
Abstract
The White-eared Night-Heron (Gorsachius magnificus, G. magnificus) is a critically endangered heron that is very poorly known and only found in southern China and northern Vietnam, with an estimated population of 250 to 999 mature individuals. However, the lack of a reference genome has hindered the implementation of conservation management efforts. In this study, we present the first high-quality chromosome-scale reference genome, which was assembled by integrating PacBio long-reads sequencing, Illumina paired-end sequencing, and Hi-C technology. The genome has a total length of 1.176 Gb, with a scaffold N50 of 84.77 Mb and a contig N50 of 18.46 Mb. Utilizing Hi-C data, we anchored 99.89% of the scaffold sequences onto 29 pairs of chromosomes. Additionally, we identified 18,062 protein-coding genes in the genome, with 95.00% of which were functionally annotated. Notably, BUSCO assessment confirmed the presence of 97.2% of highly conserved Aves genes within the genome. This chromosome-level genome assembly and annotation will be valuable for future investigating the G. magnificus’s evolutionary adaptation and conservation.
Similar content being viewed by others

Background & Summary
The White-eared Night-Heron G. magnificus is a nocturnal wader bird that is mainly distributed in southern China and northern Vietnam1,2. It belongs to the Ardeidae family and inhabits dense forests with abundant-watered areas, marshes, and reservoirs3. This species is listed as an Endangered species on the IUCN Red List4 with an estimated population of 250–999 mature individuals1 and is poorly known due to its rarity and few localities in the wild4,5. The increasing demands of humans for timber and agricultural land, intensive use of agricultural chemicals, and hunting are the main threats to its survival. It is urgent and challenging to evaluate the genetic status of G. magnificus due to conservation status and scattered localities2. Mitochondrial phylogenetic relationships suggest G. magnificus is not closely related to other members of Gorsachius but might be more closely related to heron species6. A recent phylogeny robust of herons based on ultraconserved elements revealed this species is a sister species of the African-distributed White-backed Night Heron (Gorsachius leuconotus)7. However the purpose of assembling a chromosome-level genome of an endangered night heron species has three advantages: 1) to be a high-quality reference genome for other population genomic studies in the family Ardeidae; 2) to allow comparative genomic studies of nocturnal birds to reveal local adaptation; 3) to carry out conservation genomics of this endangered species. Therefore, the availability of this genome facilitates tackling some challenges in evolution, conservation, and ecological studies8.
In this research, we have successfully generated a high-quality reference genome of G. magnificus at the chromosomal level, employing a comprehensive approach that integrates PacBio long-read sequencing, chromosome conformation capture (Hi-C) technology, and Illumina platform paired-end short-read sequencing. The assembled genome spanned a total length of 1.176 Gb, organized into 539 contigs and 79 scaffolds. The contig N50 length reached 18.46 Mb, while the scaffold N50 length was 84.77 Mb. Subsequently, 29 pairs of chromosomes with a total of length 1.175 Gb were anchored utilizing Hi-C technology, which corresponds to 99.89% of the assembled sequences. Moreover, we have identified 18,082 protein-coding genes based on de novo and homolog-based strategies, and 95% of these genes (17,177) were functionally annotated in publicly available databases including Gene Ontology, KEGG, and Pfam. Additionally, a BUSCO analysis demonstrated the completeness of 97.2% of annotated genes. This high-quality genome not only offers a reference genome for conservation genomics of G. magnificus but also facilitates phylogenomic and comparative genomic studies on a relatively understudied avian family, Ardeidae.
Methods
Ethics statement
Sample collection for scientific research purposes was in accordance with the ethical conditions in the Chinese Animal Welfare Act (20090606) and was approved by Forestry Administration of Guangdong Province, China (DFGP Project of Fauna of Guangdong-202115).
Sampling, DNA/RNA extraction, library construction, and sequencing
In our study, we gathered samples from a dead male G. magnificus specimen, which had resided at a wildlife rescue center in Shandong, Guangdong, China. In the genomic assembly of bird species, sampling female individuals offers an opportunity to obtain the W chromosome. However, we did not achieve this ideal condition for such an endangered and cryptic species. Genomic DNA was extracted from muscle tissue and utilized for whole genome sequencing and subsequent de novo assembly. Additionally, we obtained a total of nine RNA samples from various tissues within the same male individual, including the brain, lung, testis, thigh muscle, liver, pectoralis muscles, wing muscle, cardiac muscle, and eyeball, for RNA sequencing (RNA-seq) analysis.
We extracted high-molecular-weight genomic DNA from muscle samples following the instructions of CTAB (Cetyl trimethyl ammonium bromide), specifically for the purpose of de novo genome sequencing. We assessed the integrity and quality of the genomic DNA through agarose gel electrophoresis and a Qubit Fluorometer. For genome survey and polishing, we sequenced a single shotgun library with a 350-bp insert size on the Illumina NovaSeq 6000, yielding approximately 35.79 Gb (equivalent to a 30.43X coverage) of 150-bp paired-end reads (see Table 1). To facilitate genome assembly, SMRTbell libraries were created with an average 20-kb insert size using the SMRTbell Template Prep Kit 1.0 (Pacific Biosciences). Subsequently, we employed Blue Pippin (Labgene Scientific) to select fragment sizes, and we conducted library sequencing using the PacBio platform, utilizing single molecule real-time (SMRT) sequencing (PacBio RSII) technology, which generated a total of 128.52 Gb (equivalent to a 109.29X coverage) of data (see Table 1). For Hi-C sequencing, we fixed muscle tissue from the same male individual intended for de novo genome sequencing with 37% formaldehyde. Following a 10-minute incubation at room temperature, we halted the cross-linking reaction with 2.5% formaldehyde. We then collected the precipitated cells for Hi-C library preparation. A single Hi-C library was constructed and subjected to paired-end sequencing with 150 bp reads on the Illumina NovaSeq 6000 Sequencing System, resulting in a total of 110.6 Gb (94.05X coverage) of data (see Table 1). For RNA sequencing, we extracted total RNA from nine different tissues using the RiboPureTM RNA Purification Kit (Ambion®) and assessed its integrity with the RNA Nano 6000 Assay Kit on the Bioanalyzer 5400 system (Agilent Technologies, CA, USA). Following the manufacturer’s instructions, we constructed RNA libraries. All libraries were subjected to 150-bp paired-end sequencing on the Illumina NovaSeq 6000, and after adapter trimming and quality filtering using fastp (v 0.23.2)9 with default parameters, we obtained a total of 57.6 Gb of high-quality RNA sequencing data (see Table 2).
Genome assembly
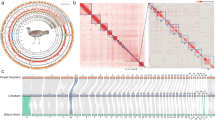
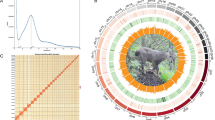
The chromosome-scale reference genome was assembled by combining PacBio long reads, Illumina short reads, and Hi-C sequencing data. Firstly, We obtained a genome size estimation of 1,181.27Mb and a heterozygosity rate of 0.397% by jellyfish (v 2.3.0)10 and GenomeScope (v 2.0.0)11 using Illumina short reads (Fig. 2). Then, we generated the initial draft contig assemblies based on high-coverage PacBio long reads data using wtdbg2 (v 2.5) and wtpoa-cns (v 1.1)12, the draft genome of the G. magnificus contained 539 contigs with a contig N50 of 18.46 Mb and a total length of 1.176 Gb (Table 3). Subsequently, PacBio long-read and additional Illumina paired-end short reads were applied to polish the draft genome following the wtdbg2 pipeline. To join contigs into scaffolds, 3D-DNA (v 190716)13 was used to produce the initial chromosomal results by aided by Hi-C data. These scaffolds were roughly reviewed and adjusted using Juicebox (v 1.11.08)14 and then further polishing the assembly using 3D-DNA. Finally, we obtained the chromosome-level assembly genome consisting of 79 scaffolds with an N50 of 84.77 Mb, and 99.89% genome was reordered and anchored onto 29 pairs of chromosomes (Table 3), with their lengths ranging from 60.39 kb to 218.43 Mb (Fig. 1, Table 4). The GC contents of the final assembled genome were 42.88% (Fig. 1, Table 3).

Characterization of assembled Gorsachius magnificus genome. From inner to outer layers for circle figure (right): Collinearity of different chromosomes, Distribution of SNPs, Genes abundance, Chromosomes, Density of GC content. (Left figure provided by Liao Zhikai).
To assess the quality of genome assembly and annotation, we used BUSCO (v 5) (Benchmarking Universal Single-Copy Orthologs)15 with aves_odb10 contains 8,338 single-copy orthologs as a reference to evaluate the completeness of the genome, the BUSCO research for assessment of the genome completeness showed that 96.6% of the BUSCO genes were complete, 0.7% were fragmented, and 2.7% were missing (Table 3).
Repeat sequences and genome annotation
To annotate the genome of the G. magnificus, we identified 122.4 Mb of repetitive sequences, accounting for 10.41% of the genome by a combination of homology-based and de novo-based identification, manual curation, and classification16 (Table 3). Repeatmasker (v 4.1.2)17 was used to search homology sequence from the Repbase library (v 20181026) for Aves, and de novo prediction was performed using Repeatmodeler (v 2.0.8)18, we furthermore used an additional method, EDTA (v 2.0.1)19, to annotate LTRs. This method combines the raw predictions of LTRharvest (v 1.1)20, LTR_FINDER_parallel (v 1.2)21, and LTR_retriever (v 2.9)22.
Next, we utilized three methods to predict protein-coding genes: transcriptome-based prediction, homology-based prediction, and de novo prediction. First, we assembled the transcriptome data from different tissues using Trinity (v 2.13.2)23. The homologous gene sets were obtained from the protein and transcript sequences of 15 proximate bird genomes (Table S1). Then, we performed Maker (v 3.01.03)24 for de novo prediction, and GeMoMa (v 1.7.1)25 for homology-based prediction to identify the protein-coding genes. Finally, we generated non-redundant gene sets from the three data sets. We also used GeMoMa to predict the UTR regions of protein-coding genes based on transcriptome sequences.
The functional annotation of protein-coding genes was constructed by mapping gene sets to protein databases Gene Ontology (GO)26 and Kyoto Encyclopedia of Genes and Genomes (KEGG)27 using eggNOG-mapper28.
We also used the combination of de novo, homolog-based, and transcript-based methods, 18,062 protein-coding genes (Genes) were predicted, and 97% of predicted regions with UTR region. A total of 17,177 (95% of Genes) were successfully annotated with at least one function term by searching against functional databases (Gene Ontology, KEGG, Pfam). we also used BUSCO (v 5) to evaluate the completeness of the annotation, The analysis for assessment of annotation completeness revealed a complete recall of 97.2% (83.9% single-copy; 13.3% duplicated) of genes, 0.9% fragmented, and 1.2% missing (Table 3).
Data Records
The Raw data of PacBio are deposited into NCBI SRA with accession number SRR26858085, and the Illumina WGS, Hi-C, and RNA-seq sequencing data were stored under accession numbers SRX22552595- SRX2255260729. The genome assembly has been deposited in the GeneBank database under the accession number JAXBDB00000000030. The genome annotations are available from the Figshare repository31.
Technical Validation
The assembled genome of has a size of 1.176 Gb, and its scaffold N50 is 84.77 Mb. This is very close to the estimated size of 1.181 Gb from our kmer-analysis (Fig. 2). The Hi-C heatmap displays a well-organized interaction pattern within the chromosomal regions (Fig. 3). Notably, 99.89% of the genome bases have been anchored onto 29 pairs of chromosomes (Figs. 1, 3, Table 4). The genome assembly quality assessment shows 96.6% completeness using BUSCO, with the protein-coding sequences achieving 97.2% completeness (Table 3).

K-mer frequency and genome size evaluation of Gorsachius magnificus genome.

Hi-C interactive heatmap of genome-wide of Gorsachius magnificus. The coordinates in the figure indicate genome length. The deeper red means a stronger interaction between the genomics regions.
We used the sequence alignment method to evaluate possible contamination and the completeness of the genome assembly. The Burrows-Wheeler Aligner (BWA, v 0.7.6)32 was used to map Illumina short reads to the assembled genome with default parameters. Importantly, the relationship between sequencing depth and GC content distribution revealed no deviations from the expected levels, allaying concerns about contamination or sequencing biases (Fig. 4). Additionally, our mapping results indicate that 99.03% of reads were successfully mapped, and the coverage rate was approximately 99.77% (Table 5), confirming the alignment consistency between the reads and the assembled genome. Compared to other assembled avian genomes, the main measures like the scaffolds N50 and the number of genes are very close to G. gallus and a recently published chromosome-level genome A. baeri) (Table 6).

The average sequencing depth and the distribution of GC content of Gorsachius magnificus. The specific GC content and sequencing depth were calculated in each window with a 1000bp size, corresponding to a dot in the figure. The Y-axis and left histogram represent the average sequencing depth, the peak value of the left histogram is about 25. The X-axis and top histogram distribution represent the GC content, the peak value of the top histogram is about 0.4.
Code availability
Genome assembly:
1. jellyfish: parameters: -m 19 -s 100M
2. GenomeScope: all parameters were set as default
3. wtdbg2: parameters: -x sq -g 1G -K 2000 –edge-min 4 -p 19 -S 4 -L 5000 –tidy-reads 8000
4. wtpoa-cns: all parameters were set as default
5. 3D-DNA: all parameters were set as default
6. Juicebox: all parameters were set as default
7. BUSCO: parameters: -l busco_downloads/lineages/aves_odb10
Genome annotation:
1. RepeatMasker: parameters: -poly -xsmall -engine ncbi -no_is
2. Repeatmodele: parameters: -engine ncbi
3. EDTA: parameters: –species others –step all –anno 1 -t 30 –rmout RepeatMasker.out
4. Trinity: parameters: –seqType fq –SS_lib_type RF –normalize_reads
5. Maker: all parameters were set as default
6. GeMoMa: GeMoMaPipeline parameters: GeMoMa.p = 20 GeMoMa.c = 0.3 AnnotationFinalizer.r = NO; AnnotationFinalizer parameters: u = YES;
7. eggNOG-mapper: all parameters were set as default
Whole genome alignment:
1. BWA: all parameters were set as default
The parameters not mentioned analysis modules in our study were used as default parameters.
References
Birds of The World. White-eared Night-Heron. https://birdsoftheworld.org/bow/species/wenher1/cur/introduction (2020).
BirdLife International. https://www.birdlife.org/ (2023).
Hu, J. & Liu, Y. Unveiling the conservation biogeography of a data-deficient endangered bird species under climate change. PLoS ONE 9, e84529, https://doi.org/10.1371/journal.pone.0084529 (2014).
Fellowes, J. R. et al. Status update on White-eared night heron Gorsachius magnificus in South China: Nycticorax magnifica Ogilvie-grant, 1899, Ibis (7) 5: 586. Bird Conserv. Int. 11, 101–111, https://doi.org/10.1017/s0959270901000193 (2001).
IUCN Red Data Book. The IUCN Red List of Threatened Species (2023).
Zhou, X., Yao, C., Lin, Q., Fang, W. & Chen, X. Complete mitochondrial genomes render the Night Heron genus Gorsachius non-monophyletic. J. Ornithol. 157, 505–513, https://doi.org/10.1007/s10336-015-1297-z (2016).
Hruska, J. P. et al. Ultraconserved elements resolve the phylogeny and corroborate patterns of molecular rate variation in herons (Aves: Ardeidae). Ornithology 140, ukad005, https://doi.org/10.1093/ornithology/ukad005 (2023).
Bock, D. G., Liu, J. Q., Novikova, P. & Rieseberg, L. H. Long-read sequencing in ecology and evolution: Understanding how complex genetic and epigenetic variants shape biodiversity. Mol. Ecol. 32, 1229–1235, https://doi.org/10.1111/mec.16884 (2023).
Chen, S., Zhou, Y., Chen, Y. & Gu, J. Fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890, https://doi.org/10.1101/274100 (2018).
Marçais, G. & Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27, 764–770, https://doi.org/10.1093/bioinformatics/btr011 (2011).
Ranallo-Benavidez, T. R., Jaron, K. S. & Schatz, M. C. Genomescope 2.0 and smudgeplot for reference-free profiling of polyploid genomes. Nat. Commun. 11, 1432, https://doi.org/10.1038/s41467-020-14998-3 (2020).
Ruan, J. & Li, H. Fast and accurate long-read assembly with wtdbg2. Nat. Methods 17, 155–158, https://doi.org/10.1038/s41592-019-0669-3 (2020).
Dudchenko, O. et al. De novo assembly of the Aedes aegyptigenome using Hi-C yields chromosome-length scaffolds. Science 356, 92–95, https://doi.org/10.1126/science.aal3327 (2017).
Durand, N. C. et al. Juicebox provides a visualization system for Hi-C contact maps with unlimited zoom. Cell Syst. 3, 99–101, https://doi.org/10.1016/j.cels.2015.07.012 (2016).
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V. & Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212, https://doi.org/10.1093/bioinformatics/btv351 (2015).
Platt, R. N., Blanco-Berdugo, L. & Ray, D. A. Accurate transposable element annotation is vital when analyzing new genome assemblies. Genome Biol. Evol. 8, 403–410, https://doi.org/10.1093/gbe/evw009 (2016).
Nishimura, D. RepeatMasker. Biotech Softw. & Internet Rep. 1, 36–39, https://doi.org/10.1089/152791600319259 (2000).
Flynn, J. M. et al. RepeatModeler2 for automated genomic discovery of transposable element families. Proc. Natl. Acad. Sci. 117, 9451–9457, https://doi.org/10.1101/856591 (2020).
Ou, S. et al. Benchmarking transposable element annotation methods for creation of a streamlined, comprehensive pipeline. Genome Biol. 20, 1–18, https://doi.org/10.1186/s13059-019-1905-y (2019).
Ellinghaus, D., Kurtz, S. & Willhoeft, U. Ltrharvest, an efficient and flexible software for de novo detection of ltr retrotransposons. BMC Bioinform. 9, 1–14, https://doi.org/10.1186/1471-2105-9-18 (2008).
Ou, S. & Jiang, N. Ltr_finder_parallel: parallelization of ltr_finder enabling rapid identification of long terminal repeat retrotransposons. Mob. DNA 10, 1–3, https://doi.org/10.1186/s13100-019-0193-0 (2019).
Ou, S. & Jiang, N. Ltr_retriever: a highly accurate and sensitive program for identification of long terminal repeat retrotransposons. Plant Physiol. 176, 1410–1422, https://doi.org/10.1104/pp.17.01310 (2018).
Grabherr, M. G. et al. Full-length transcriptome assembly from RNA-seq data without a reference genome. Nat. Biotechnol. 29, 644–652, https://doi.org/10.1038/nbt.1883 (2011).
Holt, C. & Yandell, M. MAKER2: an annotation pipeline and genome-database management tool for second-generation genome projects. BMC Bioinform. 12, 1–14, https://doi.org/10.1186/1471-2105-12-491 (2011).
Keilwagen, J., Hartung, F. & Grau, J. GeMoMa: homology-based gene prediction utilizing intron position conservation and RNA-seq data. Gene Predict. Methods Protoc. 161–177, https://doi.org/10.1007/978-1-4939-9173-0_9 (2019).
Consortium, G. O. The Gene Ontology (GO) database and informatics resource. Nucleic Acids Res. 32, D258–D261, https://doi.org/10.1093/nar/gkh036 (2004).
Kanehisa, M. & Goto, S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 28, 27–30, https://doi.org/10.1093/nar/27.1.29 (2000).
Cantalapiedra, C. P., Hernández-Plaza, A., Letunic, I., Bork, P. & Huerta-Cepas, J. eggNOG-mapper v2: functional annotation, orthology assignments, and domain prediction at the metagenomic scale. Mol. Biol. Evol. 38, 5825–5829, https://doi.org/10.1101/2021.06.03.446934 (2021).
NCBI Sequence Read Archive. https://identifiers.org/ncbi/insdc.sra:SRP472488 (2023).
Zhang, L. Aythya baeri isolate LZ_2022, whole genome shotgun sequencing project. GenBank https://www.ncbi.nlm.nih.gov/nuccore/JAKRSJ0000000000 (2023).
Zheng, C. Annotations of Gorsachius magnificus genome, Figshare, https://doi.org/10.6084/m9.figshare.24083526 (2023).
Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. ArXiv Prepr. arXiv:1303.3997 https://doi.org/10.48550/arXiv.1303.3997 (2013).
Luo, H. et al. Genomic insight into the nocturnal adaptation of the black-crowned night heron (Nycticorax nycticorax). BMC Genom. 23, 1–13, https://doi.org/10.1186/s12864-022-08904-y (2022).
NCBI RefSeq. https://www.ncbi.nlm.nih.gov/datasets/genome/GCF_016699485.2 (2021).
Zhang, L. et al. Chromosome-level genome assembly of the critically endangered baer’s pochard (Aythya baeri). Sci. Data 10, 176, https://doi.org/10.1038/s41597-023-02063-9 (2023).
Acknowledgements
This study is supported by Zhejiang Rare and Endangered Wildlife Rescue and Conservation Project (2017–2020, 2021–2025) and Zhejiang Provincial Natural Science Foundation of China under Grant No.LY13C040002 to Zhongyong Fan; Forestry Administration of Guangdong Province, China (DFGP Project of Fauna of Guangdong-202115 and Science and Technology Planning Projects of Guangdong Province-2021B1212110002) to Yang Liu, and the Interdisciplinary Innovation Team of the Chinese Academy of Sciences (CAS) “Light of West China” Program (xbzg-zdsys-202207). We appreciate the comments from the editor and two anonymous reviewers for their advice on previous versions of the manuscript.
Author information
Authors and Affiliations
Contributions
Yang Liu and Zhongyong Fan conceived and guided the idea of research. Zhongyong Fan, Shiguo Huang, and Yang Liu were assisted in funding. Aiwu Jiang, Weizhen Song, Chunsheng Xu, Xiran Qian, Yachang Cheng, and Hongzhou Lin collected samples. Yang Liu and Chenqing Zheng designed the study. Chenqing Zheng and Guoling Chen performed bioinformatic analyses. Chenqing Zheng, Qing Chen, and Guoling Chen wrote the original draft manuscript. All authors revised the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zheng, C., Chen, Q., Huang, S. et al. Chromosome-scale Genome assembly of the critically endangered White-eared Night-Heron (Gorsachius magnificus). Sci Data 11, 73 (2024). https://doi.org/10.1038/s41597-023-02894-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-023-02894-6
- Springer Nature Limited