
Abstract
Advancements in sequencing have enabled the assembly of numerous sheep genomes, significantly advancing our understanding of the link between genetic variation and phenotypic traits. However, the genome of East Friesian sheep (Ostfriesisches Milchschaf), a key high-yield milk breed, remains to be fully assembled. Here, we constructed a near-complete and gap-free East Friesian genome assembly using PacBio HiFi, ultra-long ONT and Hi-C sequencing. The resulting genome assembly spans approximately 2.96 Gb, with a contig N50 length of 104.1 Mb and only 164 unplaced sequences. Remarkably, our assembly has captured 41 telomeres and 24 centromeres. The assembled sequence is of high quality on completeness (BUSCO score: 97.1%) and correctness (QV: 69.1). In addition, a total of 24,580 protein-coding genes were predicted, of which 97.2% (23,891) carried at least one conserved functional domain. Collectively, this assembly provides not only a near T2T gap-free genome, but also provides a valuable genetic resource for comparative genome studies of sheep and will serve as an important tool for the sheep research community.
Similar content being viewed by others


Background & Summary
Selective breeding for different agricultural purposes, such as meat, wool, and milk, have established many sheep breeds with unique characteristics worldwide1. The East Friesian sheep (Ostfriesisches Milchschaf) is a highly specialized breed. The breed originates from the Frisia region of both the Netherlands and Germany, and is considered to be the world’s highest producing dairy sheep2,3. In a single lactation, the East Friesian sheep can produce 500–700 kg of milk over a period of approximately 230 days4. Additionally, East Friesian sheep have a relatively high average number of lambs per ewe, 2.25 lambs/litter, but the carcass of lambs is very lean5. In physical appearance, East Friesian sheep have many unique features. They have a relatively large body, head, face, legs, ears all clean of wool. Their most distinctive physical feature is a “rat-tail” which is thin and devoid of wool. The East Friesian sheep, renowned for its adaptability, has been successfully crossbred with breeds known for their robust ketone body composition, such as Suffolk, Dorset, and Texel. This strategic crossbreeding not only enhances the meat quality of the East Friesian sheep but also ameliorates the traits of breeds that exhibit lower milk yields and suboptimal reproductive and lambing capabilities. Hailing from the northern regions of Germany and the Friesland area in the Netherlands, the East Friesian breed has garnered international attention and has been integrated into the livestock industries of various countries, including China, the United Kingdom, and South Africa. The exploration of the breed’s genetic makeup at the molecular level presents a compelling opportunity to deepen our comprehension of the genetic underpinnings of economically significant traits in sheep, thereby contributing to the advancement of the field.
De novo genome assembly is a fundamental and powerful tool employed in the realm of molecular research. Several genomes of sheep genomes have been made publicly available in databases, including East Friesian sheep6, Tibetan sheep7, Rambouillet sheep8, and Texel sheep9. Despite the achievement of chromosome-level assembly in these sheep genomes, there still exist unidentified regions containing gaps that require further investigation and determination. A number of assemblers have been developed for long reads assembly, such as Falcon10, Flye11, Canu12, wtdbg213, NextDenovo14 and Hifiasm15. The Hifiasm method stands out for its utilization of string-overlap graphs to represent genomes, encode information for algorithmic analysis, and visually present both primary and alternative paths along a DNA sequence16. New developments in long-read sequencing technologies, such as Pacific Biosciences (PacBio) circular consensus (CCS) long-read sequencing and ultra-long ONT sequencing, has revolutionized our ability to acquire comprehensive chromosome sequences spanning from one telomere to another. With the availability of a complete genome sequence, researchers would have the opportunity to thoroughly investigate and gain a deeper understanding of genome function, regulation, and evolution17,18.
In this study, we present the first near T2T gap-free genome assembly for East Friesian sheep using a combination of PacBio high-fidelity (HiFi) long-read, Oxford Nanopore (ONT) ultra long-read, and high-throughput chromosome conformation capture (Hi-C) sequencing data. In total, we generated 321 Gb (~107X coverage) ONT reads with a N50 of 63.5 kb, 148 Gb PacBio HiFi CCS reads with a N50 of 22.1 kb (~49X coverage), and 396 Gb Hi-C data (MGISEQ paired-end reads, ~132X coverage) (Table 1). The final genome assembly of East Friesian sheep, termed as EFS v2.0, is about 2.96 Gb with a scaffold N50 of 104.10 Mb, comprising 27 chromosomes without any gaps (Table 2; Fig. 1) and 164 unplaced sequences. We observed that 94.53% of these unplaced sequences consist of repetitive elements, among which satellite sequences constitute 84.64%. Further research and refinement are needed to determine their precise genomic location and functional relevance. The EFS v2.0 assembly captured 41 telomeres and 24 centromeres (Table 3). Notably, the EFS v2.0 assembly closed 35 gaps in total compared to the previously published East Friesian genome6 (Fig. 2).

Circos plot of the EFS v2.0 genome. From inside to outside, I: GC content in nonoverlapping 1 Mb windows (histograms); II: percent coverage of repetitive sequences in nonoverlapping 1 Mb windows (heat maps); III: gene density calculated based on the number of genes in nonoverlapping 1 Mb windows (heat maps); IV: 27 super-scaffolds. Lengths are shown in Mb.

Overview of the near T2T and gap-free EFS v2.0 reference genome. The box represents the 35 closed gaps identified from GCA_018804185.1. The triangle represents the telomere region, and the circle represents the centromere region.
In the EFS v2.0 genome, repeat sequences accounted for 1.60 Gb, representing 53.98% of the assembly (Table 4). Long interspersed nuclear elements (LINE) retrotransposons (41.46%) were the most abundant component among repetitive elements, which was consistent with a previous study19 (Table 5). Gene annotation identified 24,580 protein-coding genes. Of which, 24,536 genes (99.8%) were anchored to 27 chromosomes (Fig. 1), while 44 genes anchored to unplaced scaffolds. The length and number of exons were similar to those of three other sheep breeds (Fig. 3a,b). Furthermore, the predicted proteins achieved a complete BUSCO score of approximately 98%, indicating high quality annotation (Fig. 3c). 23,891 (97.2%) protein-coding genes were successfully annotated in diverse databases, including Gene Ontology (GO), KOG, Interpro, SwissProt20, Kyoto Encyclopedia of Genes and Genomes (KEGG)21, NCBI nonredundant database (NR), and Translation of European Molecular Biology Laboratory (Trembl) (Table 6). Moreover, 17,328 (~70.5%) genes were supported by all five databases (Fig. 3d). Based on transcriptomic deep-sequencing data, we investigated gene expression level in five different tissues (Table 7). A total of 15,263 (62.2%) genes showed detectable expression levels (transcripts per million ≥ 1) in one or more of these tissues. Through structural variants analysis with the previously published East Friesian sheep6, we identified 232 newly assembled genes, among which 151 were expressed in 5 different transcriptome samples (Table 8; Fig. 4).

Quality assessment of the protein-coding genes in the EFS v2.0 assembly. (a) Comparison of exon length among four sheep gene sets. Window refers to the length of every point. (b) Comparison of exon number among four sheep gene sets. No obvious unexpected differences exist among these four organisms, indicating the high quality of gene structure annotation. (c) BUSCO assessment results of protein-coding genes in the EFS v2.0 assembly. (d) Gene function annotation results in a statistics Venn diagram using five public databases: NR, InterPro, KEGG, SwissProt and KOG.

Heatmap representation of new assembled genes. Rows represent new assembled genes, and columns represent 5 different samples. The bar in the upper right corner represents log 2 transformed TPM values. Blue and red boxes represent genes showing lower and higher expression levels, respectively. “Hea_t” represents heart, “Rum_n” represents rumen, “Sub_t” represents subcutaneous fat, “Lun_g” represents lung, and “Per_t” represents perirenal fat.
Methods
Sample collection, DNA preparation and RNA extractions
A 1-year-old female East Friesian sheep from Inner Mongolia key Lab of Bio-manufacture in Inner Mongolia autonomous region of China was chosen for DNA and RNA sequencing. The assembled sequence does not include the Y chromosome due to sampling from females. The animal was healthy, and no genetic defects were observed in it or its parents.
DNA was extracted from fresh blood specimen using the QIAGEN Blood & Cell Culture DNA Midi Kit according to the manufacturer’s instruction (QIAGEN, Germany). TRIzol (Invitrogen, Carlsbad, CA, United States) was used to extract total RNA from heart, rumen, subcutaneous fat, lung and perirenal fat tissues. The concentration of total RNA was determined using the Nano 6000 spectrophotometer Assay Kit of the Bioanalyzer 2100 system (Agilent Technologies, Santa Clara, CA, United States). The RNA purity was determined using the Qubit® RNA Assay Kit in a Qubit® 2.0 Fluorometer (Life Technologies, Camarillo, CA, United States).
Long insert libraries preparation and sequencing
The library construction and sequencing of RNA-seq full-length transcripts were conducted using a method similar to that described in Yuan, Ge et al.22, resulting in 437,807 full-length non-chimeric reads with mean length of 1,388 bp.
For the DNA PacBio long inserts libraries, the preparation was carried out in accordance with the “Using SMRTbell Express Template Prep Kit 2.0 With Low DNA Input” protocol23 provided by PacBio (Pacific Biosciences, USA). This resulted in libraries with an insert size of approximately 20 kb. Subsequently, the libraries were subjected to sequencing using PacBio Sequel II platforms operating in CCS mode. The subreads were processed through the CCS algorithm of SMRTLink (v8.0.0)24 with specific parameters: “-minPasses 3 -minPredictedAccuracy 0.99 -minLength 500”, yielding 148 Gb of PacBio’s long high-fidelity (HiFi) reads in total.
Furthermore, ultra-long DNA ONT libraries were created following the protocols detailed by Shafin et al.25. These libraries were then sequenced on the PromethION sequencer platform (Oxford Nanopore Technologies, UK). The sequencing effort resulted in the production of 8,180,779 reads, with an N50 value of 63,509 bp.
Short insert libraries preparation and sequencing
RNA-seq libraries were prepared uisng the NEBNext® Ultra™ RNA Library Prep Kit for Illumina® (NEB, Ipswich, MA, USA) following the manufacturer’s protocol. Subsequently, the RNA libraries were sequenced on a MGISEQ-2000 platform, producing 150 bp paired-end reads.
The Hi-C library was prepared using the same method described in Yin, Chen et al.26 with the same blood specimen and sequenced on a MGISEQ-2000 instrument. A total of 395 Gb of clean data were obtained from 396 Gb of sequencing data using software SOAPnuke (v2.0)27 with parameters “-n 0.01 -l 20 -q 0.1 -i -Q 2 -G 2 -M 2 -A 0.5”.
Genome assembly
With the HiFi reads, the primary contigs were assembled using Hifiasm (v 0.16.1)15 with default parameters. The Hi-C valid reads were employed to anchor contigs onto chromosomes through Juicer28 and 3d-dna pipeline29. The chromosome nomenclature was adopted for the chromosome numbering on the basis of their collinearity with 27 chromosomes of Texel sheep genome30. To achieve a near T2T gap-free reference genome assembly, gaps in the assembly genome were filled using LR_Gapcloser31 with error-corrected ONT long reads produced by NECAT32.
Annotation of repetitive sequences and identification of telomeres and centromeres
Two strategies, de novo and homolog methods, were applied to annotate repetitive sequences. RepeatModeler (v1.0.4)33 was used to identify de novo repeats, and LTR-FINDER (v1.0.7)34 was utilized to annotate long terminal repeats. DNA and protein transposable elements (TEs) were detected by RepeatMasker (v4.0.7)35 and RepeatProteinMasker (v4.0.7), respectively, based on Repbase database36. At last, tandem repeats were identified using Tandem Repeat Finder (v4.10.0)37. Referencing the methods described in sweet tea38, wild blueberry39, and rapeseed studies40, the telomeric sequences in the EFS v2.0 genome assembly were identified using quarTeT (v1.0.3)41 with the “-c animal” option. The quarTeT program comprises four modules: AssemblyMapper, GapFiller, TeloExplorer, and CentroMiner, of which, TeloExplorer is specifically utilized for identifying candidate telomeres. The telomere repeat monomer identified by quarTeT was “TTAGGG/CCCTAA”. The Centromics software (https://github.com/ShuaiNIEgithub/Centromics) was employed for centromere identification. Furthermore, we aligned the human rRNA sequences against the EFS v2.0 genome by using BLASTN with an E value of 1 × 10−5 to investigate the ribosomal DNA (rDNA) in EFS v2.0 genome. In total, we identified 1329 ribosomal RNAs (rRNAs), including 122 18S rRNA, 586 28S RNA, 59 5.8S rRNA, and 562 5S RNA.
Protein-coding genes prediction and functional annotation
Protein-coding genes in the EFS v2.0 assembly were predicted using a similar method to Fang, Mou et al.42. For the RNA-seq-based prediction approach, clean RNA-seq reads were aligned to the EFS v2.0 assembly via Hisat2 (v2.1.0)43 with parameters including–sensitive–no-discordant–no-mixed -I 1 -X 1000–max-intronlen 1000000. Additionally, 437,807 full-length transcripts were matched against the EFS v2.0 assembly using GMAP (v 2017-11-15)44. The aligned reads were then assembled using Stringtie (v1.3.5)45 using the parameters: -f 0.3 -j 3 -c 5 -g 100 -s 10000. Subsequently, TransDecoder (v5.5.0) (https://github.com/TransDecoder/TransDecoder) was invoked to identify the coding sequence with default settings. In the homolog prediction method, GeMoMa (v1.9)46 was used to detect homologous peptides across four mammalian genomes, including human (GCA_000001405.29)47, Texel sheep (GCA_000298735.2)9, Tibetan sheep (GCA_017524585.1)7, and Rambouillet sheep (GCA_016772045.1)8. Genes that had RNA-seq-based prediction support with correct structure, but were not identified via homology-based prediction, were incorporated into the gene set. Ultimately, untranslated regions and alternative splicing regions were determined using Program to Assemble Spliced Alignment48. The integrated gene set was translated into amino-acid sequences and functionally annotated by mapping against KEGG49, Swiss-Prot20, TrEMBL20, KOG50, InterPro51 and NR (NCBI Non-redundant protein) databases using BLAST (v2.2.26)52 with an E-value threshold of 1E-5. Protein domains and motifs were annotated using InterProScan53, from which GO Ontology (GO)54 was derived.
Gene expression analysis
Quality control of raw RNA-seq reads was conducted using SOAPnuke (v2.0)15. Afterwards, the clean reads were aligned to the EFS v2.0 genome using Hisat2 (v2.1.0)43, with the following parameters: ‘--phred33 -p 5 --sensitive --no-discordant --no-mixed -I 1 -X 1000’. A read count matrix was generated using featureCounts55. Gene expression levels were calculated using the transcripts per million (TPM) method.
Identification of new assembled genes
The software Syri (v1.6.3)56 was employed to detect structural variations between the EFS v2.0 genome assembly and the previously published East Friesian sheep6. A gene was classified as newly assembled if the previously published East Friesian sheep6 exhibited a deletion of at least 50 bp and the gene region had a minimum overlap of 30% with that region.
Reads coverage analysis of genome assembly
We assessed whether the long sequencing reads extended across the regions that required gap filling. Prior to this process, the genome contained eight gaps. We employed minimap257 (v 2.24) to map both the ONT and HiFi reads to the EFS v2.0 genome. Utilizing SAMtools58 (v 1.10) with the ‘-q 20’ option, we filtered out low-quality and multi-mapping reads. Subsequently, we utilized the IGV software for visualizing the high-quality alignment results.
Quality value (QV) calculations
In the realm of whole-genome sequencing, the Quality Value (QV) emerges as an essential metric for gauging the precision of nucleotide identification. The QV is derived from the Phred quality score, a measure that captures the negative logarithm of the likelihood that a given base call is erroneous. The QV is precisely calculated through the equation QV = −10 × log10 (error probability). For instance, an error probability of 0.001 equates to a QV of 30, indicating a high confidence in the correctness of the base call. Throughout the sequencing process, each nucleotide is appraised with a Phred score that is contingent upon the signal-to-noise ratio; this score is subsequently converted to a QV, thereby providing an index of the sequencing data’s fidelity. In this study, we have employed the Merqury59 software to meticulously compute the QV, ensuring robust data quality assessment.
Data Records
The DNA sequence reads of East Friesian sheep (Experiment of DNA sequencing data from ultra-long ONT library: SRR2627375660; Experiments of DNA sequencing data from Hi-C library: SRR2627376360; Experiments of DNA sequencing data from PacBio HiFi library: SRR2627376260) and RNA sequence reads of East Friesian sheep (Experiment of 5 transcriptome libraries: SRR26273757-SRR2627376160) have been deposited in the Sequence Read Archive (SRA). The genome assembly have been deposited in the GenBank database under the accession number JAWMPZ00000000061. The files of the gene structure annotation, repeat predictions and gene functional annotation have been deposited at Figshare database62.
Technical Validation
Multiple methods were employed to validate the accuracy and completeness of EFS v2.0 assembly.
Firstly, we utilized long sequencing reads to ascertain their extension across the eight gap regions (Table 9). The resulting plots confirmed comprehensive coverage of the targeted regions (Fig. 5). Secondly, the Hi-C heatmap displayed high consistency across all chromosomes, demonstrating the correct ordering and orientation of contigs in the EFS v2.0 assembly (Fig. 6). Thirdly, the EFS v2.0 assembly exhibited high collinearity with Rambouillet sheep (GCA_016772045.1)8, Tibetan sheep (GCA_017524585.1)7 and the previously published East Friesian sheep (GCA_018804185.1)6 (Fig. 7). Fourthly, the accuracy was confirmed by the high mapping rates of two type sequences on the EFS v2.0 assembly, with 99.93% of ONT reads and 100% of HiFi reads aligning to the EFS v2.0 assembly. Notably, the sequencing assembly attained a remarkable quality value (QV) score of 69.1, signifying an exceptionally low error rate of approximately 1.26 errors per 100 million bases. This level of sequencing accuracy and reliability is highly commendable and will undoubtedly facilitate subsequent genetic analysis and research. Lastly, the Benchmarking Universal Single-Copy Orthologs (BUSCO) test revealed that EFS v2.0 assembly successfully identified 97.1% of 9,226 mammalia gene sets, which exhibiting the highest level of BUSCO completeness among the four commonly used genomes (Fig. 8).

Using IGV to demonstrate the coverage of ONT and PacBio reads in the gap 1 region. The IGV images for Gap 1 through Gap 8 are available through the Figshare database62.

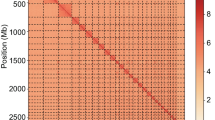
The accuracy and completeness of the EFS v2.0 genome assembly. Whole-genome Hi-C heatmap of EFS v2.0 within and between 27 chromosomes.

The identification of syntenic regions for EFS v2.0, Rambouillet sheep and Tibetan sheep was based on conducting homology searches using MCScan (Python version)63, with a minimum requirement of 30 genes per block. Macrosynteny connecting blocks of >30 one-to-one gene pairs are shown.

BUSCO plot of the several sheep genomes. C: Complete BUSCOs; S: Complete and single-copy BUSCOs; D: Complete and duplicated BUSCOs; F: Fragmented BUSCOs; M: Missing BUSCOs; n: Total BUSCO groups searched. East Friesian sheepa: GCA_018804185.1; East Friesian sheepb: EFS v2.0.
Code availability
No specific code was developed for this study. The data analyses were conducted following the manuals and protocols provided by the developers of the relevant bioinformatics tools, which are described in the Methods section along with the versions used.
References
Mohamadipoor Saadatabadi, L. et al. Signature selection analysis reveals candidate genes associated with production traits in Iranian sheep breeds. BMC Veterinary Research 17 (2021).
Li, X. et al. Whole genome re-sequencing reveals artificial and natural selection for milk traits in East Friesian sheep. Frontiers in veterinary science 9, 1034211 (2022).
Kominakis, A., Hager-Theodorides, A. L., Saridaki, A., Antonakos, G. & Tsiamis, G. Genome-wide population structure and evolutionary history of the Frizarta dairy sheep. Animal: an international journal of animal bioscience 11, 1680–1688 (2017).
Nguyen, Q. V. et al. Supplementing grazing dairy ewes with plant-derived oil and rumen-protected EPA+DHA pellets enhances health-beneficial n-3 long-chain polyunsaturated fatty acids in sheep milk. European Journal of Lipid Science and Technology 120, 1700256 (2018).
Afolayan, R. A. et al. Genetic evaluation of crossbred lamb production. 3. Growth and carcass performance of second-cross lambs. Australian Journal of Agricultural Research 58, 5 (2007).
Li, R. et al. A sheep pangenome reveals the spectrum of structural variations and their effects on tail phenotypes. Genome Research 33, 3 (2023).
Li, X. et al. Genomic analyses of wild argali, domestic sheep, and their hybrids provide insights into chromosome evolution, phenotypic variation, and germplasm innovation. Genome Res 32, 9 (2022).
Davenport, K. M. et al. An improved ovine reference genome assembly to facilitate in-depth functional annotation of the sheep genome. Gigascience 11, giab096 (2022).
NCBI Genbank. https://identifiers.org/ncbi/insdc.gca:GCA_000298735.2 (2015).
Chin, C. S. et al. Phased diploid genome assembly with single-molecule real-time sequencing. Nature methods 13, 1050–1054 (2016).
Kolmogorov, M., Yuan, J., Lin, Y. & Pevzner, P. A. Assembly of long, error-prone reads using repeat graphs. Nature Biotechnology 37, 540–546 (2019).
Koren, S. et al. Canu: scalable and accurate long-read assembly via adaptive k -mer weighting and repeat separation. Genome research 27, 722–736 (2017).
Ruan, J. & Li, H. Fast and accurate long-read assembly with wtdbg2. Nature Methods 17, 1–4 (2020).
Hu, J. et al. An efficient error correction and accurate assembly tool for noisy long reads. (bioRxiv, 2023).
Cheng, H., Concepcion, G. T., Feng, X., Zhang, H. & Li, H. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nature Methods 18, 170–175 (2021).
Marx, V. Long road to long-read assembly. Nature methods 18, 125–129 (2021).
Chen, J. et al. A complete telomere-to-telomere assembly of the maize genome. Nature Genetics 55, 1221–1231 (2023).
Nurk, S. et al. The complete sequence of a human genome. Science (New York, N.Y.) 376, 44–53 (2021).
Su, R. et al. Draft Genome of the European Mouflon (Ovis orientalis musimon). Frontiers in genetics 11, 533611 (2020).
Bairoch, A. & Apweiler, R. The SWISS-PROT protein sequence data bank and its supplement TrEMBL in 1999. Nucleic Acids Research 27, 49–54 (1999).
Kanehisa, M. & Goto, S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Research 28, 27–30 (2000).
Yuan, Z. et al. Integrative analysis of Iso-Seq and RNA-seq data reveals transcriptome complexity and differentially expressed transcripts in sheep tail fat. PeerJ 9, e12454 (2021).
PacBio. (2021). Procedure & Checklist – Preparing HiFi SMRTbell® Libraries using the SMRTbell Express Template Prep Kit 2.0. Pacific Biosciences of California, Inc. https://www.pacb.com/wp-content/uploads/Procedure-Checklist-Preparing-HiFi-SMRTbell-Libraries-using-SMRTbell-Express-Template-Prep-Kit-2.0.pdf (Updated March 2019).
Chin, C. et al. Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nature Methods 10, 563–569 (2013).
Shafin, K. et al. Nanopore sequencing and the Shasta toolkit enable efficient de novo assembly of eleven human genomes. Nature Biotechnology 38, 1044–1053 (2020).
Yin, D. et al. Gapless genome assembly of East Asian finless porpoise. Scientific Data 9 (2022).
Chen, Y. et al. SOAPnuke: a MapReduce acceleration-supported software for integrated quality control and preprocessing of high-throughput sequencing data. GigaScience 7 (2017).
Durand, N. et al. Juicer Provides a One-Click System for Analyzing Loop-Resolution Hi-C Experiments. Cell Systems 3, 95–98 (2016).
Dudchenko, O. et al. De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science 356, eaal3327 (2017).
Bahcall, O. Sheep genome. Nature Genetics 46, 665–665 (2014).
Xu, G.C. et al. LR_Gapcloser: a tiling path-based gap closer that uses long reads to complete genome assembly. GigaScience 8 (2018).
Chen, Y. et al. Efficient assembly of nanopore reads via highly accurate and intact error correction. Nature Communications 12 (2021).
Chen, N. Using RepeatMasker to Identify Repetitive Elements in Genomic Sequences. Current Protocols in Bioinformatics 5 (2004).
Xu, Z. & Wang, H. LTR_FINDER: an efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Research 35, W265–W268 (2007).
Price, A. L., Jones, N. C. & Pevzner, P. A. De novo identification of repeat families in large genomes. Bioinformatics (Oxford, England) 21(Suppl 1), i351–8 (2005).
Bao, W., Kojima, K.K. & Kohany, O. Repbase Update, a database of repetitive elements in eukaryotic genomes. Mobile DNA 6 (2015).
Benson, G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic acids research 27, 573–80 (1999).
Liu, H. et al. Chromosome-scale genome assembly of sweet tea (Lithocarpus polystachyus Rehder). Scientific Data 10 (2023).
Zeng, T. et al. The telomere-to-telomere gap-free reference genome of wild blueberry (Vaccinium duclouxii) provides its high soluble sugar and anthocyanin accumulation. in Horticulture research 10, uhad209 (2023).
Li, B. et al. A gap-free reference genome reveals structural variations associated with flowering time in rapeseed (Brassica napus). Horticulture research 10, uhad171 (2023).
Lin, Y. et al. quarTeT: a telomere-to-telomere toolkit for gap-free genome assembly and centromeric repeat identification. Horticulture Research 10, uhad127 (2023).
Fang, X. et al. The sequence and analysis of a Chinese pig genome. GigaScience 1, 16 (2012).
Kim, D., Paggi, J. M., Park, C., Bennett, C. & Salzberg, S. L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nature Biotechnology 37, 907–915 (2019).
Wu, T. D. & Watanabe, C. K. GMAP: a genomic mapping and alignment program for mRNA and EST sequence. Bioinformatics 21, 1859–75 (2005).
Kovaka, S. et al. Transcriptome assembly from long-read RNA-seq alignments with StringTie2. Genome Biology 20, 278 (2019).
Jens et al. GeMoMa: Homology-Based Gene Prediction Utilizing Intron Position Conservation and RNA-seq Data. Methods in Molecular Biology 1962, 161–177 (2019).
NCBI Genbank. https://identifiers.org/ncbi/insdc.gca:GCA_000001405.29 (2022).
Haas, B. Improving the Arabidopsis genome annotation using maximal transcript alignment assemblies. Nucleic Acids Research 31, 5654–5666 (2003).
Kanehisa, M., Sato, Y., Kawashima, M., Furumichi, M. & Tanabe, M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Research 44, D457–D462 (2015).
Korf, I. Gene finding in novel genomes. BMC bioinformatics 5, 59 (2004).
Finn, R. D. et al. InterPro in 2017—beyond protein family and domain annotations. Nucleic Acids Research 45, D190–D199 (2016).
Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. Basic local alignment search tool. Journal of Molecular Biology 215, 403–410 (1990).
Mulder, N. J. & Apweiler, R. InterPro and InterProScan: tools for protein sequence classification and comparison. Methods in molecular biology 396, 59–70 (2007).
Ashburner, M. et al. Gene Ontology: tool for the unification of biology. Nature Genetics 25, 25–29 (2000).
Liao, Y., Smyth, G. K. & Shi, W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–30 (2013).
Goel, M., Sun, H., Jiao, W.-B. & Schneeberger, K. SyRI: finding genomic rearrangements and local sequence differences from whole-genome assemblies. Genome Biology 20 (2019).
Li, H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100 (2018).
Li, H. et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics (Oxford, England) 25, 2078–2079 (2009).
Rhie, A., Walenz, B. P., Koren, S. & Phillippy, A. M. Merqury: reference-free quality, completeness, and phasing assessment for genome assemblies. Genome Biology 21 (2020).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRP464403 (2023).
Liu, Y. Ovis aries breed East Friesian sheep isolate LY-2023, whole genome shotgun sequencing project. GenBank https://identifiers.org/ncbi/insdc.gca:GCA_033439445.1 (2023).
Liu, Y. B. Annotation Files of East Friesian sheep. figshare https://doi.org/10.6084/m9.figshare.24208689.v3 (2023).
Tang, H. et al. Synteny and Collinearity in Plant Genomes. Science 320, 486–488 (2008).
Acknowledgements
This work was funded by the Inner Mongolia Autonomous Region Open Competition Projects (2022JBGS0024), China Agriculture Research System (CARS-38) and National Key Research and Development Program Projects (2021YFD1300903).
Author information
Authors and Affiliations
Contributions
Y.F.D. and H.M.Z. conceived the study. Y.B.L. was responsible for the implemented the study. Q.Y.F., S.Y.W., X.L.H., J.F.H., B.W., L.W.W and R.T. collected the samples. Q.Y.F., J.W.C., S.Y.W., Z.W, X.K.Y., T.H.S. and J.B.J. processed the samples. X.X.Y., Q.Y.F. and C.H.C. processed bioinformatics data. X.X.Y., S.Y.F., T.Z. and Y.Z. performed bioinformatics analysis. X.X.Y. and C.H.C. wrote the manuscript with significant contributions. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
You, X., Fang, Q., Chen, C. et al. A near complete genome assembly of the East Friesian sheep genome. Sci Data 11, 762 (2024). https://doi.org/10.1038/s41597-024-03581-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-024-03581-w
- Springer Nature Limited