
Abstract
Santalum album is a well-known aromatic and medicinal plant that is highly valued for the essential oil (EO) extracted from its heartwood. In this study, we present a high-quality chromosome-level genome assembly of S. album after integrating PacBio Sequel, Illumina HiSeq paired-end and high-throughput chromosome conformation capture sequencing technologies. The assembled genome size is 207.39 M with a contig N50 of 7.33 M and scaffold N50 size of 18.31 M. Compared with three previously published sandalwood genomes, the N50 length of the genome assembly was longer. In total, 94.26% of the assembly was assigned to 10 pseudo-chromosomes, and the anchor rate far exceeded that of a recently released value. BUSCO analysis yielded a completeness score of 94.91%. In addition, we predicted 23,283 protein-coding genes, 89.68% of which were functionally annotated. This high-quality genome will provide a foundation for sandalwood functional genomics studies, and also for elucidating the genetic basis of EO biosynthesis in S. album.
Similar content being viewed by others


Background & Summary
The Santalum genus (family: Santalaceae; order: Santalales), broadly known as sandalwood, contains 15 species that are typically slow-growing hemiparasitic trees whose distribution ranges widely throughout India, Australia and the Pacific Islands1. Several Santalum species contain highly-prized sesquiterpene-rich essential oil (EO) in their aromatic heartwood, including Santalum album (Indian sandalwood), S. yasi, S. spicatum, S. austrocaledonicum and S. insulare2. The economic importance of Indian sandalwood is derived from its EO, which contains a high content of α- and β-santalol, and these account for as much as 6–7% of the EO on a fresh weight basis3. Even though the broad genetic center of origin lies in tropical and temperate regions of India, Indian sandalwood (and to a lesser extent, other sandalwood species) is now extensively cultivated in South China, Sri Lanka, Indonesia, Malaysia, the Philippines and Northern Australia4. Indian sandalwood has held an important societal status in India since 1772 due to its aromatically fragrant EO5. The EO also has a long (~4,000 years) and acclaimed history in perfumery, medicine, religion, and other cultural applications6. The EO, or compounds within it, have displayed antioxidant, antitumour, antidepressive and anti-inflammatory activity7,8. Excessive demand of Indian sandalwood due to its commercial value resulted in the unsustainable exploitation of natural stands, and absent the establishment of new plantations, this led to a rapid increase in market prices9. Heartwood-derived EO was priced at more than $5,000/kg on the international market10.
As a result of the cultural and commercial importance of heartwood-derived EO, scientists have been eager to research its phytochemical characteristics. To date, most work has focused on S. album with the highest quality EO. Previous research identified over 100 terpenoids in S. album EO, about 80% of which consisted of (Z)-α-santalol and (Z)-β-santalol11. For at least a decade, attention has been paid to understanding the mechanisms associated with the biosynthetic pathway of the EO in S. album. Some key enzymes, including santalene/bergamotene synthase, SaCYP736A167 and SaCYP76Fs 37–43, have been characterized in S. album12,13,14. Others such as acetyl-CoA acetyltransferase, hydroxymethylglutaryl-CoA synthase, mevalonate kinase and phosphomevalonate kinase in the mevalonate pathway were also reported15,16. Two S. album genomes were generated based on Illumina short-read sequencing17,18. Recently, chromosome-level genome assemblies of S. album and S. yasi were documented19. However, knowledge of the genetic basis of EO biosynthesis in sandalwood trees is scarce.
A total of approximately 144.28 Gb of clean reads comprising 40.99 Gb of PacBio long reads, 37.84 Gb of Illumina reads and 65.45 Gb of Hi-C reads, were generated in this study (Table 1). A K-mer distribution analysis (K = 17) revealed that the estimated size of the S. album genome is 246.55 Mb, with a heterozygosity rate of 0.56% (Fig. S1; Table S1). A de novo assembly strategy combining PacBio long reads and Illumina paired-end reads resulted in an assembly of 207.39 Mb, with 7.33 Mb of N50 contigs (Fig. 1; Table 2; Table S2). High-quality Hi-C data were then used to further super-scaffold the genome assembly. Finally, a reference genome of S. album at the chromosome level was obtained by anchoring contigs 195.49 Mb in size into 10 chromosomes with lengths ranging between 12.54 Mb (Chr01) and 25.96 Mb (Chr02) (Figs. 2, 3; Table 3; Tables S3, S4). In terms of total length, the chromosomes accounted for 94.26% of the genome sequence, with a N50 scaffold of 18.31 Mb (Table 4), while the anchor rate was higher than that documented by Hong et al.19, with a mounting rate of 90.78% (Table 5). Mapped reads based on Illumina sequencing data to the assembled contigs accounted for as much as 96.83% of the total (Table S5). We assessed core gene statistics using Benchmarking Universal Single-Copy Orthologues (BUSCO)20 to verify the sensitivity of gene prediction and completeness. The result indicates that 94.91% of plant sets (1300 out of 1375 BUSCOs) were identified as complete (Table 6). The GC content was 37% (Table 2; Fig. S2). Collectively, these statistics and findings of the genome’s quality confirm that this chromosome-level genome assembly is complete and of high quality.

Flowchart of sequencing and assembly for the Santalum album genome.

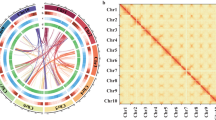
Genomic features of S. album. Circos plot from the outer to the inner layers represents the following: (a) Genomic landscape of the 10 assembled pseudo-chromosomes (Mb); (b) the GC density; (c) non-coding RNA; (d) transposable elements (TEs); (e) distribution of the density of genes and (f) syntenic blocks.

Heat map of genome-wide Hi-C intra-chromosome interactions in S. album. The image represents validation of the Hi-C-assisted pseudo-chromosome assembly by calculation of the thermal interaction correlation.
Transposable elements (TEs) are the main mechanistic drivers of genome evolution21. The Indian sandalwood genome harbored a total of 57.15 Mb of TEs, representing approximately 27.55% of the assembly (Tables S6–S8). Compared with four plant species (Malania oleifera, Vitis vinifera, Aquilaria sinensis and Oryza sativa) whose genomes were sequenced, sandalwood had the smallest genome and the lowest content of repetitive DNA (Fig. 4; Fig. S3; Table S9). Long terminal repeat (LTR) retrotransposons represented the greatest proportion of repeated content in these plants’ genomes, accounting for 16.75% of the S. album genome. Copia-LTR repeats dominate the sandalwood tree genome, contributing about 20.67 Mb (9.96%), and were 3.78-fold more abundant than Gypsy-LTR, accounting for 5.46 Mb (2.63%). In contrast, Gypsy-LTR was the most abundant repeat class in V. vinifera, A. sinensis and O. sativa and a nearly equal amount of Copia (29.51%) and Gypsy (28.15%) LTR-RTs were annotated in the M. oleifera genome.

Transposable elements in S. album. (a) Proportions of TEs among genomes of S. album, Malania oleifera, Vitis vinifera, Aquilaria sinensis and Oryza sativa. (b) Percentage of genome content comprising LTR elements for these five plant species.
After masking repeat sequences, we predicted the presence of 23,283 protein-coding genes by integrating de novo predictions, homology-based predictions, and transcriptomic data, with an average length of 3,812 bp, an average coding sequence length of 1,188 bp, and an average of 5.4 exons per gene in S. album (Fig. S4 and Tables S10, S11). About 89.68% of protein-coding genes had significant hits in several functional annotation databases (SwissProt, TrEMBL, InterPro, KEGG, Nr, COG and GO) (Fig. S5; Table S12). Among them, 1,368 genes encoding transcription factors (TFs) were predicted and classified into 58 gene families (Fig. S6). In addition, noncoding RNA (ncRNA) genes, including 65 microRNAs (miRNAs), 495 transfer RNA (tRNA), 585 ribosomal RNA (rRNA) and 257 small nuclear RNA (snRNA) genes, were identified in the genome (Table S13).
We compared the S. album assembly with sequenced genomes from 11 other plants, including M. oleifera, V. vinifera, Arabidopsis thaliana, Populus trichocarpa, A. sinensis, Cucumis sativus, Myrica rubra, Antirrhinum majus, Solanum lycopersicum, Lonicera japonica and O. sativa (Table S14). Based on gene family clustering analysis, 23,283 genes in S. album clustered into 12,430 gene families; 12,067 gene families were shared among all 12 plant species, and 344 families were unique to S. album (Fig. 5).

Comparative genomics analysis. (a) Statistics of gene families and all genes in S. album and other representative plant species. (b) Venn diagram shows the shared and unique gene families among Indian sandalwood and four other plant species.
In summary, this study presents a greatly improved sandalwood genome version, both in terms of completeness and accuracy, compared with two previously published S. album genomes that were obtained exclusively from short-reads17,18. In addition, not only is the contig N50 of our genome longer than that of a recently released genome19, the anchor rate of the genome assembly onto pseudo-chromosomes has also improved significantly. The chromosome-level S. album genome described in this paper provides a highly accurate and contiguous reference of genome sequences. Our study provides insight into the evolution of S. album, and presents a valuable genomic resource for further elucidating the genetic basis of EO biosynthesis in sandalwood trees.
Methods
Sample collection and DNA extraction
Fresh leaf tissues were collected from a 10-year-old tree grown at the South China Botanical Garden, CAS, Guangzhou, China in May, 2020 and immediately frozen in liquid nitrogen and stored at −80 °C. High-quality genomic DNA was extracted using a modified CTAB method22.
Genome sequencing and assembly
A paired-end library with insert lengths of 350 bp was constructed using the Illumina TruSeq library construction kit according to the manufacturer’s instructions. The constructed library was sequenced on the Illumina HiSeq X Ten platform (Illumina, San Diego, CA, USA) at the Beijing Genomics Institute (BGI). For PacBio sequencing, one library with a 20 kb insert size was constructed with the SMRTbell Template Prep Kit 1.0 according to the PacBio standard protocol. In brief, high-quality DNA was fragmented and concentrated. The fragments were purified by beads, damage was repaired, and resulting fragments were used as the 20 kb SMRTbell templates. PacBio long reads were sequenced for two SMRT cells on the PacBio Sequel System (Pacific Biosciences, CA, USA) at the BGI.
Errors in the PacBio SMRT sequences were initially corrected by Canu (v1.8)23 with the following parameters: corOutCoverage = 50, useGrid = true. Corrected reads were then assembled to primary contigs using SMARTdenovo (https://github.com/ruanjue/smartdenovo) with the following parameters: -c 1 -t 15 -J 5000 -k 17. The draft genome was polished using Arrow software (https://github.com/PacificBiosciences/Genomic-Consensus) based on corrected PacBio long reads with options parameters. To increase the accuracy of the assembly, Illumina short reads were recruited to correct the assembled contigs through the Pilon program (https://github.com/broadinstitute/pilon). The quality of genome assembly was assessed using BUSCO20.
Genome size estimation
The size of the S. album genome was estimated by using k-mer (k = 17) distribution analysis with Jellyfish24 using 350 bp Illumina pair-end reads. The Illumina reads were first trimmed to remove adaptors and reads with >10% ambiguous or >20% low-quality bases using the SOAPnuke package (v1.6.5)25 with parameters “-n 0.05 -l 15 -q 0.2 -Q 2 −5 1”. In this analysis, genome size = knum/kdepth, where knum is the total number of k-mers, and kdepth is the expected depth of k-mers. The size of the S. album genome was estimated as 246.55 Mb with the total number of 17-mers = ∼3.3 × 1010 and their main peak at a depth of 145.52, using GenomeScope26.
Hi-C scaffolding
About 5 g of fresh young leaf tissue from living plants was used to construct the Hi-C sequencing library. Samples were crosslinked using 37% formaldehyde to yield a 2% final concentration, mixed gently and incubated at room temperature (RT) for 10 min on plates that were gently rotated every 2 min. Then, 2.5 mL of 2.5 M glycine was added to quench the crosslinks, mixed well, incubated at RT for 5 min, then incubated on ice for 15 min to stop crosslinking completely. Cross-linked DNA was digested with MboI endonuclease overnight. The sticky ends of the digested fragments were biotinylated, diluted, and randomly ligated to each other to form chimeric junctions. Following ligation, a protease was used to remove the crosslinks and DNA was purified using the Qiagen MinElute PCR Purification Kit according to the manufacturer’s protocols. Finally, the Hi-C library with an insert size of 350 bp was constructed and sequenced on the BGISEQ-500 sequencer (BGI, Beijing China) with 2 × 150-bp reads at the BGI.
Hi-C raw reads were filtered using SOAPnuke (v1.6.5)25 with the following parameters (-n 0.05 -l 15 -q 0.2 -G -Q 2 −5 0) to obtain clean reads. Clean reads were first aligned to the contig-level sandalwood genome using Bowtie2 (v2.2.5)27 with the following parameters: “GLOBAL_OPTIONS = --very-sensitive -L 30 --score-min L, −0.6, −0.2–end-to-end –reorder” and “LOCAL_OPTIONS = --very-sensitive -L 20 --score-min L, −0.6, −0.2 --end-to-end --reorder”. The ratio of mapped reads to total reads reached 91.14% and 91.16%, respectively (Supplemental Table 1). The Hi-C sequence data obtained from global mapped reads were further qualified with HiC-Pro (v2.5.0)28, in which the unique mapped read pairs were selected. Final valid interaction pairs were obtained after removing duplicated pairs. The Hi-C data were then aligned and the misassembled reads were deleted by the Juicer pipeline29 to obtain unique reads. 3D de novo assembly (3D-DNA) software was applied to cluster, order and orient the filtered contigs onto chromosomes30. The completeness of the genome assembly was evaluated using BUSCO20. In addition, the paired-end Illumina short reads were mapped to the genome assembly using BWA-MEM (v0.7.12)31 to assess the integrity and accuracy of the genome.
Transcriptome sequencing
To facilitate gene model prediction, all tissues (leaves, flowers, fruits, heartwood and roots) were obtained from 10-year-old sandalwood trees for transcriptome sequencing. Leaves, flowers and fruits were harvested separately. Wood shavings of heartwood and root tissues were obtained using a Hagloff wood borer as described previously32. Total RNA was extracted followed an established method33. RNA (2 µg) from each sample was pooled to construct cDNA libraries, which were sequenced on BGISEQ-500 with PE100. Clean reads from all tissues were obtained by removing adaptor sequences and filtering low-quality reads with SOAPnuke25 with the following parameters: “-l 15 -q 0.5 -n 0.1”.
Genome model prediction and functional annotations
Repeat sequences in the S. album genome were annotated by integrating de novo and homology-based approaches. First, a de novo library from the assembled genome was constructed using LTR_Finder (v1.0.6)34 (http://tlife.fudan.edu.cn/ltr_finder/), Piler35 (http://www.drive5.com/piler/), and RepeatScout36 (http://www.repeatmasker.org/). Then, this de novo repeat database together with the Repbase TE library (http://www.girinst.org/repbase) were used to identify repeats by RepeatMasker (v4.0.7)37 and to identify repeat related proteins by RepeatProteinMask (v4.0.7) (http://www.repeatmasker.org/). We also annotated the non-interspersed repeat sequences, including low complexity repeats, satellites and simple repeats, with RepeatMasker (v4.0.7).
Protein-encoding gene models were predicted using ab initio, homology-based and RNA-seq-based pipelines based on the repeat-masked genome. Firstly, the protein sequences from grape (V. vinifera), Arabidopsis (A. thaliana), poplar (P. trichocarpa) and rice (O. sativa) were downloaded from the Plant Genome Database (PlantGDB; http://www.plantgdb.org/), then aligned to the sandalwood genome assembly using Blast with the following parameters: “blastall -F F -m 8 –p tblastn -e 1e-05 -a 5”. Secondly, GeneWise (v2.4.1)38 was used to align them against corresponding proteins to determine gene structures with the following parameters: “genewise -trev -sum -genesf -gff”. For the de novo prediction, Augustus39 and SNAP40 were applied. Furthermore, Genscan41 was used for de novo predictions with gene model parameters trained from Arabidopsis. Clean RNA-seq reads were aligned to the sandalwood genome with HISAT2 (v2.0.4) with the parameters “--phred64 --sensitive --no-discordant–no-mixed -I 1 -X 1000 –dta”42 and transcripts were predicted by StringTie (v1.0.4)43 with parameters “-f 0.3 -j 3 -c 5 -g 100 -s 10000 -p 8”. Finally, gene models from ab initio, homology-based, and transcriptome-based predictions were merged by EVidenceModeler software (v1.1.1)44. To validate the quality of the gene predictions, we compared the length distribution of protein-coding genes, coding sequences, exons and introns between sandalwood, grape, Arabidopsis and poplar. Completeness of the final gene set was assessed with BUSCO20. These genes were named according to the nomenclature used for Arabidopsis (Arabidopsis Genome Initiative, 2000) to indicate the relative positions of genes on the pseudo-chromosomes.
The functions of protein-coding genes were identified by BLASTP (v2.2.31) searches against SwissProt (http://www.uniprot.org/), GO (http://geneontology.org/page/go-database), KEGG (http://www.genome.jp/kegg/) and NR (http://ftp.ncbi.nlm.nih.gov/) databases. The predicted proteome was also assigned to TF families based on PlantTFDB (v5.0) (http://planttfdb.gao-lab.org/). S. album noncoding RNAs were annotated by tRNAscan-SE45 for tRNA, BLASTN for rRNA, and INFERNAL for miRNA and snRNA46.
In addition, we used OrthoFinder (v2.3.1)47 to identify gene families from S. album and 11 representative plant genomes, 10 eudicots including M. oleifera in the Santalales, V. vinifera, A. thaliana, P. trichocarpa, A. sinensis, C. sativus, M. rubra, A. majus, S. lycopersicum and L. japonica, and one monocot, O. sativa.
Data Records
The raw sequencing data have been deposited in the Genome Sequence Archive at the National Genomics Data Center (NGDC, https://ngdc.cncb.ac.cn/), Beijing Institute of Genomics, Chinese Academy of Sciences/China National Center for Bioinformation, under accession code CRA009778, including the PacBio reads48, Illumina short reads49, Hi-C Illumina reads50 and transcriptome reads51, which only these data were associated with this study. This Whole Genome Shotgun project has been deposited at DDBJ/ENA/GenBank under the accession JAXCHL00000000052. The version described in this paper is version JAXCHL010000000. The chromosome-level assembled genome sequences and annotation were deposited in the Figshare database53.
Technical Validation
To evaluate the completeness of the sandalwood assembly, we first mapped Illumina short-reads to the PacBio long read-based assembly to obtain 100% coverage. Then, BUSCO was employed to assess the assembly’s completeness. A total of 1,305 complete BUSCOs (94.91%) out of the 1,375 BUSCO groups were identified, including 1,261 complete and single-copy BUSCOs and 44 complete and duplicated BUSCOs, suggesting a remarkably complete assembly of the S. album genome. Moreover, we anchored the Hi-C data to the 10 pseudo-chromosomes, and then analyzed and visualized the Hi-C data. The paired-end Illumina short reads were mapped to the genome assembly to yield a 96.83% mapping rate. The signal intensities of interaction between the two bins were clearly divided into 10 distinct groups (Fig. 3), indicating the high-quality nature of the pseudo-chromosomes’ assembly. Finally, a list of chromosome ID conversions between assembled pseudo-chromosomes documented in this study and in a previous report19, has been compiled in Table S15.
Code availability
All bioinformatic tools used in this study followed the corresponding manuals and protocols. The versions and code/parameters of software are described in the Methods. Default parameters were employed if no detailed parameters were mentioned for the software used in this study.
References
Harbaugh, D. T. & Baldwin, B. G. Phylogeny and biogeography of the sandalwoods (Santalum, Santalaceae) repeated dispersals throughout the Pacific. Am. J. Bot. 94, 1028–1040 (2007).
Moniodis, J. et al. The transcriptome of sesquiterpenoid biosynthesis in heartwood xylem of Western Australian sandalwood (Santalum spicatum). Phytochemistry 113, 79–86 (2015).
Zhang, X. H., Teixeira da Silva, J. A., Yan, J. & Ma, G. H. Essential oils composition from roots of Santalum album L. J. Essent. Oil Bear. Pl. 15, 1–6 (2012).
Teixeira da Silva, J. A. et al. Sandalwood: basic biology, tissue culture, and genetic transformation. Planta 243, 847–887 (2016).
Mahesh, H. B. & Gowda, M. In The Sandalwood Genome: Compendium of Plant Genomes (Gowda, M. et al. (eds.), 1–5 (Springer Nature Switzerland press, 2022).
Burdock, G. A. & Carabin, I. G. Safety assessment of sandalwood oil (Santalum album L.). Food Chem. Toxicol. 46, 421–432 (2008).
Kim, T. H. et al. Antifungal and ichthyotoxic sesquiterpenoids from Santalum album heartwood. Molecules 22, 1139 (2017).
Bommareddy, A. et al. Medicinal properties of alpha-santalol, a naturally occurring constituent of sandalwood oil: review. Nat. Prod. Res. 33, 527–543 (2019).
Kumar, A. N. A., Joshi, G. & Ram, H. Y. M. Sandalwood: history, uses, present status and the future. Curr. Sci. 103, 1408–1416 (2012).
Tropical Forestry Services (TPS). TFS Sandalwood Project 2015, Indian Sandalwood. Product Disclosure Statement. Tropical Forestry Services Ltd., 169 Broadway, Nedlands WA 6009, Australia (2015).
Baldovini, N., Delasalle, C. & Joulain, D. Phytochemistry of the heartwood from fragrant Santalum species: a review. Flavour Frag. J. 26, 7–26 (2011).
Jones, C. G. et al. Sandalwood fragrance biosynthesis involves sesquiterpene synthases of both the terpene synthase (TPS)-a and TPS-b subfamilies, including santalene synthases. J. Biol. Chem. 286, 17445–17454 (2011).
Diaz-Chavez, M. L. et al. Biosynthesis of sandalwood oil: Santalum album CYP76F cytochromes P450 produce santalols and bergamotol. PLoS One 8, e75053 (2013).
Celedon, J. M. et al. Heartwood-specific transcriptome and metabolite signatures of tropical sandalwood (Santalum album) reveal the final step of (Z)-santalol fragrance biosynthesis. Plant J. 86, 289–299 (2016).
Niu, M. Y. et al. Cloning and expression analysis of mevalonate kinase and phosphomevalonate kinase genes associated with the MVA pathway in Santalum album. Sci. Rep. 11, 16913 (2021).
Niu, M. Y. et al. Cloning, characterization, and functional analysis of acetyl-CoA C-acetyltransferase and 3-hydroxy-3-methylglutaryl-CoA synthase genes in Santalum album. Sci. Rep. 11, 1082 (2021).
Mahesh, H. B. et al. Multi-omics driven assembly and annotation of the sandalwood (Santalum album) genome. Plant Physiol. 176, 2772–2788 (2018).
Dasgupta, M. G., Ulaganathan, K., Dev, S. A. & Balakrishnan, S. Draft genome of Santalum album L. provides genomic resources for accelerated trait improvement. Tree Genet. Genomes 15, 34 (2019).
Hong, Z. et al. Chromosome-level genome assemblies from two sandalwood species provide insights into the evolution of the Santalales. Commun Biol 6, 587 (2023).
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V. & Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212 (2015).
Bennetzen, J. L. & Wang, H. The contributions of transposable elements to the structure, function, and evolution of plant genomes. Annu. Rev. Plant Biol. 65, 505–530 (2014).
Porebski, S., Bailey, L. G. & Baum, B. R. Modification of a CTAB DNA extraction protocol for plants containing high polysaccharide and polyphenol components. Plant Mol. Biol. Rep. 15, 8–15 (1997).
Koren, S. et al. Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 27, 722–736 (2017).
Marcais, G. & Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27, 764–770 (2011).
Chen, Y. X. et al. SOAPnuke: a MapReduce acceleration-supported software for integrated quality control and preprocessing of high-throughput sequencing data. Gigascience 7, 1–6 (2018).
Vurture, G. W. et al. GenomeScope: fast reference-free genome profiling from short reads. Bioinformatics 33, 2202–2204 (2017).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012).
Servant, N. et al. HiC-Pro: an optimized and flexible pipeline for Hi-C data processing. Genome Biol. 16, 259 (2015).
Durand, N. C. et al. Juicer provides a one-click system for analyzing loop-resolution Hi-C experiments. Cell Syst. 3, 95–98 (2016).
Dudchenko, O. et al. De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science 356, 92–95 (2017).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Zhang, X. H. et al. Identification and functional characterization of three new terpene synthase genes involved in chemical defense and abiotic stresses in Santalum album. BMC Plant Biol. 19, 115 (2019).
Kolosova, N. et al. Isolation of high-quality RNA from gymnosperm and angiosperm trees. Biotechniques 36, 821–824 (2004).
Xu, Z. & Wang, H. LTR_FINDER: an efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Res. 35, W265–W268 (2007).
Edgar, R. C. & Myers, E. W. PILER: identification and classification of genomic repeats. Bioinformatics 21(Suppl 1), i152–i158 (2005).
Price, A. L., Jones, N. C. & Pevzner, P. A. De novo identification of repeat families in large genomes. Bioinformatics 21(Suppl 1), i351–i358 (2005).
Smit, A. F. A., Hubley, R. & Green, P. RepeatMasker Open-3.0. 1996–2010. (2010).
Birney, E., Clamp, M. & Durbin, R. GeneWise and Genomewise. Genome Res. 14, 988–995 (2004).
Stanke, M. et al. AUGUSTUS: ab initio prediction of alternative transcripts. Nucleic Acids Res. 34, W435–W439 (2006).
Johnson, A. D. et al. SNAP: a web-based tool for identification and annotation of proxy SNPs using HapMap. Bioinformatics 24, 2938–2939 (2008).
Burge, C. & Karlin, S. Prediction of complete gene structures in human genomic DNA. J. Mol. Biol. 25, 78–94 (1997).
Kim, D., Langmead, B. & Salzberg, S. L. HISAT: a fast spliced aligner with low memory requirements. Nat. Methods 12, 357–360 (2015).
Pertea, M. et al. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 33, 290–295 (2015).
Haas, B. J. et al. Automated eukaryotic gene structure annotation using EVidenceModeler and the program to assemble spliced alignments. Genome Biol. 9, R7 (2008).
Lowe, T. M. & Eddy, S. R. tRNAscan-SE a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 25, 955–964 (1997).
Griffiths-Jones, S. et al. Rfam: annotating non-coding RNAs in complete genomes. Nucleic Acids Res. 33, D121–D124 (2005).
Emms, D. M. & Kelly, S. OrthoFinder: phylogenetic orthology inference for comparative genomics. Genome Biol. 20, 238 (2019).
NGDC Genome Sequence Archive https://ngdc.cncb.ac.cn/gsa/browse/CRA009778/CRX582846 (2023).
NGDC Genome Sequence Archive https://ngdc.cncb.ac.cn/gsa/browse/CRA009778/CRX582847 (2023).
NGDC Genome Sequence Archive https://ngdc.cncb.ac.cn/gsa/browse/CRA009778/CRX582848 (2023).
NGDC Genome Sequence Archive https://ngdc.cncb.ac.cn/gsa/browse/CRA009778/CRX582849 (2023).
Zhang, X. H. et al. Santalum album TX1, whole genome shotgun sequencing project. GenBank https://identifiers.org/ncbi/insdc.gca:GCA_034195605.1 (2023).
Zhang, X. H. et al. Improved chromosome-level genome assembly of Indian sandalwood (Santalum album). figshare https://doi.org/10.6084/m9.figshare.23694729.v1 (2023).
Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (32171841, 32371925 and 31870666), Funding by Science and Technology Projects in Guangzhou (E33309), National Key Research & Development Program of China (2021YFC3100400, 2022YFC3103700) and Guangdong Key Areas Biosafety Project (2022B1111040003).
Author information
Authors and Affiliations
Contributions
X.H.Z. and M.Z.L. conceived and designed the project. X.H.Z., M.Z.L. and X.H.C. performed the experiments and analyzed the data. Z.B., Y.L., Y.P.X., L.F., K.L.W. and S.J.Z. performed part of the work and provided technical assistance. X.H.Z. wrote the manuscript. S.G.J., R.J.W., H.R., J.A.T.S. and G.H.M. revised and edited the manuscript. J.A.T.S. provided scientific advice and guidance. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhang, X., Li, M., Bian, Z. et al. Improved chromosome-level genome assembly of Indian sandalwood (Santalum album). Sci Data 10, 921 (2023). https://doi.org/10.1038/s41597-023-02849-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-023-02849-x
- Springer Nature Limited