
Warburgia ugandensis and Saururus chinensis are two of the most important medicinal plants in magnoliids and are widely utilized in traditional Kenya and Chinese medicine, respectively. The absence of higher-quality reference genomes has hindered research on the medicinal compound biosynthesis mechanisms of these plants. We report the chromosome-level genome assemblies of W. ugandensis and S. chinensis, and generated 1.13 Gb and 0.53 Gb genomes from 74 and 27 scaffolds, respectively, using BGI-DIPSEQ, Nanopore, and Hi-C sequencing. The scaffold N50 lengths were 82.97 Mb and 48.53 Mb, and the assemblies were anchored to 14 and 11 chromosomes of W. ugandensis and S. chinensis, respectively. In total, 24,739 and 20,561 genes were annotated, and 98.5% and 98% of the BUSCO genes were fully represented, respectively. The chromosome-level genomes of W. ugandensis and S. chinensis will be valuable resources for understanding the genetics of these medicinal plants, studying the evolution of magnoliids and angiosperms and conserving plant genetic resources.
Similar content being viewed by others

Background & Summary
Among angiosperms, Amborellales, Nymphaeales and Austrobaileyales (collectively referred to as ANA grade) are followed by the rapid diversification of the remaining angiosperms or mesangiosperms1,2. The major mesangiosperm lineages are the eudicot, monocot and magnoliid clades3. Among the magnoliid clades, there are four orders, namely Canellales, Piperales, Magnoliales and Laurales. Long read sequencing has enabled chromosome-level assembly of many important plant genomes4,5,6. Although the number of sequenced magnoliid genomes has been increasing recently3,7,8,9,10,11,12,13,14, no Canellales genomes have been published. Similarly, only a few Piperales genomes have been published. In addition, there are many unanswered questions about the early diversification of mesangiosperm and the molecular mechanisms that have contributed to diversification and evolution within lineages15,16,17,18,19,20,21,22. Warburgia ugandensis is a medicinal plant belonging to Canellales, magnoliid.While, Saururus chinensis is a Piperales medicinal plant, athough a genome version of S. chinensis has been released23, deciphering their genomes will provide valuable and complementary genomic resources for future investigations into systematic evolution and medicinal components in the magnoliids.
W. ugandensis, a member of the Canellaceae, Canellales, is widely used for its pharmacological properties. The medicinal effectiveness of W. ugandensis is mainly associated with abundant terpenoids, particularly drimane and coloratane-type sesquiterpenoids, as well as fatty acid derivatives in the leaf and bark tissues24,25,26. The ever-increasing global demand for W. ugandensis and its use in treating and managing various diseases has led to overexploitation of this species, coupled with the difficulty of introducing it into temperate and cold regions, resulting in a drastic decrease in its population size15,27. W. ugandensis is therefore listed as a vulnerable species by the International Union for Conservation of Nature and Natural Resources (IUCN)28. Conservation strategies and rapid propagation techniques need to be implemented to protect this “miraculous species” from extinction. However, studies on the molecular characterization and biosynthesis of terpenoids and unsaturated fatty acids in W. ugandensis are relatively limited. Little is known about its genetic background, and its karyotype has not been previously reported. Therefore, genome sequencing is crucial for understanding the genetic background of this species, and can in turn lay a solid foundation for the development of its medicinal value and species conservation.
S. chinensis, with a chromosome number of 22 (2n = 2x = 22), is a perennial aquatic herb belonging to the Saururaceae, Piperales, and has been listed in the 2020 edition of Pharmacopoeia of the People’s Republic of China29. It is not only used as an ornamental aquatic plant but also has a long history of traditional medicinal use in China. It has significant analgesic, hypoglycemic, hepatoprotective, anti-angiogenesis, antioxidant, and anti-inflammatory30,31,32 properties. A comprehensive review of the taxonomic classification, morphology, and geographical distribution of Saururaceae plants revealed that Saururaceae is an early-branching family of Piperales and a stable component of ancient herbaceous plants33. However, despite the publication of a version of the S. chinensis genome by previous researchers, further research on the pharmacological components of S. chinensis requires a higher quality genome and additional transcriptome data from different tissues as a research foundation. Overall, genomics-based approaches for S. chinensis can provide valuable insights not only into the origin and early evolution of flowering plants (angiosperms), but also into how to directly and significantly modify one or more genes in the genomes of medicinal species, or to identify effective genetic markers and genes for molecular breeding23.
In the present study, we constructed high-quality genome assemblies for W. ugandensis and S. chinensis using the integration strategy of short reads (BGI-DIPSEQ sequencing), long reads (nanopore sequencing) and Hi-C reads (proximity ligation chromatin conformation capture). The final assembled genomes were 1.13 Gb and 533.01 Mb in length with scaffold N50 values of 82.97 Mb and 48.53 Mb for W. ugandensis and S. chinensis, respectively. A total of 1.12 Gb (99.49%) and 531.46 Mb (99.59%) of assembled genome sequences were successfully anchored on 14 and 11 chromosomes, respectively (Table 1). A total of 24,739 protein-coding genes were predicted for W. ugandensis, and 20,561 protein-coding genes for S. chinensis (Table 2). The percentage of functionally annotated genes in the W. ugandensis and S. chinensis accounted for as high as 99.94% and 99.93%, respectively (Table 2).
Methods
Sample preparation, DNA/RNA extraction, library construction and sequencing
Samples of W. ugandensis were collected from plants grown in the greenhouse of Wuhan Botanical Garden, and S. chinensis was collected from the lakeside of South China National Botanical Garden. For second-generation short-read library construction and sequencing, DNA was extracted using the CTAB (cetyltrimethylammonium bromide) method34 on fresh young leaves. The library was sequenced on the BGI-DIPSEQ platform35, generating ~149 Gb and ~137 Gb of 100 bp paired-end reads with an insert size of ~250 bp for W. ugandensis and S. chinensis, respectively. For subsequent analyses, such as genome size estimation and ONT assembly polishing, only high-quality reads were used.
For ONT library construction and sequencing36, after grinding fresh young leaf tissues of W. ugandensis and S. chinensis in liquid nitrogen, extraction was performed. With the LSK108 kit (SQK-LSK108, Oxford), we generated the library and performed sequencing on the Nanopore GridION X5 sequencer using five flow cells. The base calling was performed using Guppy (version 4.0.11) in the MinKNOW package. There were ~132 Gb (118x) and 65 Gb (123x) of raw data for W. ugandensis and S. chinensis, respectively, in total available for assembly (Table 1).
We collected fresh young leaves, mature leaves, stems close to the apical meristem, stems far from the apical meristem, rhizomes, root tissues, budding flowers, full-blooming flowers and flowers nearing the senescence stage of S. chinensis and the fresh young leaves of W. ugandensis for transcriptome sequencing, three biological replicates for each sample. Total RNA was extracted using the TIANGEN Kit with DNase I and then processed using the NEBNextUltraTM RNA Library Prep Kit to create a pair-end library with a 250 bp insert size. The RNA libraries were subsequently sequenced on the BGI-DIPSEQ platform. After the filtering of low-quality data by the Trimmomatic (version 0.39)37 with the parameters: ILLUMINACLIP:adapter.fa:2:30:10 LEADING:5 TRAILING:5, 6 Gb of 100 bp paired-end data for each tissue was used for later analysis.
Hi-C library construction and sequencing
The construction of Hi-C libraries was performed by utilizing the DpnII restriction enzyme and following the method developed by BGI QingDao Institute38. The chromatin digested with DpnII was labeled at the ends with biotin-14-dATP (Thermo Fisher Scientific, Waltham, MA, USA). Subsequently, the DNA was extracted, purified, and sheared using Covaris S2 (Covaris, Woburn, MA, USA). Hi-C libraries were subjected to sequencing on a BGI-DIPSEQ platform, generating ~290 Gb (258x) and ~100 Gb (187x) of data with 100 bp paired-end reads (Table 1). Hi-C data enabled the identification of 14 chromosomes for W. ugandensis and 11 for S. chinensis, which was consistent with the reported chromosome numbers of S. chinensis23.
Genome size, heterozygosity and ploidy level evaluation
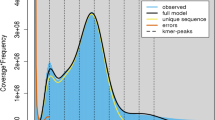
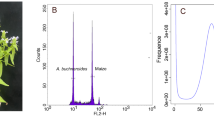
Two approaches were used to estimate the size of W. ugandensis genome: flow cytometry and k-mer spectral analysis of 60x BGI-DIPSEQ short reads. The flow cytometry technique was conducted with Liriodendron as the reference, generating 1.16 Gb of W. ugandensis (Fig. 1a). Additionally, we used the k-mer frequencies with the size of 17 to assess the genome size from short BGI-DIPSEQ reads. According to the results of 17-mer frequency distribution analysis with GenomeScope 239, a 1.16 Gb genome size of W. ugandensis was estimated (Fig. 1c and Table 1). While k-mer analysis estimated the S. chinensis genome size to be 555 Mb (Fig. 1d and Table 1), which is similar in size to 553 Mb obtained through flow cytometry analysis by Xue et al.23. To minimize the sequencing error rate, strict quality control was performed using SOAPfilter (version 2.2)40. To estimate the heterozygosity of the genomes, we used the Genome Analysis Toolkit (GATK) 4.2.3.0 for variant-calling of whole-genome short-read data, resulting in the heterozygosity values of 0.24% and 2.50% for W. ugandensis and S. chinensis, respectively. Given that the ploidy of W. ugandensis is unclear, we used Smudgeplot (https://github.com/KamilSJaron/smudgeplot) to estimate its ploidy and found that it may be diploid (Fig. 1b).

Genome sizes and ploidy levels estimated by flow cytometry experiment, smugeplot and survey analyses. (a) Flow cytometry experiment of W. ugandensis conducted with Liriodendron chinense as the reference (b) Ploidy assessment of W. ugandensis using a Smugeplot. (c,d) Genome survey based on k-mer (k = 17) analysis of W. ugandensis and S. chinensis, respectively.
Genome assembly and assessment of the assembly quality
The raw long reads obtained from ONT were used for de novo assembly using the NextDenovo assembler (version 2.2, https://github.com/Nextomics/NextDenovo) with the parameters: read_ cutoff = 1 k, seed_cutoff = 26,766 (W. ugandensis) and 16,118 (S. chinensis). The NextPolish (version 1.3.0, https://github.com/Nextomics/NextPolish)41 was subsequently applied to polish the initial draft assembled contigs with six rounds (two rounds with ONT long reads and four rounds with short reads). Purge dups (version 1.2.3)42 was then used to select contigs of S. chinensis to retain for the haploid assembly by taking into account mapped read coverage using short read and Minimap2 alignments43.
Hi-C paired-end reads were trimmed to remove low-quality bases and adapter sequences using Trimmomatic (version 0.39)37. To calculate the contact frequency, all the filtered reads were aligned to contig assembly using Juicer (https://github.com/aidenlab/juicer, version 3)44. Then, 3D-DNA (version 180922)45 pipeline was subsequently run with two iterative rounds for misjoining correction (-r2), and other parameters were set to the default values. Manual checking and refinement of the draft assembly were carried out with Juicebox assembly tools (version 1.11.08)46 (Fig. 2a,b).

Overview of the chromosomal features of two magnoliid genomes. (a,b) Hi-C interaction heatmaps of 14 chromosomes of W. ugandensis and 11 chromosomes of S. chinensis. (c,d) Circos plots of W. ugandensis and S. chinensis. The concentric circles from the outermost to the innermost regions show the chromosome and megabase values, (I) Pseudo-chromosomes, (II) gene density, (III) GC content, (IV) repeat density, (V) LTR density, (VI) LTR Copia density, (VII) LTR Gypsy density and (VIII) inter-chromosomal synteny (calculated in non-overlapping 200 Kb sliding windows).
The assembly evaluations for the genomes are provided as follows: First, mapping of the 1,614 conserved core eukaryotic genes from the BUSCO dataset (embryophyta_odb10, BUSCO v5)47, resulted in 98.5% and 98.0% (Table 1) of the core eukaryote genes recovered for the majority of the W. ugandensis and S. chinensis genome assemblies, respectively. We then mapped the RNA reads to the draft assemblies to evaluate the RNA reads mapping rate using Hisat248, with the mapping rate > 93%. Taken together, these results indicated good genome assembly qualities for this sequenced species.
In this study, a comparison of the genome of S. chinensis with a previously published genome revealed a similar genome size, but the current study identified a smaller number of gaps in the genome (132 gaps, size: 30,702 bp) compared to the previous version (804 gaps, size: 80,400 bp). Additionally, this study presented a lower number of contigs (75 contigs) with a higher contig N50 (14.96 Mb) compared to the previous version (842 contigs, Contig N50: 1.40 Mb). Moreover, the fragmented BUSCOs (F) and missing BUSCOs (M) in the genome were reduced in this study (BUSCO (F + M): 2.89) compared to the previous version (BUSCO (F + M): 5.76). The assembled genome of S. chinensis in our study is of high quality, potentially due to the large amount and deep depth of the short reads, long reads and Hi-C reads used by us. In addition, we used additional tissues and libraries for the transcriptome.
Repetitive element annotation
Repeat sequences in the genomes were identified using a combination of de novo and homology-based approaches. For de novo approaches, we used LTR_retriever49, LTR_FINDER (version 1.0.7)50, and RepeatModeler251 to construct a new repeat library and later RepeatMasker (version 4.0.6)52 was used to annotate the repeat elements. Finally, tandem repeats were searched across the genome using the software Tandem Repeats Finder (version 4.07)53. For homology-based approaches, repeat elements were predicted by employing a combination of homology-based comparisons in RepeatMasker (version 4.0.5) and RepeatProteinMask52. Both W. ugandensis and S. chinensis displayed moderate quantity repetitive elements, which accounted for 54.83% and 54.81% of assemblies, respectively (Fig. 2c,d and Table 2), while the percentage of Copia elements was 5.09% and 5.82%, respectively (Table 2).
Protein-coding gene prediction
The prediction of protein-coding gene sets was inferred using de novo gene prediction, homology-based annotation and evidence-based gene prediction. In the De novo approach, gene prediction was performed on a repeat-masked genome using Augustus (version 3.0.3)54, GlimmerHMM (version 3.0.1)55, and SNAP (version 11/29/2013)56. We analyzed the repeat-masked genomes of W. ugandensis and S. chinensis to predict coding genes, respectively. In the homology comparisons, homologous gene prediction was achieved by comparing the amino acid sequences of Amborella trichopoda, Arabidopsis thaliana, Oryza sativa, four related species (Aristolochia fimbriata, Chimonanthus salicifolius, Liriodendron chinense, and Litsea cubeba) using GeMoMa (version 1.3.1)57, and uniprot database (release 2021_04). By comparing with protein sequences covering the complete genome, TBLASTN (version 2.2.18) (e-value cutoff: 1e-5)58 was used to predict putative homologous genes. Then, GeneWise (version 2.2.0)59 was utilized to process the alignment regions and obtain precise exon and intron information. In the RNA-seq-based prediction approach, gene prediction was carried out by aligning the clean RNA-seq reads generated in this study against the assembled genomes using Hisat2 (version 2.0.4)48. cDNAs were identified through a genome-guided approach using StringTie (version 1.2.2)60 and then mapped back to the genome using PASA (version 2.3.3)61. The resulting cDNA sequences assembly from Trinity (version 2.6.6)62 were aligned to the W. ugandensis and S. Chinensis genome sequences using BLAT (version 34 × 12)63, respectively. After predicting genes using the aforementioned three methods, a non-redundant gene set was generated through BRAKER264 pipeline. A total of 24,739 and 20,561 protein-coding genes were predicted in W. ugandensis and S. chinensis, individually. As shown in Fig. 3, we can also observe that the mRNA length distribution, CDS length distribution, intron length, and exon number of W. ugandensis and S. chinensis in this study are similar to the distribution characteristics of other Magnoliid species genomes (Aristolochia fimbriata65, Cinnamomum kanehirae66, Aristolochia contorta67) published previously. However, the mRNA length distribution and CDS length distribution of the previously published S. chinensis genome are significantly higher than the percentage of other close related species in magnoliids, including the S. chinensis genome from this study. Moreover, the fragmented BUSCOs (F) and missing BUSCOs (M) in the gene set were reduced in this study (BUSCO (F + M): 4.77) compared to the previous version (BUSCO (F + M): 8.98). Our statistics estimated that we assembled a high-quality genome for S. chinensis (Table 1, Fig. 3).

Comparison of the distribution of gene elements for each gene among the six magnoliid species. (a) mRNA length. (b) CDS length. (c) Exon length. (d) Intron length. The x-axis represents the length and the y-axis represents the density of genes. Saururus Chinensis-p refers to the genome published previously.
Functional annotation
The protein-coding genes were subjected to functional annotation by performing sequences against similarity and domain conservation. Initially, a homolog search against public protein databases was conducted using BLASTP (e-value cutoff: 1e-5) to identify protein-coding genes with the following filtering criteria: -tophit 5, amino acid identify > 0.3, and match length cutoff > 0.5. The following public protein databases were used: SwissProt (release-2020_05)68, KEGG (59.3)69, TrEMBL (release-2020_05)68 and NCBI non-redundant protein NR database (20201015). Subsequently, InterProScan (version 5.28-67.0)70 was used to provide functional annotation by detecting and classifying domains and motifs. Finally, the annotation rates for W. ugandensis and S. chinensis were found to be 99.94% and 99.93% respectively (Table 3).
Phylogenetic analyses
The protein-coding genes of 17 of representative species combining the two Magnoliid genomes were selected for gene families analysis using OrthoFinder (version 2.3.14)71 with default parameters, among which A. trichopoda was set as an outgroup. Totally, 601 low-copy Orthogroups were used for phylogenetic tree construction. The protein sequences from the 601 low-copy Orthogroups were extracted and aligned by using MAFFT (version 7.310)72. The aligned sequences were concatenated into a super matrix and subsequently input into IQ-TREE (version 1.6.1)73 with “-bb2000-alrt 1000” to construct phylogenetic tree. The topology revealed a robust topology and supported sister relationship between magnoliids and Chloranthus (bootstrap support = 100), which together formed a sister group relationship (bootstrap support = 100) with eudicots and monocots (Fig. 4b). Magnoliid comprises four orders and no genome was available for any species of the Canellales order. What sets our research apart from previous angiosperm phylogenetic trees is that it is the first time we have combined the genomes of all four orders of Magnoliid with those of monocotyledonous plants, dicotyledonous plants, and outgroups to construct a comprehensive phylogenetic tree. This approach enhances the persuasiveness of our results.

Phylogenetic tree among 19 of representative species. A phylogenetic tree among 17 representative species, combining the two Magnoliid genomes in this study, was constructed using the maximum likelihood method.
Data Records
The Nanopore, Hi-C, BGI-DIPSEQ and RNA sequencing data that were used for the genome assembly and annotation have been deposited in the Genome Sequence Archive in National Genomics Data Center (NGDC) Genome Sequence Archive (GSA) database with the accession number CRA01416274 under the BioProject accession number PRJCA02241375. All the genomic sequencing raw data were also deposited in the CNGB Nucleotide Sequence Archive (CNSA) under accession numbers CNP000458676 and CNP000330977 for W. ugandensis and S. chinensis, respectively. The final contigs and chromosome assemblies are submitted to the NCBI under the accession number GCA_035236585.178, GCA_035235625.179 of W. ugandensis and S. chinensis, respectively. The contigs and chromosome-scale genome assemblies were also deposited in the Genome Sequence Archive80 in the National Genomics Data Center81 (CNCB/NGDC) under the BioProject accession PRJCA018454, with accession numbers GWHDQZE0000000082 and GWHDQZF0000000083 for W. ugandensis and S. chinensis, respectively. The annotation files are available in figshare84. All the other data generated or analyzed during this study are included in this article.
Technical Validation
Completeness assessment was performed using BUSCO (Bench-marking Universal Single-Copy Orthologs) version 3.0.147 with the Embryophyta odb10 database. Among the 1,614 core Embryophyta genes, 98.50% and 98.00% were identified in the W. ugandensis and S. chinensis, respectively (Table 1). To further evaluate the completeness of the assembled genome, we performed short-read mapping using clean raw data. In total, 99.64% and 96.33% of them were properly paired with W. ugandensis and S. chinensis, respectively. The transcriptome sequences were assembled by using Bridger tool85, then mapping to the scaffold assembly was performed by using the BLAT software63, 93.32% and 99.64% of them were paired with W. ugandensis and S. chinensis, respectively on average. The BUSCO analysis was again performed after the Hi-C assembly which gave similar results as those of the ONT genome assemblies.
Code availability
No specific script was used in this work. The codes and pipelines used in data processing were all executed according to the manual and protocols of the corresponding bioinformatic tools (Table S1). The software versions are described in the Methods section.
References
Moore, M. J., Bell, C. D., Soltis, P. S. & Soltis, D. E. Using plastid genome-scale data to resolve enigmatic relationships among basal angiosperms. Proceedings of the National Academy of Sciences 104, 19363–19368 (2007).
Qiu, Y. et al. The earliest angiosperms: evidence from mitochondrial, plastid and nuclear genomes. Nature 402, 404–407 (1999).
Hu, L. et al. The chromosome-scale reference genome of black pepper provides insight into piperine biosynthesis. Nature Communications 10, 4702 (2019).
Sahu, S. K. & Liu, H. Long-read sequencing (method of the year 2022): The way forward for plant omics research. Molecular Plant 16, 791–793 (2023).
Guo, X. et al. Chloranthus genome provides insights into the early diversification of angiosperms. Nature Communications 12, 6930 (2021).
Wang, S. et al. The chromosome-scale genomes of Dipterocarpus turbinatus and Hopea hainanensis (Dipterocarpaceae) provide insights into fragrant oleoresin biosynthesis and hardwood formation. Plant Biotechnology Journal 20, 538–553 (2022).
Chen, J. et al. Liriodendron genome sheds light on angiosperm phylogeny and species–pair differentiation. Nature Plants 5, 18–25 (2019).
Chaw, S. M. et al. Stout camphor tree genome fills gaps in understanding of flowering plant genome evolution. Nature Plants 5, 63–73 (2019).
Martha, R. A. et al. The avocado genome informs deep angiosperm phylogeny, highlights introgressive hybridization, and reveals pathogen-influenced gene space adaptation. Proceedings of the National Academy of Sciences 116, 17081–17089 (2019).
Chen, Y. et al. The Litsea genome and the evolution of the laurel family. Nature Communications 11, 1675 (2020).
Strijk, J. S. et al. Chromosome-level reference genome of the soursop (Annona muricata): A new resource for Magnoliid research and tropical pomology. Molecular Ecology Resources 21, 1608–1619 (2021).
Shang, J. et al. The chromosome-level wintersweet (Chimonanthus praecox) genome provides insights into floral scent biosynthesis and flowering in winter. Genome Biology 21, 200 (2020).
Lv, Q. et al. The Chimonanthus salicifolius genome provides insight into magnoliid evolution and flavonoid biosynthesis. The Plant Journal 103, 1910–1923 (2020).
Dong, S. et al. The genome of Magnolia biondii Pamp. provides insights into the evolution of Magnoliales and biosynthesis of terpenoids. Horticulture Research 8, 38 (2021).
WHO traditional medicine strategy 2002–2005. World Health Organization (2002).
Soltis, D. E. & Soltis, P. S. Nuclear genomes of two magnoliids. Nature Plants 5, 6–7 (2019).
James H, L. M. et al. One thousand plant transcriptomes and the phylogenomics of green plants. Nature 574, 679–685 (2019).
Wickett, N. J. et al. Phylotranscriptomic analysis of the origin and early diversification of land plants. Proceedings of the National Academy of Sciences 111, E4859–E4868 (2014).
Huang, C. H. et al. Resolution of brassicaceae phylogeny using nuclear genes uncovers nested radiations and supports convergent morphological evolution. Molecular Biology and Evolution 33, 394–412 (2016).
Yang, L. et al. Phylogenomic insights into deep phylogeny of angiosperms based on broad nuclear gene sampling. Plant Communications 1, 100027 (2020).
Li, H. et al. Origin of angiosperms and the puzzle of the Jurassic gap. Nature Plants 5, 461–470 (2019).
Yang, Y. et al. Prickly waterlily and rigid hornwort genomes shed light on early angiosperm evolution. Nature Plants 6, 215–222 (2020).
Xue, J. Y. et al. The Saururus chinensis genome provides insights into the evolution of pollination strategies and herbaceousness in magnoliids. The Plant Journal 113, 1021-1034 (2023).
Abuto, J. O. & Murono, D. A. Interaction effects of sites, samples, plant parts and solvent types on antimicrobial activity of the Kenyan populations of Warburgia ugandensis (Sprague). (2018).
Frum, Y., Viljoen, A. M., Drewes, S. E. & Houghton, P. J. In vitro 5-lipoxygenase and anti-oxidant activities of Warburgia salutaris and drimane sesquiterpenoids. South African Journal of Botany 71, 447–449 (2005).
Maobe, M. A. G. & Nyarango, R. M. Fourier transformer infra-red spectrophotometer analysis of Urtica dioica medicinal herb used for the treatment of diabetes, malaria and pneumonia in Kisii region, Southwest Kenya. (2013).
Denis, O., Richarh, K., Motlalepula, G. M. & Kang, Y. M. A review on the botanical aspects, phytochemical contents and pharmacological activities of Warburgia ugandensis. Journal of Medicinal Plants Research 12, 448–455 (2018).
Howard, G. et al. Warburgia ugandensis subsp. longifolia. The IUCN Red List of Threatened Species 2020: e.T32874A2826016. https://doi.org/10.2305/IUCN.UK.2020-2.RLTS.T32874A2826016.en (2020).
Pharmacopoeia of the People’s Republic of China. (National Pharmacopoeia Commission, 2020).
Kim, R. G. et al. Inhibition of methanol extract from the aerial parts of Saururus chinensis on lipopolysaccharide-induced nitric oxide and prostagladin E2 production from murine macrophage RAW 264.7 cells. Biological and Pharmaceutical Bulletin 26, 481–486 (2003).
Cho, H. Y., Cho, C. W. & Song, Y. S. Antioxidative and anti-inflammatory effects of Saururus chinensis methanol extract in RAW 264.7 macrophages. Journal of Medicinal Food 8, 190–197 (2005).
Yoo, H. J. et al. Anti-inflammatory, anti-angiogenic and anti-nociceptive activities of Saururus chinensis extract. Journal of Ethnopharmacology 120, 282–286 (2008).
Jiang, Q. & Lin, H. Karyotype analysis of Houttuynia cordata and Saururus chinensis effusus. Journal of Shenzhen University Science and Engineering 22, 349–353 (2005).
Sahu, S. K., Thangaraj, M. & Kathiresan, K. DNA extraction protocol for plants with high levels of secondary metabolites and polysaccharides without using liquid nitrogen and phenol. ISRN Molecular Biology 2012, 205049 (2012).
Huang, J. et al. A reference human genome dataset of the BGISEQ-500 sequencer. GigaScience 6, gix024 (2017).
Cherf, G. M. et al. Automated forward and reverse ratcheting of DNA in a nanopore at 5-Å precision. Nature Biotechnology 30, 344–348 (2012).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
Lieberman Aiden, E. et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 326, 289–293 (2009).
Ranallo-Benavidez, T. R., Jaron, K. S. & Schatz, M. C. GenomeScope 2.0 and Smudgeplot for reference-free profiling of polyploid genomes. Nature Communications 11, 1432 (2020).
Luo, R. et al. SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler. GigaScience 1, 2047-217X-1-18 (2012).
Hu, J., Fan, J., Sun, Z. & Liu, S. NextPolish: a fast and efficient genome polishing tool for long-read assembly. Bioinformatics 36, 2253–2255 (2020).
Guiglielmoni, N., Houtain, A., Derzelle, A., Van Doninck, K. & Flot, J. F. Overcoming uncollapsed haplotypes in long-read assemblies of non-model organisms. BMC Bioinformatics 22, 303 (2021).
Li, H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100 (2018).
Durand, N. C. et al. Juicer provides a one-click system for analyzing loop-resolution Hi-C experiments. Cell Systems 3, 95–98 (2016).
Dudchenko, O. et al. De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science 356, 92–95 (2017).
Durand, N. C. et al. Juicebox provides a visualization system for Hi-C contact maps with unlimited zoom. Cell Systems 3, 99–101 (2016).
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V. & Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212 (2015).
Kim, D., Langmead, B. & Salzberg, S. L. HISAT: a fast spliced aligner with low memory requirements. Nature Methods 12, 357–360 (2015).
Ou, S. & Jiang, N. L. T. R. _retriever: LTR_retriever: a highly accurate and sensitive program for identification of long terminal repeat retrotransposons. Plant Physiology 176, 1410–1422 (2018).
Xu, Z. & Wang, H. LTR_FINDER: an efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Research 35, W265–W268 (2007).
Flynn, J. M. et al. RepeatModeler2 for automated genomic discovery of transposable element families. Proceedings of the National Academy of Sciences 117, 9451–9457 (2020).
Maja, T. G. & Chen, N. S. Using RepeatMasker to identify repetitive elements in genomic sequences. Current Protocols in Bioinformatics 25, 4.10.1–4.10.14 (2009).
Benson, G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Research 27, 573–580 (1999).
Stanke, M., Steinkamp, R., Waack, S. & Morgenstern, B. AUGUSTUS: a web server for gene finding in eukaryotes. Nucleic Acids Research 32, W309–W312 (2004).
Majoros, W. H., Pertea, M. & Salzberg, S. L. TigrScan and GlimmerHMM: two open source ab initio eukaryotic gene-finders. Bioinformatics 20, 2878–2879 (2004).
Korf, I. Gene finding in novel genomes. BMC Bioinformatics 5, 59 (2004).
Keilwagen, J. et al. Using intron position conservation for homology-based gene prediction. Nucleic Acids Research 44, e89–e89 (2016).
Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. Basic local alignment search tool. Journal of Molecular Biology 215, 403–410 (1990).
Birney, E., Clamp, M. & Durbin, R. GeneWise and Genomewise. Genome research 14, 988–995 (2004).
Pertea, M. et al. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nature Biotechnology 33, 290–295 (2015).
Haas, B. J. et al. Improving the Arabidopsis genome annotation using maximal transcript alignment assemblies. Nucleic Acids Research 31, 5654–5666 (2003).
Grabherr, M. G. et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nature Biotechnology 29, 644–652 (2011).
Kent, W. J. BLAT–the BLAST-like alignment tool. Genome research 12, 656–664 (2002).
Brůna, T., Hoff, K. J., Lomsadze, A., Stanke, M. & Borodovsky, M. BRAKER2: automatic eukaryotic genome annotation with GeneMark-EP+ and AUGUSTUS supported by a protein database. NAR Genomics and Bioinformatics 3, lqaa108 (2021).
Qin, L. et al. Insights into angiosperm evolution, floral development and chemical biosynthesis from the Aristolochia fimbriata genome. Nature Plants 7, 1239–1253 (2021).
Xiong, B. et al. Genome of Lindera glauca provides insights into the evolution of biosynthesis genes for aromatic compounds. iScience 25, 104761 (2022).
Cui, X. et al. Chromosome-level genome assembly of Aristolochia contorta provides insights into the biosynthesis of benzylisoquinoline alkaloids and aristolochic acids. Horticulture Research 9, uhac005 (2022).
Boeckmann, B. et al. The SWISS-PROT protein knowledgebase and its supplement TrEMBL in 2003. Nucleic Acids Research 31, 365–370 (2003).
Kanehisa, M. & Goto, S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Research 28, 27–30 (2000).
Quevillon, E. et al. InterProScan: protein domains identifier. Nucleic Acids Research 33, W116–W120 (2005).
Emms, D. M. & Kelly, S. OrthoFinder: solving fundamental biases in whole genome comparisons dramatically improves orthogroup inference accuracy. Genome biology 16, 157 (2015).
Katoh, K., Kuma, K. I., Toh, H. & Miyata, T. MAFFT version 5: improvement in accuracy of multiple sequence alignment. Nucleic Acids Research 33, 511–518 (2005).
Minh, B. Q. et al. IQ-TREE 2: new models and efficient methods for phylogenetic inference in the genomic era. Molecular Biology and Evolution 37, 1530–1534 (2020).
NGDC Genome Sequence Archive https://ngdc.cncb.ac.cn/gsa/browse/CRA014162 (2024).
NGDC BioProject https://ngdc.cncb.ac.cn/bioproject/browse/PRJCA022413 (2024).
CNGB Nucleotide Sequence Archive https://db.cngb.org/search/project/CNP0004586/ (2023).
CNGB Nucleotide Sequence Archive https://db.cngb.org/search/project/CNP0003309/ (2023).
NCBI GenBank https://identifiers.org/ncbi/insdc.gca:GCA_035236585.1 (2023).
NCBI GenBank https://identifiers.org/ncbi/insdc.gca:GCA_035235625.1 (2023).
Chen, T. et al. The genome sequence archive family: toward explosive data growth and diverse data types. Genomics, Proteomics & Bioinformatics 19, 578–583 (2021).
Database resources of the national genomics data center, china national center for bioinformation in 2022. Nucleic Acids Research 50, D27–D38 (2022).
NGDC Genome Warehouse https://ngdc.cncb.ac.cn/gwh/Assembly/65976/show (2023).
NGDC Genome Warehouse https://ngdc.cncb.ac.cn/gwh/Assembly/65975/show (2023).
Fang, D. M. The genome and gene sequence ofWarburgiaandSaururus, Figshare, https://doi.org/10.6084/m9.figshare.23735505.v1 (2023).
Chang, Z. et al. Bridger: a new framework for de novo transcriptome assembly using RNA-seq data. Genome Biology 16, 30 (2015).
Acknowledgements
This research was supported by the funding from the National Natural Science Foundation of China (Grant No. 32000171) awarded to X. Guo. This work is also supported by China National GeneBank (CNGB; https://www.cngb.org/).
Author information
Authors and Affiliations
Contributions
X.G., H.L., G.H. and L.L. led and designed this project. L.L. and D.F. conceived the study. L.L. and S.K.S. wrote the manuscript. F.W., Q.L. and X.G., participated in data analyses, and generated the figures. Y.S. and J.K. conducted the HI-C Library. S.K.S., S.L., H.L. and G.H., revised and edited the manuscript. L.L., D.F. and M.L. assisted in uploading the scientific research data of this article to the designated website. All authors read and approved the final paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Luo, L., Fang, D., Wang, F. et al. The chromosome-level genomes of the herbal magnoliids Warburgia ugandensis and Saururus chinensis. Sci Data 11, 554 (2024). https://doi.org/10.1038/s41597-024-03229-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-024-03229-9
- Springer Nature Limited