
Abstract
Plagiognathops microlepis is an economic freshwater fish in the subfamily Xenocyprinae of Cyprinidae. It is widely distributed in the freshwater ecosystem of China, with moderate economic value and broad development prospects. However, the lack of genomic resources has limited our understanding on the genetic basis, phylogenetic status and adaptive evolution strategies of this fish. Here, we assembled a chromosome-level reference genome of P. microlepis by integrating Pacbio HiFi long-reads, Illumina short-reads and Hi-C sequencing data. The size of this genome is 1004.34 Mb with a contig N50 of 38.80 Mb. Using Hi-C sequencing data, 99.59% of the assembled sequences were further anchored to 24 chromosomes. A total of 578.91 Mb repeat sequences and 28,337 protein-coding genes were predicted in the current genome, of which, 26,929 genes were functionally annotated. This genome provides valuable information for investigating the phylogeny and evolutionary history of cyprinid fishes, as well as the genetic basis of adaptive strategies and special traits in P. microlepis.
Similar content being viewed by others


Background & Summary
The smallscale yellowfin, Plagiognathops microlepis, belongs to the cyprinid subfamily Xenocyprinae. It is a small to medium-sized fish that inhabits the middle-to-bottom layers of the water and is widely distributed in freshwater ecosystems of China1. This fish feeds on humus, organic debris and algae, therefore it is often used as a tool fish to purify water and control algal bloom, playing an important role in freshwater systems2,3. With the advantages of delicious taste, fast growth and few diseases, P. microlepis has been domesticated into an aquaculture variety4,5,6. Due to its unique dietary characteristics, when co-cultured with other fish species, P. microlepis does not affect the growth of its companions. Instead, it can help to increase yield and purify water, showing broad development prospects7,8. In recent years, the artificial seedling breeding and stock enhancement and releasing of P. microlepis have been carried out in many regions of China9. Studies on the culture technique and models7, population structure10, nutritional composition8, biochemistry and toxicology2,3, and the roles in water quality improvement of this fish11 have also been conducted. However, due to the lack of genetic data resources, researches on the evolutionary adaptation strategies and the molecular mechanisms of excellent traits in P. microlepis are still scarce, which limit our understanding and effective utilization of this fish.
In phylogenetic studies, the subfamily Xenocyprinae is a branch of Cyprinidae with fewer species. These species distribute widely and discretely in East Asia (especially in China) and have a long history, making it possible to evaluate species and populations differentiation under historical environment changes in East Asia12. Currently, the widely accepted classification view is that this subfamily includes 10 species in 4 genera13. However, different views have emerged on the phylogenetic positions and evolutionary history of some fish within this subfamily as phylogenetic studies increasing. Some studies considered P. microlepis as the only one species in Plagiognathops1,14, while others suggest that Plagiognathops is not a valid genus and this fish should be classified into the genus Xenocypris13,15. Both views have been supported by phylogenetic evidences based on different molecular markers16. Nevertheless, most of these studies were conducted based on mitochondrial or several nuclear genes, resulting in inconsistent results. With the development of sequencing technology, phylogenetic studies have entered the era of omics. However, in the subfamily Xenocyprinae, only the genome of Pseudobrama simoni has recently been deciphered17. Therefore, to provide a high-quality genome of P. microlepis is also essential for conducting phylogenetic analysis at the genomics level and elucidating the controversy in the validity of the genus Plagiognathops.
In this work, we constructed a chromosome-level reference genome of P. microlepis by integrating 33.62 Gb of HiFi reads, 44.58 Gb of short reads, and 99.57 Gb of Hi-C reads. The assembled size of this genome was 1004.34 Mb, and 1004.12 Mb of which was anchored to 24 chromosomes with a contig N50 length of 39.98 Mb. This genome contains about 57.64% (578.91 Mb) of repeat elements and 26,929 protein-coding genes. In addition, we also assembled two chromosome-level haplotypes, Haploid A (997.69 Mb) and Haploid B (995.24 Mb), with contig N50 lengths of 36.21 Mb and 33.97 Mb respectively. The completeness (>97.0% complete BUSCO), consistency (>99.8% mapping ratio) and consensus quality values (>47.5) of all the 3 assemblies were estimated to be high. Overall, this genome will provide a reference for phylogenetic, adaptative evolutionary and genetic basis studies on P. microlepis and other cyprinid fishes, which may also provide valuable information for the regulation and restoration of freshwater ecosystems. And the assembled haplotype genomes can serve as a baseline for studies on allele-specific expression or conservation genomics.
Methods
Ethics statement
This work was approved by the Care and Use of Laboratory Animals in Yangtze River Fisheries Research Institute, Chinese Academy of Fishery Sciences (Wuhan, China).
Sampling and genome survey
A healthy female P. microlepis (body weight: 539.79 g) was collected from the original breeding farm in Suizhou, Hubei Province, China (Fig. 1). After anesthesia with MS222 (0.05% in concertation), the muscle, heart, liver, brain, gill and spleen tissues were immediately sampled and frozen in liquid nitrogen, then transferred to −80 °C for further use. High-quality genomic DNA (gDNA) of muscle was extracted with a modified cetyltrimethyl ammonium bromide (CTAB) method18, while the total RNAs were isolated from all tissues using the Omega Bio-tek’s E.Z.N.A.® Total RNA Kit I (R6834, Omega, USA). The quality and concentration of DNA (and RNA) were tested by 0.75% (and 1.5%) agarose gel electrophoresis, NanoDrop One spectrophotometer (Thermo Fisher Scientific) and Qubit 3.0 Fluorometer (Life Technologies, Carlsbad, CA, USA).

Morphological characters of the Plagiognathops microlepis used for genome sequencing.
For genome survey, libraries with 350 bp insert size were constructed using the Nextera DNA Flex Library Prep Kit (Illumina, San Diego, CA, USA) and sequenced with PE-150 paired-end strategy on Illumina Novaseq 6000 platform. After obtaining raw data (44.58 Gb), the sequencing adaptors and low-quality reads were filtered using the Fastp (v 0.21.0) tool. The 19-mer frequency depth distribution was constructed using Jellyfish (v 2.2.10)19, and the genome size was subsequently estimated with Jellyfish and Genomescope (v 2.0)20. Finally, based on the obtained 43.72 Gb clean data, the estimated genome size for P. microlepis was 949.39 Mb with a heterozygous ratio of 0.52 (Fig. S1).
PacBio and Hi-C based whole-genome sequencing
For PacBio sequencing, the qualified high-quality gDNA was sheared to 15–20 Kbp and a SMART bell library was constructed according to the manufacturer’s instructions, followed by sequenced on PacBio Sequel II system in CCS mode. Raw subreads were obtained after filtering polymerase reads, and then processed using SMARTLink (v 8.0) with the parameter “–min-passes = 3–min-rq = 0.99” to generate HiFi reads. As a result, a total of 33.62 Gb HiFi reads were obtained with a mean read length of 17.82 Kb (Table 1).
A Hi-C library was constructed by cross-linking the muscle tissue, digesting with Dpn II restriction enzyme, biotinylating 5′ overhang, blunt-end ligation, and shearing the DNA into 300–700 bp size21. Hi-C sequencing was performed on Illumina Novaseq 6000 platform with PE-150 strategy, and 99.57 Gb of raw data were obtained. After filtering with fastp (v 0.21.0), 98.31 Gb of Hi-C clean reads were retained (Table 1). All of the above sequencing was performed in the Wuhan Benagen Technology Co., Ltd (Wuhan, China).
De novo assembly and Hi-C assembly
In order to generate the monoploid and two haplotype-resolved assembly, the Hi-C integration strategy was used for de novo assembly. HiFi long reads and Hi-C short reads were submitted simultaneously to HiFiasm (v 0.16.1)22 to improve the accuracy of assembly and haplotype construction. Haplotypic duplications in the assembly were removed using purge_dups (v1.2.5, parameter: -f 0.9)23. As a result, a preliminary monoploid assembly of 1004.34 Mb and two haploid assemblies of 997.69 Mb (Haploid A) and 995.24 Mb (Haploid B) were yielded, whose contig N50 lengths were 38.80 Mb, 36.21 Mb and 33.97 Mb, respectively (Table 2). The size of this genome is slightly larger than that of survey result and the previously assembled genome of P. simoni’s (940.9 Mb) in Xenocyprinae.
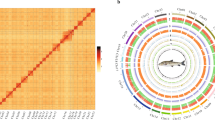
For the chromosome-level assembly, clean Hi-C reads were mapped to the preliminary assembled genome and filtered using Jucier (v1.6)24 with default parameters. Only valid interaction pairs were retained for further analysis. As a diploid, the chromosome number of P. microlepis is reported to be 2n = 48 based on karyotype analysis9,25,26. Therefore, we used the software 3D-DNA (v 180419)27 and Jucier to scaffold the genome onto 24 chromosomes, followed by using Juicebox (v 1.11.08)28 to manually adjust and orient the chromosomes and draw Hi-C interaction heatmap of contigs. Ultimately, 99.98% and 99.59% of the preliminary assembled contigs were anchored to the chromosomes of monoploid (24 chromosomes) and two haplotypes (48 chromosomes) respectively, and a chromosome-level genome of P. microlepis with haplotype-resolution was therefore obtained (Fig. 2, S2 and Table 3). In the assembled monoploid and two haplotypes, 19 and 24 chromosomes were found gap-free (contained only one contig for each chromosome).

Circos plot (a) and Hi-C interaction heatmap (b) showing the features and interactions among chromosomes of the assembled P. microlepis genome. Tracks from outer to inner layers represent the 24 chromosomes (1), gene density (2), repeat elements density (3), GC content (4) and links of intragenomic syntenic blocks within 100Kbp sliding windows.
The mitochondrial genome of P. microlepis was also assembled using MitoZ (v 3.6) and Getorganelle (v 1.7.1a) based on the short reads. The obtained circular mitochondrial genome is 11,619 bp in size, with 13 protein-coding genes, 22 tRNAs and 2 rRNAs (Fig. S3).
Genome annotation
For repeat annotation of the P. microlepis genome, de novo prediction was firstly conducted using RepeatModeler (v 1.0.11, parameters: BuildDatabase -name mydb, RepeatModeler -database mydb -pa 10)29 to detect repetitive elements. LTR sequences were predicted and deduplicated with LTR_FINDER_parallel (parameters: -threads 16 -harvest_out -size 1000000 -time 300) and LTR_retriever (v 2.9.0). These de novo predicted sequences were merged with the RepBase library (v 20181026), and RepeatMasker (v 4.0.9, parameters: -nolow -no_is -norna -parallel 2) and RepeatProteinMask (v 4.0.9) were subsequently employed to identify repeat elements and TE_protein class repeat sequences30. Finally, all the predicted results were merged together and deduplicated, and 578.91 Mb of repeat sequences were identified, accounting for 57.64% of the assembled genome. The most abundant element among these repetitive sequences was DNA transposon, encompassed 31.55% (316.89 Mb) of the assembled genome (Table 4 and Fig. 3a).

The repeat elements distribution and identified protein-coding genes in P. microlepis genome. (a) Distribution of divergence rate for transposable elements in the genome. (b) Veen diagram showing the number of shared and unique genes annotated with different databases.
Three prediction methods were used for the structural annotation of protein-coding genes, including transcript mapping, ab initio prediction and homologous gene alignment. In the RNA-seq based method, 2 μg of qualified RNA from each tissue was equally pooled and an RNA-seq library was prepared with NEBNext® Ultra™ RNA Library Prep Kit (#E7530L, NEB, USA). The constructed library was sequenced on the Illumina Novaseq 6000 platform to obtain 150 bp paired-end reads. After sequencing, the obtained data (11.48 Gb) were filtered with Fastp (v0.21.0), aligned against the genome with Hisat2 (v2.1.0)31 and assembled with Stringtie (v2.1.4)32. The assembled transcripts ORF were predicted using TransDecoder (v 5.1.0). The ab initio prediction was conducted using Augustus (v3.3.2, parameter: –uniqueGeneId = true–noInFrameStop = true–gff3 = on–strand = both)33, Genscan (v1.0)34 and GlimmerHMM (v3.0.4, parameter: -f -g)35 after repeat elements were masked from the genome. In the homology-based prediction, the protein sequences of Danio rerio (GCF_000002035.6), Ctenopharyngodon idellus (GCF_019924925.1), Megalobrama amblycephala (GCF_018812025.1), P. simoni17 and Hypophthalmichthys molitrix (unpublished data, provided by our lab) were downloaded from NCBI database or GigaDB (http://GigaDB.org) and mapped to the genome using tblastn (v 2.7.1, parameter: -t 16 -q 7). Transcripts and coding regions in P. microlepis genome were then predicted using Exonerate (v 2.4.0, parameter: -model protein2genome -showtargetgff 1)36. Finally, all the gene sets predicted by three methods were integrated using MAKER (v 2.31.10, parameter: maker_exe.ctl maker_opts.ctl maker_bopts.ctl -ignore_nfs_tmp -fix_nucleotides)37, and incomplete genes and genes with too short CDS (<150 bp) were also removed, resulting in a non-redundant reference gene set including 28,337 protein-coding genes. The average exon number, exon length and CDS length in each gene were 9.35, 300.06 bp and 1,644.51 bp, respectively (Table 5).
Functional annotation of protein-coding genes was carried out by aligning the predicted sequences against entries in Uniprot and NR databases using Diamond (v 2.0.11.149, parameter: -e-value 1e-5)38. Gene motifs and domains were searched using InterProScan (v 5.52–86.0, parameter: -goterms -pa -dp -verbose -cpu 20)39 and Hmmscan (v 3.3.2, parameter: -E 0.01). The GO terms for genes were obtained from the corresponding InterPro or Uniprot entry. Pathway annotation was performed using Diamond and KOBAS (v3.0) against the KEGG database. At last, a total of 26,929 protein-coding genes were functionally annotated, representing 95.03% of all the predicted genes (Fig. 3b, Table 6).
For non-coding RNA annotation, tRNAs in the genome were searched using tRNAscan-SE (v 2.0.12, parameters: -E -j tRNA.gff -o tRNA.result -f tRNA.struct -thread 16)40 based on the structural characteristics of tRNA, rRNAs were predicted using RNAmmer (v 1.2, parameters: -S euk -m tsu,lsu,ssu)41, and ncRNA sequences were searched using INFERNAL (v 1.1.4, parameter: -cut_ga -rfam -nohmmonly -fmt 2)42 based on the Rfam database. Ultimately, 2,442 miRNAs, 9,677 tRNAs, 4,455 rRNAs and 1,058 snRNAs were identified (Table 7). Based on the annotation results, syntenic blocks among the 24 chromosomes were identified using MCScanX (https://github.com/wyp1125/MCScanx, parameter: -a -e 1e-5 -s 5), and a circular diagram showing the distribution of gene and repeat density, GC content and synteny in the genome was generated using circlize package43 (Fig. 2a).
Chromosomal synteny analysis
To compare the structural characteristics of the genomes, as well as to verify the accuracy of our assemblies, genomic synteny analysis was performed between P. microlepis and two related species, H. molitrix and M. amblycephala. Similar gene pairs and syntenic blocks between any two of the genomes were determined and visualized using Last (v1170)44 and JCVI (v0.9.13)45. The result showed a high degree of collinearity across the three genomes, in which the structures of most chromosomes in P. microlepis remained unchanged compared with those of H. molitrix and M. amblycephala (Fig. 4). Such high consistency also indicated the high quality of our assembled and annotated genome.

Genomic synteny among P. microlepis, Megalobrama amblycephala and Hypophthalmichthys molitrix.
Data Records
The raw sequencing data reported in this paper have been deposited in NCBI Sequence Read Archive (SRA) database under the accession number SRR2788402746, SRR2788402847, SRR2788402948 and SRR2788403049. The assembled nuclear and mitochondrial genomes are available in GenBank with the accessions GCA_040144785.150 and PP836169.151. The genome annotation results have been deposited in the figshare database52.
Technical Validation
Quality evaluation of the genome assembly and annotation
Completeness of the assembled genome was evaluated using BUSCO (v 5.3.0, parameter: -m prot -c 40 -long -f)53 with actinopterygii_odb10 database, and 97.6%, 97.6% and 97.3% complete BUSCOs were found in the assembled monoploid, Haploid A and Haploid B genomes (Table 8), manifesting high completeness of our assembled genomes. The consistency was estimated by mapping the Illumina short-reads to the assembled genomes using BWA (v 0.7.17). As a result, high mapping rates against the three assemblies (99.87%~99.88%) and high coverages (>99.94%) were also found (Table 8). Using Merqury54, the consensus quality value (QV) of genomes representing per-base consensus accuracy were estimated to be 48.11, 48.01 and 47.93 for the assembled monoploid, haploid A and haploid B genomes, respectively. For the quality evaluation of chromosomes, strong interactive signals were found along the diagonals of Hi-C heatmaps, and no obvious noises were found at other areas (Fig. 2b and S2), supporting the precision of chromosome assembly. Finally, to verify the accuracy of haploid splitting, the assembled Haploid A was aligned to Haploid B and similar syntenic blocks between them were also shown. The result showed high similarity and high synteny between the two haploids, indicating high splitting accuracy (Fig. S4).
BUSCO analysis was also performed to validate the quality of genome annotation, and the result revealed that 95% of the identified BUSCOs (including 93.5% single-copy and 1.5% duplicated genes) were complete (Table 8). In addition, the length distributions of genes, CDSs, introns and exons in the genomes of P. microlepis, D. rerio, P. simoni, H. molitrix, M. amblycephala and C. idellus were also compared and found to be similar (Fig. S5), indicating the reliability of our genome annotation.
Code availability
No custom codes were used in this study. All commands and pipelines used in data processing were executed according to the manual and protocols of the corresponding software. The parameters for different software and tools were specified in the Methods section, and default parameters were used for those without detailed parameters.
References
Hu, Y., Zhou, Q., Song, Y., Chen, D. & Li, Y. Complete mitochondrial genome of the smallscale yellowfin, (Teleostei: Cypriniformes: Cyprinidae). Mitochondrial DNA 26, 463–464, https://doi.org/10.3109/19401736.2013.830298 (2015).
Peng, X., Zhao, L., Liu, J., Guo, X. & Ding, Y. Comparative transcriptome analyses of the liver between Xenocypris microlepis and Xenocypris davidi under low copper exposure. Aquatic Toxicology 236, 1–9, https://doi.org/10.1016/j.aquatox.2021.105850 (2021).
Wu, H., Zhang, Y., Zhou, C. & Gao, Y. Influence of temperature on physiological characteristics of Plagiognathops microlepis Bleeker. Environmental Engineering 33, 23–89, https://doi.org/10.13205/j.hjgc.201505006 (2015).
Wang, W., Teng, S. & Ma, Q. High-yield and high-profit rearing technique for smallscale yellowfin. Scientific Fish Farming 12, 11 (2021).
Du, C., Han, Y., Shi, Y. & Zhu, J. Cryopreservation of Plagiognathops microlepis sperm. Cryobiology 85, 105–112 (2018).
Xiong, B. & Lv, G. Preliminary studies on reproduction biology of Xenocypris microlepis. Journal of Hydroecology 31, 76–81 (2010).
Administrative Board of the Tao-Yuan-Ho Reservoir, H. P. S. o. I. A., Laboratory of Fish Genetics Breeding, Institute of Hydrobiology, Hupei Province. Culture experiment and relevant biological studies on Plagiognathops microlepis. ACTA HYDROBIOLOGICA SINICA, 421–438 (1975).
Hu, Z., Liu, G., Dong, Y., Tong, H. & Li, S. Analysis and evaluation of nutritive composition in muscles of Plagiognathops microlepis. Freshwater Fisheries 48, 62–68, https://doi.org/10.13721/j.cnki.dsyy.2018.01.010 (2018).
Zhang, L., Zhou, J., Zhang, T. & He, L. Analysis of morphological, biochemical and genetic characteristics of Xenocypris microlepis of the Yangtze River. Chinese Fishery Quality and Standards 8, 29–35 (2018).
Qiao, D. et al. Genetic variation of mt DNA-Cyt b gene in three populations of Plagiognathops microlepis. Journal of Lake Sciences 23, 813–820 (2011).
Gao, Y. et al. Eutrophicated water quality improvement by combination of different organisms. Chinese Journal of Environmental Engineering 11, 3555–3563 (2017).
Xiao, W., Zhang, Y. & Liu, H. Molecular systematics of Xenocyprinae (Teleostei: Cyprinidae): Taxonomy, biogeography, and coevolution of a special group restricted in east Asia. Molecular Phylogenetics and Evolution 18, 163–173, https://doi.org/10.1006/mpev.2000.0879 (2001).
Liu, H. “Xenocyprinae” in Fauna Sinica, OsteichthyesCypriniformes II. Editor Y. Y. Chen (Beijing: Science). 208–223 (1998).
Zhao, Y., Kullander, F., Kullander, S. O. & Zhang, C. A review of the genus Distoechodon (Teleostei: Cyprinidae), and description of a new species. Environmental Biology of Fishes 86, 31–44, https://doi.org/10.1007/s10641-008-9421-z (2009).
Li, L. et al. Molecular systematics of Xenocyprinae (Cypriniformes, Cyprinidae). ACTA HYDROBIOLOGICA SINICA 47, 628–636 (2023).
Zhang, Z., Li, J., Zhang, X., Lin, B. & Chen, J. Comparative mitogenomes provide new insights into phylogeny and taxonomy of the subfamily Xenocyprinae (Cypriniformes: Cyprinidae). Frontiers in Genetics 13, 1–10, https://doi.org/10.3389/fgene.2022.966633 (2022).
Fan, G. et al. Genomic data of Pseudobrama simoni. GigaScience, https://doi.org/10.5524/102191 (2020).
Shan, G., Jin, W., Lam, E. & Xing, X. Purification of total DNA extracted from activated sludge. Journal of Environmental Sciences 20, 80–87, https://doi.org/10.1016/S1001-0742(08)60012-1 (2008).
Marcais, G. & Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27, 764–770, https://doi.org/10.1093/bioinformatics/btr011 (2011).
Ranallo-Benavidez, T. R., Jaron, K. S. & Schatz, M. C. GenomeScope 2.0 and Smudgeplot for reference-free profiling of polyploid genomes. Nature Communications 11, 1432, https://doi.org/10.1038/s41467-020-14998-3 (2020).
Lieberman-Aiden, E. et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 326, 289–293, https://doi.org/10.1126/science.1181369 (2009).
Cheng, H. Y. et al. Haplotype-resolved assembly of diploid genomes without parental data. Nature Biotechnology 40, 1332–1335, https://doi.org/10.1038/s41587-022-01261-x (2022).
Guan, D. F. et al. Identifying and removing haplotypic duplication in primary genome assemblies. Bioinformatics 36, 2896–2898, https://doi.org/10.1093/bioinformatics/btaa025 (2020).
Durand, N. C. et al. Juicer provides a one-click system for analyzing loop-resolution Hi-C experiments. Cell Systems 3, 95–98, https://doi.org/10.1016/j.cels.2016.07.002 (2016).
Zhou, M., Kang, Y., Li, Y. & Zhou, D. Studies on the silver-stained karyotypes of 7 species in Cyprinidae (pisces). Zoological Research 9, 225–229 (1988).
Li, K., Li, Y., Zhou, M. & Zhou, D. Studies on the karyotypes of Chinese Cyprinid fishes II. karyotypes of four species of Xenocyprininae. ACTA Zoologica Sinica 29, 207–213 (1983).
Dudchenko, O. et al. De novo assembly of the genome using Hi-C yields chromosome-length scaffolds. Science 356, 92–95, https://doi.org/10.1126/science.aal3327 (2017).
Durand, N. C. et al. Juicebox provides a visualization system for Hi-C contact maps with unlimited zoom. Cell Systems 3, 99–101, https://doi.org/10.1016/j.cels.2015.07.012 (2016).
Flynn, J. M. et al. RepeatModeler2 for automated genomic discovery of transposable element families. PNAS 117, 9451–9457, https://doi.org/10.1073/pnas.1921046117 (2020).
Tarailo-Graovac, M. & Chen, N. Using RepeatMasker to identify repetitive elements in genomic sequences. Current Protocals in Bioinformatics, 4.10.11-14.10.14, https://doi.org/10.1002/0471250953.bi0410s25 (2009).
Kim, D., Paggi, J. M., Park, C., Bennett, C. & Salzberg, S. L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nature Biotechnology 37, 907–915, https://doi.org/10.1038/s41587-019-0201-4 (2019).
Kovaka, S. et al. Transcriptome assembly from long-read RNA-seq alignments with StringTie2. Genome Biology 20, https://doi.org/10.1186/s13059-019-1910-1 (2019).
Stanke, M., Diekhans, M., Baertsch, R. & Haussler, D. Using native and syntenically mapped cDNA alignments to improve denovo gene finding. Bioinformatics 24, 637–644, https://doi.org/10.1093/bioinformatics/btn013 (2008).
Burge, C. & Karlin, S. Prediction of complete gene structures in human genomic DNA. Journal of Molecular Biology 268, 78–94, https://doi.org/10.1006/jmbi.1997.0951 (1997).
Delcher, A. L., Bratke, K. A., Powers, E. C. & Salzberg, S. L. Identifying bacterial genes and endosymbiont DNA with Glimmer. Bioinformatics 23, 673–679, https://doi.org/10.1093/bioinformatics/btm009 (2007).
Slater, G. S. & Birney, E. Automated generation of heuristics for biological sequence comparison. BMC Bioinformatics 6, https://doi.org/10.1186/1471-2105-6-31 (2005).
Holt, C. & Yandell, M. MAKER2: an annotation pipeline and genome-database management tool for second-generation genome projects. BMC Bioinformatics 12, https://doi.org/10.1186/1471-2105-12-491 (2011).
Buchfink, B., Xie, C. & Huson, D. H. Fast and sensitive protein alignment using DIAMOND. Nature Methods 12, 59–60, https://doi.org/10.1038/nmeth.3176 (2015).
Blum, M. et al. The InterPro protein families and domains database: 20 years on. Nucleic Acids Research 49, D344–D354, https://doi.org/10.1093/nar/gkaa977 (2021).
Chan, P., Lin, B., Mak, A. J. & Lowe, T. M. tRNAscan-SE 2.0: improved detection and functional classification of transfer RNA genes. Nucleic Acids Research 49, 9077–9096, https://doi.org/10.1093/nar/gkab688 (2021).
Lagesen, K. et al. RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic acids research 35, 3100–3108, https://doi.org/10.1093/nar/gkm160 (2007).
Nawrocki, E. P. & Eddy, S. R. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 29, 2933–2935, https://doi.org/10.1093/bioinformatics/btt509 (2013).
Gu, Z. G., Gu, L., Eils, R., Schlesner, M. & Brors, B. Circlize implements and enhances circular visualization in R. Bioinformatics 30, 2811–2812, https://doi.org/10.1093/bioinformatics/btu393 (2014).
Frith, M. C., Hamada, M. & Horton, P. Parameters for accurate genome alignment. BMC Bioinformatics 11, https://doi.org/10.1186/1471-2105-11-80 (2010).
Tang, H. B. et al. Synteny and collinearity in plant genomes. Science 320, 486–488, https://doi.org/10.1126/science.1153917 (2008).
NCBI Sequence Read Archive. https://identifiers.org/ncbi/insdc.sra:SRR27884027 (2024).
NCBI Sequence Read Archive. https://identifiers.org/ncbi/insdc.sra:SRR27884028 (2024).
NCBI Sequence Read Archive. https://identifiers.org/ncbi/insdc.sra:SRR27884029 (2024).
NCBI Sequence Read Archive. https://identifiers.org/ncbi/insdc.sra:SRR27884030 (2024).
NCBI GenBank. https://identifiers.org/ncbi/insdc.gca:GCA_040144785.1 (2024).
NCBI GenBank. https://identifiers.org/ncbi/insdc:PP836169.1 (2024).
Wu, Y. The genome annotation of Plagiognathops microlepis. Figshare. https://doi.org/10.6084/m9.figshare.25002110 (2024).
Manni, M., Berkeley, M. R., Seppey, M., Simao, F. A. & Zdobnov, E. M. BUSCO update: novel and streamlined workflows along with broader and deeper phylogenetic coverage for scoring of eukaryotic, prokaryotic, and viral genomes. Molecular Biology and Evolution 38, 4647–4654, https://doi.org/10.1093/molbev/msab199 (2021).
Rhie, A., Walenz, B. P., Koren, S. & Phillippy, A. M. Merqury: reference-free quality, completeness, and phasing assessment for genome assemblies. Genome Biology 21 (2020).
Acknowledgements
We thank Dr. Zhixiong Zhou and Lin Chen in Xiamen University for their kindly helps in data analysis. This work was financially supported by Central Public-interest Scientific Institution Basal Research Fund, CAFS (NO. 2023TD36), the earmarked fund for CARS (CARS-45), National Freshwater Aquatic Germplasm Resource Center (FGRC18537).
Author information
Authors and Affiliations
Contributions
H.L. and G.Z. conceived and supervised this study. H.S. and X.L. collected the samples and extracted the genomic DNA. Y.W. designed the experiment, performed data analysis and drafted the manuscript. H.L. revised this manuscript. All authors have read and approved the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wu, Y., Sha, H., Luo, X. et al. Chromosome-level genome assembly of Plagiognathops microlepis based on PacBio HiFi and Hi-C sequencing. Sci Data 11, 802 (2024). https://doi.org/10.1038/s41597-024-03645-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-024-03645-x
- Springer Nature Limited