
Abstract
Knoxia roxburghii is a well-known medicinal plant that is widely distributed in southern China and Southeast Asia. Its dried roots, known as hongdaji in traditional Chinese medicine, are used to treat a range of diseases, including cancers, carbuncles, and ascites. In this study, we report a de novo chromosome-level genome sequence for this diploid plant, which has a length of approximately 446.30 Mb with a contig N50 size of 42.26 Mb and scaffold N50 size of 44.38 Mb. Approximately 99.78% of the assembled sequences were anchored to 10 pseudochromosomes and 3 gapless assembled chromosomes were included in this assembly. A total of 24,507 genes were annotated, along with 68.92% of repetitive elements. Overall, our results will facilitate further active component biosynthesis for K. roxburghii and provide insights for future functional genomic studies and DNA-informed breeding.
Similar content being viewed by others
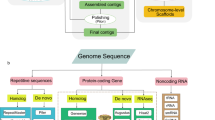

Background & Summary
Knoxia roxburghii (Sprengel) M. A. Rau (2n = 20, homotypic synonym: Knoxia valerianoides Thorel ex Pitard), a perennial herb naturally distributed in southern China and Southeast Asia, is a member of the Rubiaceae family and the Knoxia genus1. The dried roots of K. roxburghii, known as hongdaji in Chinese medicine, exhibit a significant therapeutic effect in treating cancer, carbuncles, diarrhoea, ascites, chronic pharyngitis, and schizophrenia2. Additionally, the plant is a crucial ingredient in various Chinese herbal formulations, such as ZiJinDing, which has been shown to possess antitumour properties by modern pharmacology3. Phytochemical studies have revealed that K. roxburghii is rich in anthraquinones, triterpenoids, lignans, coumarins, sitosterols, and other important compounds4,5. Anthraquinones, such as 3-hydroxymoridone, knoxiadin, and damnacanthal, are considered key active components of K. roxburghii, exhibiting diverse biological activities including anticancer, antibacterial, anticoagulant, and antiviral effects6,7. Triterpenoids, which are a significant component of K. roxburghii, have anti-inflammatory, anticancer, and antioxidant effects. They are primarily responsible for reducing inflammation and swelling in K. roxburghii8,9.
In recent years, the wild populations of K. roxburghii in China have been facing an increased risk of extinction due to a surge in market demand10. Additionally, seed germination and emergence rates for this species are less than 1% under natural conditions, and it exhibits a protracted maturation period11. K. roxburghii has been categorized as a first-class protected wild Chinese herbal medicine, and its production area has been prohibited from being utilized12. As a result, artificially cultivated K. roxburghii has become the primary source of medicinal materials. Nevertheless, the cultivation process is plagued by southern blight and leaf spot, which have severely limited the plant’s production13. Therefore, there is an urgent need for the breeding of promising new K. roxburghii varieties to tackle this issue.
Whole‐genome-level studies can provide insights for enhancing medicinal material quality, molecular breeding, wild resource conservation, and functional gene discovery and utilization of plants14,15,16. However, to date, no whole-genome sequence of K. roxburghii has been reported. In the present study, by using DNBSEQ sequencing, single-molecule real-time sequencing, and high-throughput chromosome conformation capture sequencing (Hi-C) sequencing technologies, we provide a de novo high-quality chromosome-level genome sequence for K. roxburghii. The 99.78% genome sequence is anchored to 10 chromosomes, with a total length of 446.30 Mb and scaffold N50 of 44.38 Mb. Transposable elements accounted for 68.92% (307.60 Mb) of the assembled genome sequence, with long terminal repeats (LTRs) being the dominant type. The LTR retrotransposon burst was estimated to have occurred approximately 0.2 million years ago. Phylogenetic analysis revealed that Copia and Gypsy elements could be grouped into eight and five lineages, respectively. The reference genome information obtained herein constitutes a valuable resource for promoting genetic improvement and elucidating the biosynthesis of active ingredients in this medicinal plant.
Methods
Sample collection and sequencing
For genomic DNA extraction, fresh leaves of K. roxburghii were collected from Chuxiong (N24°58′, E101°28′) in Yunnan Province, China. Additionally, stems, roots, buds, and leaves were gathered to perform transcriptome sequencing. The materials were immediately preserved in liquid nitrogen, transported to the laboratory, and stored at −80 °C. High-quality genomic DNA was extracted from leaves using the DNeasy Plant Mini Kit (QIAGEN, Valencia, California, USA). Total RNA was extracted from each sample using the Directzol RNA kit (Zymo Research, Irvine, CA, USA) following the manufacturer’s instructions.
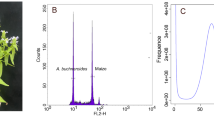
For short-reads sequencing, paired‐end DNBSEQ libraries were constructed using the NextEra DNA Flex Library Prep Kit (Illumina, San Diego, CA, USA) with an insert size of 350 bp and sequenced on the DNBSEQ-T7 platform (MGI Tech, Shenzhen, China). A quality assessment of the short sequencing reads was conducted using fastp v. 0.21.017 with default parameters. This process involved the removal of adapter sequences, contaminants, PCR duplicates, and reads with a low-quality base percentage exceeding 30%. A total of 107.86 Gb clean short reads (251.78 × coverage) were generated and used for subsequent data processing. The genome size was estimated to be 428.39 Mb, with a heterozygosity of 1.23% and repetitive content of 46.86% based on previous K-mer distribution analyses18.
For PacBio sequencing, the libraries were constructed with an insert size of 15 kb using the SMRTbell Template Prep Kits (Pacific Biosciences of California, Inc., CA, USA) and sequenced in CCS mode on the PacBio Sequel II platform (continuous long reads (CLR) sequencing mode). After trimming the low-quality reads and adaptor sequences from the raw data, approximately 52.85 Gb of long reads were generated, covering approximately 124 × of the estimated genome size.
For Hi-C sequencing, the library was prepared according to the protocol described by Lieberman-Aiden19 et al. DNA was purified from proteins and randomly sheared into fragments of 300–700 bp in size. The resulting Hi-C library was sequenced on the Illumina NovaSeq 6000 sequencing platform using paired-end 150 bp reads. The raw data from Hi-C sequencing were processed using fastp. A total of 36.14 Gb (84.36 × coverage) of clean reads were obtained.
For Oxford Nanopore Technologies (ONT) sequencing, all RNA samples of the same quantity were mixed for PCR-cDNA library construction using the Ligation Sequencing Kit (SQK-LSK109) and sequenced on the PromethION sequencer (Oxford Nanopore Technologies, Oxford, UK). NanoFilt v. 2.8.020 (parameters: –q 7 –l 100 –headcrop 30 –minGC 0.3) was used to process the RNA-seq data. Finally, a total of 6.2 Gb of full-length RNA-seq data were obtained for genome annotation.
Genome and chromosome assembly
The contig-level genome of K. roxburghii was assembled using Hifiasm v. 0.14.221 with default parameters. Two rounds of error correction were performed based on PacBio sequencing and Illumina NovaSeq sequencing data using NextPolish v. 1.3.122 (parameters: sgs_options = –max_depth 200 lgs_options = –min_read_len 1k –max_read_len 100k –max_depth 100 lgs_minimap2_options = –x map-ont) and Pilon v. 1.2323 (parameters:–fix all–changes), respectively. The heterozygous sequences were removed by using the Purge_haplotigs pipeline v. 1.0.424. The high, mid, and low cut-off read depth parameters were set to 170, 55, and 5, respectively, to remove haplotigs. Consequently, the genome assembly contained 446.30 Mb in 19 contigs with a contig N50 of 42.26 Mb, and the GC content of the genome was 35.98% (Table 1).
The Hi-C clean data were mapped to the draft genome using HiCUP v. 0.8.225 (parameters: –format sanger –longest 800 –shortest 150 –nofill N), followed by filtration to remove unmapped reads, invalid pairs, and PCR amplification-induced repetitive sequences. ALLHiC v. 0.9.826 (parameters: –e GATC –k 10) was utilized to cluster the contigs into chromosomal groups, with subsequent sorting and orientation. The interactions between contigs were converted into a specific binary file using 3D-DNA v. 18041927 and Juicer v. 1.628. Then, the visual correction of the assembly was finalized using JuiceBox v. 1.11.0829 based on the intensity of chromosome interaction. Additionally, very short contigs without any interaction relationships were placed in the “unassigned” category. The final chromosomal-level genomic sequence was obtained by using 100 N to fill the gaps. Finally, 99.78% of the initial assembled sequences were anchored to 10 pseudo-chromosomes with lengths ranging from 42.02 Mb to 48.32 Mb (Fig. 1a, Table 2). The total length of the genome assembly was 446.30 Mb, with a scaffold N50 of 44.38 Mb (Table 1).

Overview of the genomic features of Knoxia roxburghii. (a) Genomic features of K. roxburghii. Tracks from outside to inside (a–e) are as follows: chromosomes, gene density, repeat sequence density, GC content, and collinearity between the chromosomes; (b) Hi-C interaction heatmap for the K. roxburghii genome showing interactions among the ten chromosomes.
Genome annotations
Three gene prediction methods, namely de novo-based, RNA-seq-based, and homologue-based, were combined to identify gene structures. For de novo‐based prediction, gene prediction was performed using AUGUSTUS v. 3.2.330 and GlimmerHMM v. 3.0.431 with default parameters. In the RNA-seq-based approach, the full-length sequence underwent alignment to the reference genome using Minimap2 v. 2.1732 (parameters: –ax map-ont –xsplice –G 1000000). Subsequently, the alignment results were used as inputs in StringTie v. 1.3.333 for genome-based transcript assembly, and coding regions were then predicted using TransDecoder v. 2.0 (http://transdecoder.github.io). Homology‐based predictions were performed with protein sequences from five reference species: Arabidopsis thaliana34, Coffea arabica35, Coffea canephora36, Leptodermis oblonga37, and Mitragyna speciosa38. The results of the three methods were integrated using MAKER v. 2.31.1039. Overall, a total of 24,507 genes have been successfully predicted, with an average gene length, average coding-sequence length, average exon length, and average exon number per gene of 4036.6 bp, 1205.64 bp, 318.24 bp, and 5.14, respectively (Table 3).
Gene functions were assigned to the protein-coding gene models and compared to the National Center for Biotechnology Information (NCBI) Non-redundant protein (NR) (ftp://ftp.ncbi.nih.gov/pub/nrdb/), the Universal Protein Knowledgebase (UniProt) database40, and the Kyoto Encyclopedia of Genes and Genomes (KEGG) database41 using diamond v. 2.0.11.14942 (parameters: –evalue 1e-5). The motifs and domains were identified using InterProScan v. 5.52-86.043 against multiple publicly available databases including ProDom44, PRINTS45, Pfam46, SMRT47, PANTHER48, and PROSITE49. A total of 24,236 genes (94.85% of the predicted protein-coding genes) were annotated using the above databases. Specifically, approximately 90.88%, 91.06%, 25.34%, 92.88%, 70.87%, and 69.22% were annotated in UniProt, Nr, KEGG, InterPro, GO, and Pfam, respectively.
The identification of transfer RNAs (tRNAs) was performed using tRNAscan-SE v. 2.0.750. Other non-coding RNAs (ncRNAs), such as microRNAs (miRNAs) and small nuclear RNAs (snRNAs), were identified using Infernal v. 1.1.251 by searching against the Rfam database52. Lastly, the number of rRNAs, snRNAs, miRNAs, and tRNAs predicted from K. roxburghii genome were 1,053, 550, 81, and 387, respectively (Table 4).
Transposable elements and annotation of repeat sequences
Repetitive elements were identified through transposable element annotation using the Extensive de novo TE Annotator (EDTA) program v. 2.0.153 (parameters:–sensitive 1–anno 1). The insertion time was calculated using the LTR_retriever54 with default parameters. TEsorter v. 1.355 (parameters: -db rexdb) was used to classify the clade level of LTR-RTs and extract LTR-RT protein domains. MAFFT v. 7.47556 (parameters:–auto) was utilized to align LTR-RT sequences, and a phylogenetic tree was constructed using IQ‐TREE v. 2.2.2.657 (parameters: –bb 1000).
Based on the high-quality reference genome in this study, 307.60 Mb of repetitive sequences of K. roxburghii were predicted (Table 6). Among the integrated results, 33.56% (149.76 Mb) of the sequences were long terminal repeat (LTR) retrotransposons, with LTR/Copia elements being the dominant class (28.71% of the whole genome, 128.15 Mb), followed by LTR/Gypsy elements (2.79% of the whole genome, 12.47 Mb). To investigate the evolutionary history of transposable elements (TEs) in the K. roxburghii genome, a distribution plot of identity values between genomic copies and their consensus sequences was generated. The distributions of LTRs showed a peak at 89% identity, which was larger than the peaks of the other TE types, indicating that LTR-retrotransposons were recently transposed in the genome of K. roxburghii (Fig. 2a). Additionally, the genome contained 3,394 LTR-RTs, and the LTR retrotransposon burst was estimated to have occurred approximately 0.2 million years ago (Fig. 2b). For LTR/Gypsy and LTR/Copia, phylogenetic trees revealed that repeat elements were organized into different clades and expanded in clusters (Fig. 2c,d).

Repeat sequence analysis of the Knoxia roxburghii genome. (a) Distribution of sequence identity values between genomic copies and consensus repeats in the K. roxburghii genome. (b) Distribution of sequence identity values between genomic copies and consensus repeats in the K. roxburghii genome. (c) Phylogenetic tree of Ty1/Copia-type retrotransposons. (d) Phylogenetic tree of Ty3/Gypsy-type LTR retrotransposons.
Data Records
The BGI short reads, PacBio HiFi long-reads, Hi-C reads, and RNA-Seq data have been deposited in the NCBI Sequence Read Archive with accession numbers SRR2577737258, SRR2578793459, SRR2495841360, and SRR2577516761. The genome assembly has been deposited in DDBJ/ENA/GenBank under the accession number JAUECX00000000062. The chromosomal assembly and dataset of gene annotation have been deposited in the FigShare database at https://doi.org/10.6084/m9.figshare.2354256663.
Technical Validation
The integrity of the genome assembly was assessed using the sequence identity method. Reads from a small-fragment library were specifically selected and aligned to the assembled genome using BWA v. 0.7.17-r118864. The alignment rate of all small fragment reads to the genome was approximately 99.60%, and the coverage rate was approximately 99.49%, indicating consistency between the reads and the assembled genome.
We performed a Benchmarking Universal Single-Copy Orthology (BUSCO) v. 4.1.465 analysis based on the embryophyta_odb10 database to assess the completeness of the assembly, which indicated that 97.50% of the complete BUSCOs were present in the assembly (Table 5). Furthermore, 99.78% of the scaffolds were successfully anchored to the 10 chromosomes. The accuracy of the chromosome assembly was indirectly confirmed by examining the Hi-C heatmap, which revealed a well-organized interaction contact pattern along the diagonals within and around the chromosome region (Fig. 1b). This observation provides additional support for the precision of the chromosome assembly.
To validate the predicted genes, we performed a BUSCO analysis. The analysis revealed a high reliability of the annotated results, as approximately 98.40% of the complete BUSCOs were identified (Table 5). The annotation results were considered acceptable since the number of predicted genes and structural characteristics of the K. roxburghii genome were consistent with those of the genomes of closely related species.
Code availability
All software and pipelines were executed according to the manual and protocols of the published bioinformatics tools. The version and code/parameters of the software have been detailed and described in Methods. No custom code was used during the compilation of the dataset.
References
Wu, Z. Y., Raven, P. H. & Hong, D. Y. Flora of China (Science Press, 2011).
National Pharmacopoeia Commission of China. Pharmacopoeia of the People’s Republic of China (China Medical Science Press, 2020).
Zou, C. D. Knoxia valerianoides as the main syndrome differentiation prescription for the treatment of 40 cases of schizophrenia. Henan Tradit. Chin. Med. 31, 1429–1431 (2011).
Wu, C. J., Wang, J. L., Chen, J. P., Zhao, J. & Li, Y. Simutaneous determination of three anthraquinone components in Knoxia valerianoides by HPLC. China Pharm. 20, 1120–1122 (2017).
Zhao, F. et al. Anthraquinones from the roots of Knoxia valerianoides. China J. Chin. Mater. Med. 36, 2080–2086 (2011).
Hong, Y. L. et al. Anthraquinones and triterpenoids from roots of Knoxia roxburghii. China J. Chin. Mater. Med. 39, 4230–4233 (2014).
Chen, X. J. et al. 3-Hydroxymorindone from Knoxia roxburghii (Spreng.) M. A. Rau induces ROS-mediated mitochondrial dysfunction cervical cancer cells apoptosis via inhibition of PI3K/AKT/NF-κB signaling pathway. J. Funct. Foods 103, 105498 (2023).
Zhao, F. et al. Non-anthraquinone constituents from the roots of Knoxia valerianoides. China J. Chin. Mater. Med. 37, 2980–2986 (2011).
Chen, X. J. et al. Extracts of Knoxia roxburghii (Spreng.) M. A. Rau induce apoptosis in human MCF-7 breast cancer cells via mitochondrial pathways. Molecules 27, 6435 (2022).
Guo, Q. Y. et al. Preliminary study on introduction and cultivation of Knoxia valeriaides in mid-altitude area. Res. Pract. Chin. Med. 30, 8–11 (2016).
He, M. J., Hu, T. S., Huang, J. J. & Wei, X. J. Observation of ecological environment and biological characteristics of Knoxia valeriaides. Chin. Wild Plant Res. 2, 12–14 (1994).
Huang, J. L. Priority development of rare medicinal herb: Knoxia roxburghii. Technol. Mark. 03, 32–33 (2006).
Guo, Q. Y., Zhao, J. N. & Pu, H. T. Diagnosis and control techniques for the main diseases of the endangered Yi medicine red mahogany. Rural Pract. Technol. 11, 29–30 (2018).
Bohra, A., Chand, J. U., Godwin, I. D. & Kumar Varshney, R. Genomic interventions for sustainable agriculture. Plant Biotechnol. J. 18, 2388–2405 (2020).
Kersey, P. J. Plant genome sequences: past, present, future. Curr. Opin. Plant Biol. 48, 1–8 (2019).
Bock, D. G. et al. Genomics of plant speciation. Plant Commun. 4, 100599 (2023).
Chen, S., Zhou, Y., Chen, Y. & Gu, J. Fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890 (2018).
Pu, X. Y. et al. Estimation of genome size of Knoxia roxburghii by flow cytometry and genome survey. Mol. Plant Breed. 1, 1–13 (2023).
Lieberman-Aiden, E. et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 326, 289–93 (2009).
Wouter, D. C., Svenn, D., Darrin, T. S., Marc, C. & Christine, V. B. NanoPack: visualizing and processing long-read sequencing data. Bioinformatics 15, 2666–2669 (2018).
Cheng, H. Y., Concepcion, G. T., Feng, X. W., Zhang, H. W. & Li, H. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat. Methods 18, 170–175 (2021).
Hu, J., Fan, J., Sun, Z. & Liu, S. NextPolish: a fast and efficient genome polishing tool for long-read assembly. Bioinformatics 36, 2253–2255 (2020).
Walker, B. J. et al. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One 19, e112963 (2014).
Roach, M. J., Schmidt, S. A. & Borneman, A. R. Purge Haplotigs: allelic contig reassignment for third-gen diploid genome assemblies. BMC Bioinf. 19, 460 (2018).
Wingett, S. et al. HiCUP: pipeline for mapping and processing Hi-C data. F1000Research 4, 1310 (2015).
Zhang, X., Zhang, S., Zhao, Q., Ming, R. & Tang, H. Assembly of allele-aware, chromosomal-scale autopolyploid genomes based on Hi-C data. Nat. Plants 5, 833–845 (2019).
Dudchenko, O. et al. De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science 356, 92–95 (2017).
Durand, N. C. et al. Juicer provides a one-click system for analyzing loop-resolution Hi-C experiments. Cell Syst. 3, 95–98 (2016).
Durand, N. C. et al. Juicebox provides a visualization system for Hi-C contact maps with unlimited zoom. Cell Syst. 3, 99–101 (2016).
Stanke, M., Steinkamp, R., Waack, S. & Morgenstern, B. AUGUSTUS: a web server for gene finding in eukaryotes. Nucleic Acids Res. 32, W309–W312 (2004).
Majoros, W. H., Pertea, M. & Salzberg, S. L. TigrScan and GlimmerHMM: two open source ab initio eukaryotic gene-finders. Bioinformatics 20, 2878–2879 (2004).
Li, H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100 (2018).
Pertea, M. et al. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 33, 290–295 (2015).
Swarbreck, D. et al. The Arabidopsis information resource (TAIR): gene structure and function annotation. Nucleic Acids Res. 36, D1009–D1014 (2008).
Zimin, A. V. et al. Coffea arabica cultivar Caturra red isolate CCC135-36, whole genome shotgun sequencing project. GenBank https://identifiers.org/ncbi/insdc:RHJU00000000 (2018).
Denoeud, F., Wincker, P. & Lashermes, P. Coffea canephora strain DH200=94, whole genome shotgun sequencing project. GenBank https://identifiers.org/ncbi/insdc:CBUE000000000 (2015).
Guo, X., Wang, R. & Wang, Z. Leptodermis oblonga isolate Rui-Jiang Wang 3514, whole genome shotgun sequencing project. GenBank https://identifiers.org/ncbi/insdc:VMRK00000000 (2021).
Naktang, C. Mitragyna speciosa cultivar kratom01, whole genome shotgun sequencing project. GenBank https://identifiers.org/ncbi/insdc:JAMWEH000000000 (2022).
Holt, C. & Yandell, M. MAKER2: an annotation pipeline and genome-database management tool for second-generation genome projects. BMC Bioinf. 12, 1–14 (2011).
Apweiler, R. et al. UniProt: the universal protein knowledgebase. Nucleic Acids Res. 32, D115–D119 (2004).
Kanehisa, M. & Goto, S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30 (2000).
Buchfink, B., Xie, C. & Huson, D. H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 12, 59–60 (2015).
Jones, P. et al. InterProScan 5: genome-scale protein function classification. Bioinformatics 30, 1236–1240 (2014).
Bru, C. et al. The ProDom database of protein domain families: more emphasis on 3D. Nucleic Acids Res. 33, D212–D215 (2005).
Attwood, T. K. The PRINTS database: a resource for identification of protein families. Briefings Bioinf. 3, 252–263 (2002).
Finn, R. D. et al. Pfam: the protein families database. Nucleic Acids Res. 42, D222–D230 (2014).
Schultz, J., Copley, R. R., Doerks, T., Ponting, C. P. & Bork, P. SMART: A web-based tool for the study of genetically mobile domains. Nucleic Acids Res. 28, 231–234 (2000).
Mi, H. et al. The PANTHER database of protein families, subfamilies, functions and pathways. Nucleic Acids Res. 33, D284–D288 (2005).
Hulo, N. et al. The PROSITE database. Nucleic Acids Res. 34, D227–D230 (2006).
Chan, P. P., Lin, B. Y., Mak, A. J. & Lowe, T. M. tRNAscan-SE 2.0: improved detection and functional classification of transfer RNA genes. Nucleic Acids Res. 49, 9077–9096 (2021).
Nawrocki, E. P. & Eddy, S. R. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 29, 2933–2935 (2013).
Nawrocki, E. P. et al. Rfam 12.0: updates to the RNA families database. Nucleic Acids Res. 43, D130–D137 (2015).
Ou, S. J. et al. Benchmarking transposable element annotation methods for creation of a streamlined, comprehensive pipeline. Genome Biol. 20, 275 (2019).
Ou, S. J. & Jiang, N. LTR_retriever: a highly accurate and sensitive program for identification of long terminal repeat retrotransposons. Plant Physiol. 176, 1410–1422 (2018).
Zhang, R. G. et al. TEsorter: an accurate and fast method to classify LTR retrotransposons in plant genomes. Horticulture Res. 9, uhac017 (2022).
Katoh, K., Kuma, K., Toh, H. & Miyata, T. MAFFT version 5: improvement in accuracy of multiple sequence alignment. Nucleic Acids Res. 33, 511–518 (2005).
Minh, B. Q. et al. IQ‐TREE 2: New models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 37, 1530–1534 (2020).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR25777372 (2023).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR25787934 (2023).
NCBI Sequence Read Archive, https://identifiers.org/ncbi/insdc.sra:SRR24958413 (2023).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR25775167 (2023).
Zhang, Y. M. Knoxia roxburghii isolate Krox-001, whole genome shotgun sequencing project. GenBank https://identifiers.org/ncbi/insdc:JAUECX000000000 (2023).
Zhang, YM. Genome annotations ofKnoxia roxburghii, Figshare, https://doi.org/10.6084/m9.figshare.23542566 (2023).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V. & Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212 (2015).
Acknowledgements
This work was supported by the Major Science and Technology Project of Yunnan Province (202102AA310037, 202102AE090031), National Natural Science Foundation of China (82260739), the Yunnan Provincial Science and Technology Department – Applied Basic Research Joint Special Funds of Yunnan University of Traditional Chinese Medicine (202001AZ07000-015, 202101AZ070001-005).
Author information
Authors and Affiliations
Contributions
G.L. and B.Q. designed the study. Y.Z., F.Z., L.J. and Y.P. performed the experiments and analyzed the data. Y.Z., F.Z., L.J. and T.Z. wrote the paper. T.Z., G.L. and B.Q. revised the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhang, Y., Zhang, F., Jin, L. et al. A chromosome-level genome assembly of the Knoxia roxburghii (Rubiaceae). Sci Data 10, 803 (2023). https://doi.org/10.1038/s41597-023-02725-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-023-02725-8
- Springer Nature Limited