (2015).
Parks, D. H. et al. GTDB: an ongoing census of bacterial and archaeal diversity through a phylogenetically consistent, rank normalized and complete genome-based taxonomy. Nucleic Acids Res. 50, D785–D794, https://identifiers.org/ncbi/insdc.sra:SRP423788 (2022). NCBI Sequence Read Archive(2023).
Wang, J. P., Wu, Y. H., Zhou, J., Bing, H. J. & Sun, H. Y. Carbon demand drives microbial mineralization of organic phosphorus during the early stage of soil development. Biol. Fertil. Soils 52, 825–839 (2016).
Li, D. H., Liu, C. M., Luo, R. B., Sadakane, K. & Lam, T. W. MEGAHIT: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 31, 1674–1676 (2015).
Gurevich, A., Saveliev, V., Vyahhi, N. & Tesler, G. QUAST: quality assessment tool for genome assemblies. Bioinformatics 29, 1072–1075 (2013).
Uritskiy, G. V., DiRuggiero, J. & Taylor, J. MetaWRAP-a flexible pipeline for genome-resolved metagenomic data analysis. Microbiome 6, 158 (2018).
Wu, Y. W., Simmons, B. A. & Singer, S. W. MaxBin 2.0: an automated binning algorithm to recover genomes from multiple metagenomic datasets. Bioinformatics 32, 605–607 (2016).
Kang, D. W. D., Froula, J., Egan, R. & Wang, Z. MetaBAT, an efficient tool for accurately reconstructing single genomes from complex microbial communities. PeerJ 3, e1165 (2015).
Alneberg, J. et al. Binning metagenomic contigs by coverage and composition. Nature Methods 11, 1144–1146 (2014).
Olm, M. R., Brown, C. T., Brooks, B. & Banfield, J. F. dRep: a tool for fast and accurate genomic comparisons that enables improved genome recovery from metagenomes through de-replication. ISME J. 11, 2864–2868 (2017).
Chaumeil, P. A., Mussig, A. J., Hugenholtz, P. & Parks, D. H. GTDB-Tk: a toolkit to classify genomes with the Genome Taxonomy Database. Bioinformatics 36, 1925–1927 (2020).
Letunic, I. & Bork, P. Interactive Tree Of Life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 49, 293–296 (2021).
NCBI Bioproject. https://identifiers.org/ncbi/bioproject:PRJNA1131620 (2024).
Nucleotide Sequence Archive. https://identifiers.org/ncbi/insdc.sra:SRP517910 (2024).
Figshare. https://doi.org/10.6084/m9.figshare.26139184.v1 (2024).
Acknowledgements
This work was supported by the National Key R&D Program of China (2022YFC2805505; 2022YFC2805400; 2022YFC2805304), the Innovational Fund for Scientific and Technological Personnel of Hainan Province (KJRC2023C37), and the International Partnership Program of Chinese Academy of Sciences for Big Science (183446KYSB20210002). We thank the pilots of the deep-sea HOV “Shenhaiyongshi”, the crew of the R/V “Tan Suo Yi Hao” for their professional service during the cruise of TS14. We would like to thank the Institutional Center for Shared Technologies and Facilities of IDSSE, CAS for measurements of the water chemistry.
Author information
Authors and Affiliations
Contributions
Y.Z.: Data analysis, manuscript writing. H.J.: Experimental Design, manuscript editing.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhang, Y., Jing, H. Metagenome sequencing and 107 microbial genomes from seamount sediments along the Yap and Mariana trenches. Sci Data 11, 887 (2024). https://doi.org/10.1038/s41597-024-03762-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-024-03762-7
- Springer Nature Limited