
Abstract
The fruit fly Zeugodacus tau (Diptera: Tephritidae) is a major pest of melons and other cucurbits in Southeast Asia. In this study, we used Illumina, Nanopore, and Hi-C sequencing technologies to assemble a reference genome of Z. tau at the chromosomal level. The assembled genome was 421.79 Mb and consisted of six chromosomes (one X-chromosome + five autosomes). The contig N50 was 4.23 Mb. We identified 20,922 protein-coding genes, of which 17,251 (82.45%) were functionally annotated. Additionally, we found 247 rRNAs, 435 tRNAs, 67 small nuclear RNAs, and 829 small RNAs in the genome. Repetitive elements accounted for 55.30 Mb (13.15%) of the genome. This high-quality genome assembly is valuable for evolutionary and genetic studies of Z. tau and its relative species.
Similar content being viewed by others


Background & Summary
The tau fruit fly Zeugodacus tau (Diptera: Tephritidae) is a polyphagous pest that has invaded many regions worldwide, causing serious agricultural losses1. This species was previously classified in the subgenus Zeugodacus of the genus Bactrocera. Recently, the subgenus Zeugodacus was elevated to the genus level2. Species of Zeugodacus are considered more harmful than those of Bactrocera due to their high adaptability and invasive ability3. Zeugodacus tau has been listed as a quarantine species in many regions and countries, including China, the United States, Indonesia, Pakistan, and Japan4,5. Currently, Z. tau is distributed in most regions of southern China. It is generally present in tropical and subtropical Asia, sub-equatorial Africa, Australia, the Solomon Islands, and the South Pacific region3,6. Field monitoring has shown that Z. tau continues to expand to the high-latitude areas. However, there is limited data on historical records, and this species’ origin and colonization history remain unknown. Genetic studies may help reveal the adaptation and predict the future dispersal of this species. Due to the lack of genome data, studies on the invasion and genetics of Z. tau have been limited to the mitochondrial level7. Obtaining genomic data for this worldwide invasive insect could aid in controlling the spread of this pest and provide information on other invasive species.
In this study, we assembled a chromosome-level genome of Z. tau using a combination of Nanopore long-read, Illumina short-read sequencing, and chromosome conformation capture (Hi-C) technologies. We then performed structural and functional annotation on the obtained genome, incorporating transcriptome data from all developmental stages of Z. tau. This high-quality reference genome of Z. tau serves as a valuable resource for understanding the genetics, ecology, and evolution of Z. tau and providing information on the environmental adaptability and invasion mechanism of Tephritidae pests.
Methods
Sample preparation and genomic DNA sequencing
Zeugodacus tau samples were collected from Guangxi, China. They were reared for approximately nine generations in the laboratory under the following conditions: temperature of 27 ± 1 °C, relative humidity of 65 ± 5%, and a photoperiod of 14 L:10D. For genome sequencing, one pupa with unknown sex was used for the Nanopore library and Illumina library. Genomic DNA was extracted using the CTAB method and purified using a Blood and Cell Culture DNA Midi Kit (QIAGEN, Germany). The purity of the extracted DNA was determined using 0.75% agarose gel electrophoresis, and the concentration was assessed using a Qubit 2.0 Fluorometer from Thermo Fisher Scientific, USA. An Illumina paired-end (PE) library was constructed with an insert size of approximately 350 bp using the TruSeq Nano DNA HT Sample Preparation Kit (Illumina, San Diego, California, USA). The library was sequenced on the Illumina NovaSeq 6000 platform to generate paired-end reads of 150 bp. A total of 24.18 Gb (57.33 × coverage) of clean data was generated (Table 1). A long-insert library was also constructed using the same genomic DNA but with the SQK-LSK108 1D Ligation Sequencing Kit (Oxford Nanopore Technologies, Kidlington, Oxford, UK). This library was sequenced on the Nanopore PromethION sequencer at GrandOmics. The sequencing resulted in 51.67 Gb (122.50 × coverage) of long-reads, with an N50 length of 22,320 bp and an average length of 14,781.67 bp (Table 1).
Hi-C library preparation and sequencing
Two pupae with unknown sex were used to create the Hi-C library to capture genome-wide chromatin interactions. Chromatin digestion was carried out using the restriction enzyme MboI. The Hi-C samples were then extracted through biotin labeling, flat-end ligation, and DNA purification. The Hi-C library was sequenced using the Illumina NovaSeq platform with paired-end 150-bp reads. A total of 110.05 Gb (260.90 × coverage) of clean data were generated (Table 1).
Transcriptome sequencing
For transcriptome sequencing, we collected three groups of samples. Each group consisted of five larvae, five pupae, five male and female adults, respectively, along with approximately 100 eggs. We extracted total RNA using the TRIzol reagent (Thermo Fisher Scientific, USA). Paired-end libraries were constructed using the VAHTSTM mRNA-seq V2 Library Prep Kit (Vazyme, Nanjing, China). The libraries were then sequenced on the Illumina NovaSeq 6000 platform with PE reads of 150 bp for subsequent genome annotation. A total of 84.20 Gb (199.72 × coverage) of clean data were generated (Table 1).
Estimation of genomic characteristics
The K-mer method was utilized to survey the genome features of Z. tau with the Illumina short reads. The k-mer count histogram was calculated from Illumina short reads using Jellyfish8 version 2.2.10 with the parameters: ‘count -m 21 -C -s 5 G’. Genome size, heterozygosity, and duplication rate were estimated using GenomeScope9 version 1.0. The 21-mer analysis estimated the genome size of Z. tau to be approximately 548.38 Mb, with a high degree of duplication (1.12%) and heterozygosity (0.97%) (Fig. 1).
Genome assembly
The Nanopore long reads were corrected and assembled into contigs using NextDenovo version 1.2.5 (https://github.com/Nextomics/NextDenovo) with parameters: ‘read_cutoff = 1k, genome_size = 400 m, pa_correction = 20, nextgraph_options = -a 1’. These contigs were then polished for three iterations using NextPolish version 1.4.010 with the parameters: ‘genome_size = auto, sgs_options = -max_depth 100 -bwa, rerun = 3’. Subsequently, polished sequences were assembled into a chromosomal level based on Hi-C reads using Juicer version 1.6 with default parameters and 3D-DNA (3D de novo assembly, version 180922) pipelines11,12,13 with a modified parameter of ‘--editor-repeat-coverage 10’. The scaffolds were ordered manually using Juicebox version 1.11.08 (https://github.com/aidenlab/Juicebox) to obtain the final chromosome assembly. Syntenic blocks between chromosomes of Z. tau and Drosophila melanogaster14 were detected using MCScan15 based on the genome assembly and annotation results

The estimated characteristics of Zeugodacus tau genome based on Illumina short-read data using 21-mers count histogram. Genome size was estimated to be 548.38 Mb, with a duplication rate of 1.12% and heterozygosity rate of 0.97%.
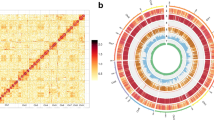
The Z. tau genome has a G + C content of approximately 35.54% (Table 2). At the contig level, we assembled the Z. tau genome into 424.74 Mb, consisting of 231 contigs. The contig N50 is 4.23 Mb (Table 2). These contigs were assembled into 421.79 Mb at the chromosomal level, with a scaffold N50 of 77.26 Mb. The chromosome level assembly includes six scaffold groups, with the longest group being 80.04 Mb and the shortest group being 10.74 Mb (Table 2, Fig. 2a). Synteny analysis reveals a highly conserved gene order with small-scale rearrangements and translocations between Z. tau and Drosophila melanogaster (Fig. 2b). The karyotype of Z. tau is 2n = 12, consisting of one pair of heteromorphic sex chromosomes (XX in females, XY in males) and five pairs of autosomes16,17. Zt_Chr3 is potentially the X chromosome, showing conserved synteny with the X chromosome of D. melanogaster18 (Fig. 2b).

Genome-wide all-by-all Hi-C interaction identified six pseudo-chromosome linkage groups of Zeugodacus tau (Zt) genome (a) and its synteny between Drosophila melanogaster (Dm) genome (b).
Repeat element and non-coding RNA annotation
Repetitive elements and transposable element families in the assembled genome were detected both by RepeatMasker version 4.0.719 against the Diptera repeats within RepBase Update (http://www.girinst.org) with parameters of ‘-e ABBlast, -species Diptera’. And ab initio predicted with the program RepeatModeler version open-1.0.8 (https://www.repeatmasker.org/RepeatModeler/). Most non-coding RNAs (ncRNA) were annotated by aligning the genomic sequence against RFAM (http://rfam.xfam.org/) with version 1.1.220 with the parameter of ‘-e ABBlast’. Transfer RNA (tRNA) was predicted by tRNAscanSE v.1.3.121 with default parameters, and ribosome RNA (rRNA) was predicted by RNAmmer-1.222 with parameters: ‘-S euk, -multi’.
In the Z. tau genome, a total of 55.30 Mb sequences (13.15%) were identified as repetitive elements (Table 3). We predicted 247 rRNAs, 435 tRNAs, 67 small nuclear RNAs, and 829 small RNAs in the Z. tau genome based on Rfam databases (Table 4).
Gene and functional predictions
Protein-coding genes were annotated using homolog-based, RNA-seq-based, and ab initio methods in the Maker genome annotation pipeline version 3.01.0423 with three iterations. The transcriptome of Z. tua was first assembled using StringTie version 1.3.3b24 and PASA version 2.0.225. based on the FASTA files of final chromosome assembly and using the transcriptome sequencing reads as input data with default parameters. Homologous genes of D. melanogaster14 and the transcripts were used to train ab initio predicting models for Augustus version 3.4.026 with default parameters and SNAP version 2006-07-2827 with the parameters of ‘-categorize 1000, -export 1000, -plus’. The results were used for the next round of model training and annotation. Three rounds of Maker annotations were conducted. The annotated genes were improved by PASA and then filtered based on gene expression evidence and functional annotation. Genes with fragments per kilobase per million (FPKM) value greater than 0 in any RNA-seq data were retained for further analysis. Functions of protein-coding genes, Gene Ontology (GO), and Kyoto Encyclopedia of Genes and Genomes (KEGG) items were annotated using the eggNOG-Mapper version 2.1.9 in the Expected eggNOG DB version 5.0.228 with the parameters of ‘--tax_scope auto, --go_evidence experimental, --target_orthologs all, --seed_ortholog_evalue 0.001, --seed_ortholog_score 60 --override’. A total of 20922 protein-coding genes were annotated in the chromosome-level assembly, in which 17251 genes (82.45%) were functionally annotated.
Data Records
The Z. tau genome project was deposited at NCBI under BioProject No. PRJNA84388129. Genomic Nanopore sequencing data were deposited in the Sequence Read Archive at NCBI under accession number SRR1953691830. Genomic Illumina sequencing data were deposited in the Sequence Read Archive at NCBI under accession SRR2610745231. Hi-C sequencing data were deposited in the Sequence Read Archive at NCBI under accession number SRR2610595232. RNA-seq data were deposited in the Sequence Read Archive at NCBI under accession number SRR26086842-SRR2608685633,34,35,36,37,38,39,40,41,42,43,44,45,46. The final chromosome assembly was deposited in GenBank at NCBI under accession number GCA_031772095.147. The genome annotation files are available in Figshare under a DOI number of https://doi.org/10.6084/m9.figshare.c.6843474.v248.
Technical Validation
We evaluated the accuracy of the final genome assembly by aligning Illumina short reads to the Z. tau genome using BWA-MEM version 0.7.1721 (https://github.com/lh3/bwa). The analysis revealed that 98.73% of the short reads were successfully mapped to the Z. tau genome.
To assess the completeness of the Z. tau genome, we conducted analysis using BUSCO version 5.2.249 with the insecta-odb10 database, which consists of 1,367 genes. The BUSCO analysis showed that for the contig level and chromosome level assemblies, 99.7% of the evaluated single-copy genes were identified as complete (single-copied gene: 99.1%, duplicated gene: 0.6%). Additionally, for all protein-coding genes and functionally annotated protein-coding genes, 97.6% (single-copied gene: 96.9%, duplicated gene: 0.7%) and 97.5% (single-copied gene: 96.8%, duplicated gene: 0.7%) were identified as complete, respectively (Table 5).
Usage Notes
All data analyses were conducted following the manual and protocols of the published bioinformatic tools. The version and parameters of the software have been described in the Methods section.
Code availability
No custom scripts or code were used in this study.
References
Noman, M. S., Shi, G., Liu, L. J. & Li, Z. H. Diversity of bacteria in different life stages and their impact on the development and reproduction of Zeugodacus tau (Diptera: Tephritidae). Insect Sci 28, 363–376, https://doi.org/10.1111/1744-7917.12768 (2021).
Virgilio, M., Jordaens, K., Verwimp, C., White, I. M. & De Meyer, M. Higher phylogeny of frugivorous flies (Diptera, Tephritidae, Dacini): localised partition conflicts and a novel generic classification. Mol Phylogenet Evol 85, 171–179, https://doi.org/10.1016/j.ympev.2015.01.007 (2015).
Singh, S. K., Kumar, D. & Ramamurthy, V. V. Biology of Bactrocera (Zeugodacus) tau (Walker) (Diptera: Tephritidae). Entomol. Res. 40, 259–263 (2010).
Ohno, S., Tamura, Y., Haraguchi, D. & Kohama, T. First detection of the pest fruit fly, Bactrocera tau (Diptera: Tephritidae), in the field in Japan: evidence of multiple invasions of Ishigaki Island and failure of colonization. Appl. Entomol. Zool. 43, 541–545, https://doi.org/10.1303/aez.2008.541 (2008).
Shi, W., Kerdelhue, C. & Ye, H. Genetic structure and colonization history of the fruit fly Bactrocera tau (Diptera: Tephritidae) in China and Southeast Asia. J Econ Entomol 107, 1256–1265, https://doi.org/10.1603/ec13266 (2014).
Liu, H., Wang, X., Chen, Z. & Lu, Y. Characterization of cold and heat tolerance of Bactrocera tau (Walker). Insects 13, 329, https://doi.org/10.3390/insects13040329 (2022).
Jaleel, W., Lu, L. H. & He, Y. R. Biology, taxonomy, and IPM strategies of Bactrocera tau Walker and complex species (Diptera; Tephritidae) in Asia: a comprehensive review. Environ Sci Pollut R 25, 19346–19361 (2018).
Marcais, G. & Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27, 764–770, https://doi.org/10.1093/bioinformatics/btr011 (2011).
Vurture, G. W. et al. GenomeScope: fast reference-free genome profiling from short reads. Bioinformatics 33, 2202–2204, https://doi.org/10.1093/bioinformatics/btx153 (2017).
Hu, J., Fan, J., Sun, Z. & Liu, S. NextPolish: a fast and efficient genome polishing tool for long-read assembly. Bioinformatics 36, 2253–2255, https://doi.org/10.1093/bioinformatics/btz891 (2020).
Li, H. & Durbin, R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26, 589–595, https://doi.org/10.1093/bioinformatics/btp698 (2010).
Dudchenko, O. et al. De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science 356, 92–95, https://doi.org/10.1126/science.aal3327 (2017).
Durand, N. C. et al. Juicer provides a one-click system for analyzing loop-resolution Hi-C experiments. Cell Syst 3, 95–98, https://doi.org/10.1016/j.cels.2016.07.002 (2016).
NCBI Genome, https://identifiers.org/ncbi/insdc.gca:GCA_000001215.4 (2014).
Wang, Y. et al. MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res 40, e49, https://doi.org/10.1093/nar/gkr1293 (2012).
Baimai, V., Phinchongsakuldit, J., Sumrandee, C. & Tigvattananont, S. Cytological evidence for a complex of species within the taxon Bactrocera tau (Diptera: Tephritidae) in Thailand. BIOL J LINN SOC 69, 399–409, https://doi.org/10.1111/j.1095-8312.2000.tb01213.x (2000).
Gouvi, G. et al. The chromosomes of Zeugodacus tau and Zeugodacus cucurbitae: a comparative analysis. Front. Ecol. Evol. 10, 1–12, https://doi.org/10.3389/fevo.2022.854723 (2022).
Chang, C. H. & Larracuente, A. M. Heterochromatin-enriched assemblies reveal the sequence and organization of the Drosophila melanogaster Y chromosome. Genetics 211, 333–348, https://doi.org/10.1534/genetics.118.301765 (2019).
Tarailo Graovac, M. & Chen, N. Using RepeatMasker to identify repetitive elements in genomic sequences. Curr Protoc Bioinformatics 4, 1–4, https://doi.org/10.1002/0471250953.bi0410s25 (2009).
Weisman, C. M., Murray, A. W. & Eddy, S. R. Mixing genome annotation methods in a comparative analysis inflates the apparent number of lineage-specific genes. Curr Biol 32, 2632–2639, https://doi.org/10.1016/j.cub.2022.04.085 (2022).
Lowe, T. M. & Eddy, S. R. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res 25, 955–964, https://doi.org/10.1093/nar/25.5.955 (1997).
Lagesen, K. et al. RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res 35, 3100–3108, https://doi.org/10.1093/nar/gkm160 (2007).
Cantarel, B. L. et al. MAKER: an easy-to-use annotation pipeline designed for emerging model organism genomes. Genome Res 18, 188–196, https://doi.org/10.1101/gr.6743907 (2008).
Pertea, M. et al. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat Biotechnol 33, 290–295, https://doi.org/10.1038/nbt.3122 (2015).
Pereira, C. G. Rajesh & Srivastava, Mani. PASA: a software architecture for building power aware embedded systems. IEEE Trans. Embed. Syst. 10(2), 123–135 (2002).
Stanke, M. & Waack, S. Gene prediction with a hidden Markov model and a new intron submodel. Bioinformatics 19, 215–225, https://doi.org/10.1093/bioinformatics/btg1080 (2003).
Korf, I. Gene finding in novel genomes. BMC Bioinformatics 5, 59, https://doi.org/10.1186/1471-2105-5-59 (2004).
Huerta, C. J. et al. Fast genome-wide functional annotation through orthology assignment by eggNOG-Mapper. Mol Biol Evol 34, 2115–2122, https://doi.org/10.1093/molbev/msx148 (2017).
NCBI Broproject, https://identifiers.org/ncbi/bioproject:PRJNA843881 (2022).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR19536918 (2022).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR26107452 (2023).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR26105952 (2023).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR26086848 (2023).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR26086856 (2023).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR26086850 (2023).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR26086855 (2023).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR26086852 (2023).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR26086846 (2023).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR26086853 (2023).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR26086844 (2023).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR26086854 (2023).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR26086843 (2023).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR26086845 (2023).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR26086849 (2023).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR26086851 (2023).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR26086847 (2023).
NCBI Genome https://identifiers.org/insdc.gca:GCA_031772095.1 (2023).
Wang, Y. T., Wei, S. J. & Zhou, Z. S. Chromosome-level genome assembly of an agricultural pest Zeugodacus tau (Diptera: Tephritidae). Figshare. Collection. https://doi.org/10.6084/m9.figshare.c.6843474.v2 (2023).
Manni, M., Berkeley, M. R., Seppey, M., Simão, F. A. & Zdobnov, E. M. BUSCO Update: Novel and Streamlined Workflows along with Broader and Deeper Phylogenetic Coverage for Scoring of Eukaryotic, Prokaryotic, and Viral Genomes. Mol. Biol. Evol. 38, 4647–4654, https://doi.org/10.1093/molbev/msab199 (2021).
Acknowledgements
This study was supported by the National Key Research and Development Program of China (No.2022YFC2601400), the National Nature Science Foundation (Grant No: 32102205), the Nanfan special project, CAAS (Grant No: ZDXM2312), and the Program of Beijing Academy of Agriculture and Forestry Sciences (Grant No: JKZX202208).
Author information
Authors and Affiliations
Contributions
S.W. and Z.S. designed the study and led the research. J.Y., X.G., H.C., Y.Z. and Z.T. contribute to the materials of this study. J.C., Y.W., W.S. and L.C. analyzed the data. Y.W., L.C., W.M. and W.S. contribute to the genome assembly and annotation. Y.W. and S.W. wrote the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wang, YT., Cao, LJ., Chen, JC. et al. Chromosome-level genome assembly of an agricultural pest Zeugodacus tau (Diptera: Tephritidae). Sci Data 10, 848 (2023). https://doi.org/10.1038/s41597-023-02765-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-023-02765-0
- Springer Nature Limited