
Abstract
The melon fly, Zeugodacus cucurbitae (Coquillett), is an important destructive pest worldwide. Functional studies of the genes associated with development and reproduction during different life stages are limited in Z. cucurbitae. There have yet to be comprehensive transcriptomic resources for genetic and functional genomic studies to identify the molecular mechanisms related to its development and reproduction. In this study, we comprehensively sequenced the transcriptomes of four different developmental stages: egg, larva, pupa, and adults. Using the Illumina RNA-Seq technology, we constructed 52 libraries from 13 stages with four biological replicates in each and generated 435.61 Gb clean reads. We comprehensively characterized the transcriptomes with high-coverage mapping to the reference genome. A total of 13,760 genes were mapped to the reference genome, and another 4481 genes were characterized as new genes. Finally, 14,931 genes (81.85%) were functionally annotated against six annotation databases. This study provides the first comprehensive transcriptome data of all developmental stages of Z. cucurbitae, and will serve as a valuable resource for future genetic and functional studies.
Measurement(s) | RNA • sequence_assembly • sequence feature annotation • gene expression |
Technology Type(s) | RNA sequencing • sequence assembly process • sequence annotation • Expectation-Maximization(EM) algorithm |
Factor Type(s) | biological replicate • developmental stage • sex |
Sample Characteristic - Organism | Zeugodacus cucurbitae |
Machine-accessible metadata file describing the reported data: https://doi.org/10.6084/m9.figshare.11698254
Similar content being viewed by others


Background & Summary
The melon fly, Zeugodacus cucurbitae (Coquillett) (Diptera, Tephritidae), is an important pest of cucurbit crops1. Although this species is thought to be native to India, it is now widely distributed in tropical, sub-tropical and temperate regions worldwide, including the Asia-Pacific and Africa1,2. Z. cucurbitae attacks more than 130 plant hosts from 39 families, among which the bitter gourd, snap melon and muskmelon are the preferred hosts3. Because of its powerful invasive ability, Z. cucurbitae has been defined as a category A fruit fly, with polyphagous, highly destructive characteristics4. Hence, although the Z. cucurbitae genome is available5, few functional genomic and genetic studies have been carried out comparing it to other species of tephritidae fruit flies. Functional studies of the key genes underlying mechanisms associated with Z. cucurbitae development, behavior, and reproduction are limited. Gene expression during development, which are useful for various analyses, have yet to be characterized.
Transcriptome sequencing is an efficient and low-cost method by which to explore gene expression patterns at multiple developmental stages6,7, tissues in insects8,9. Many insect transcriptomes have been obtained using next-generation sequencing techniques for functional genomic studies10,11. For example, in Ceratitis capitata, a conserved Maleness-on-the-Y gene was isolated to be involved in the male sex determination by RNA-Seq.12. Comprehensive studies of gene expression during various developmental stages in many important insect pests, e.g. the oriental fruit fly Bactrocera dorsalis13,14 and the ladybird Henosepilachna vigintioctopunctata6, have been performed to explore the molecular mechanisms underlying metamorphic development. In C. capitata, the gene expression at the specific embryogenesis stage was also obtained by transcriptomic sequencing technology15. In addition, gene expression has been studied in the fat bodies, ovaries, and testes of B. dorsalis to identify critical genes involved in female and male gonad-specific roles8,16,17. Comparative transcriptomic analyses have also been used to characterize the differential expression of genes putatively associated with fecundity during the maturation of B. dorsalis and C. capitata adults18,19. Transcriptomes have also been sequenced to investigate the mechanisms of the insect response to environmental stressors, including insecticides20 and microbial infection21,22. Thus, transcriptome sequencing allows us to determine the gene expression mechanisms underlying certain biological functions and to identify potential targets associated with pest control.
Many transcriptomes have already been published and published in the National Center of Biotechnology Information (NCBI) Sequence Read Archive (SRA) database, which is the most popular repository of raw RNA-Seq data. For example, more than 1398 samples of 47 projects have been published and open accessible for another insect pest species in the Tephritidae pest, B. dorsalis. However, for Z. cucurbitae, only 23 samples from five projects have been released (before August, 2019), and four of the five projects were sequenced for genome annotation and provided a fundamental reference regarding protein-coding genes of Z. cucurbitae5,23, but no more direct gene expression was exhibited, and no more biological replicates were performed. The remaining project, from 2016, was sequenced only from middle-aged adults and focused on proteins associated with chemosensory perception24, in which a small number (13) of odorant binding proteins (OBPs) were identified. In homologous B. dorsalis fruit fly, 49 OBPs were identified and characterized25. There is considerable gene expression information left to collect. The fruit fly is a holometabolous insect, and its life cycle has four developmental stages. In addition to the differences gene expression across these stages, complex biochemical processes take place within the larval development, pupal development, and sex maturation during the adult stage. For example, in larval development, the juvenile hormone and ecdysone are the most important effectors, and they cooperate with other genes and pathways to regulate the molting process26,27. Comparative transcriptomes were performed within the adult stage to explore the candidate genes involved in fecundity of B. dorsalis18,19. Complex biochemical processes also take place during the pupa stage in which the tissues were remodeled, e.g. SP95 involved in integument remodeling28. Gene expression during the specific time strictly in the process of the molting from old larva to the early pupa was also critical to successful molting, e.g. the wondering stage14. Although four transcriptomes were released, they are not a good resource because of the lack of biological replicates. The genome genetic sequence information is of great help for gene expression annotation, e.g. C. capitata and Z. cucurbitae5,29. A global gene expression profile for Z. cucurbitae has yet to be produced for assessment of gene expression throughout development are therefore still inaccessible.
In this study, we performed the first deep transcriptomic sequencing of multiple different developmental stages of Z. cucurbitae by RNA-seq, including 13 stages, and 52 libraries (four biological replicates in each stage) (Fig. 1, Online-only Table 1). We generated a high-coverage dataset for genome-wide gene expression profiling. We successfully mapped 13,760 genes to the reference genome, and identified 4481 novel genes. In total, 14,931 genes (81.85%) were functionally annotated against the NCBI non- redundant (NR), Swiss-Prot, Pfam, Gene Ontology (GO), Cluster of Orthologous Group (COG), and Kyoto Encyclopedia of Genes and Genomes (KEGG) databases (Table 1). Four biological replicates of each stage provide a reliable evaluation of the gene expression during all the life stages. These data can also be used to analyze gene expression during metamorphosis to elucidate the molecular network, as well as the gene expression during the larval stages, pupal stage, and during the sexual maturation of adults. In addition, the comparative analysis of gene expression with other tephritidae fruit flies can also be performed, e.g. B. dorsalis, C. capitata, in which such data are available14,30. Our work represents a valuable resource, and will be helpful for future studies of functional genes associated with metamorphic development and reproduction.

Schematic work flow of the transcriptomes from the sample preparation to sequencing. (a) Sample collection of Zeugodacus cucurbitae, and the 13 stages analyzed in this study are shown as photos. (b) Schematic overview of the sample collection, sequencing and analysis.
Methods
Insect culture, sample collection
Z. cucurbitae were collected as pupae from Haikou, Hainan Province, China, in 2016, and were reared at 26–27 °C with 70 ± 5% relative humidity and a photoperiod of 14:10 h light: dark. Similar to B. dorsalis artificial diet31, Z. cucurbitae larvae were reared on an artificial diet consists of bitter gourd, corn, yeast powder, wheat flour, and sucrose; adults were fed a mixture of sucrose, yeast powder, and honey. Samples were collected at several different developmental stages: egg (~100 individuals < 8 h post-laying), 1st instar larva (100 individuals at 1-d-old), 2nd instar larva (20 individuals at 3-d-old), 3rd instar larva (10 individuals at 7-d-old), pupa (three individuals at 1-, 5-, and 9-d-old, representing the early-, middle-, and late-stage of pupae respectively), virgin female and male adults (three individuals at 1-, 5-, and 9-d-old) (Fig. 1a). As preliminary experiments showed that adults were sexually mature at 9-day-old, the time points 1-, 5-, and 9-d-old represented the newly-emerged, vitellogenic, and sex-matured stages, respectively. Four biological replicates of each stage were prepared.
Total RNA isolation
Total RNA was extracted from the 13 stages with TRIZol reagent (Invitrogen, Carlsbad, CA, USA), following the manufacturer’s instructions. The concentration and purity of total RNA samples were firstly quantified using a NanoDrop One (Thermo Fisher Scientific, Madison, WI, USA), and were re-determined using a Bioanalyser 2100 (Agilent Technologies, Palo Alto, CA, USA) before library construction.
Library construction and sequencing
The transcriptome libraries were constructed using the TruSeq RNA sample preparation kit (Illumina, San Diego, CA, USA), following the standard instructions. In brief, all the mRNA was isolated using the polyA selection method with oligo (dT) magnetic beads (Illumina), and then fragmented using fragmentation buffer. Double-stranded cDNA was synthesized using a SuperScript double-stranded cDNA synthesis kit (Invitrogen, Carlsbad, CA, USA) with buffer, dNTPs, random hexamer primers, DNA polymerase I, and RNase H. The synthesized cDNA was subjected to end-repair, phosphorylation, and ‘A’ base addition following the Illumina library construction protocol. Each library was size selected for cDNA target fragments (200–300 bp) on 2% low range ultra-agarose gels, then PCR amplified using Phusion DNA polymerase (NEB, Ipswich, MA, USA). Finally, paired-end RNA sequencing libraries were sequenced with the Illumina HiSeq Xten (2 × 150 bp read length). The raw data files obtained from high-throughput sequencing were converted to the original sequence.
Preprocessing of sequencing data
We constructed 52 libraries and produced 3,013,315,500 clean reads (435,605,241,804 bp) (Online-only Table 1). The raw paired-end reads were trimmed and quality controlled using SeqPrep (https://github.com/jstjohn/SeqPrep) and Sickle (https://github.com/najoshi/sickle) with default parameters of the sequencing analyzer32. The quality of each base was evaluated by FASTX (Fig. 2a,b). The error bases with N and low quality reads were removed (Fig. 2c–f). Clean reads were aligned to the reference genome using HISAT2 (http://ccb.jhu.edu/software/hisat2/index.shtml)33. The mapping criteria were as follows: sequencing reads should be uniquely matched to the genome, with up to two mismatches allowed but without insertions or deletions. The mapping ratios were calculated as an index of the sequence quality. The quality assessment of the transcriptomic sequencing was evaluated using RSeQC 2.3.6 package. The highest coverage paths were assembled and constructed as transcript isoforms. All clean reads those were not mapping to the transcriptional regions were assembled into new transcripts based on overlaps using StringTie (http://ccb.jhu.edu/software/stringtie)34.

Quality assessment of the RNA-Seq. Example of the base quality of the raw reads (a) and clean reads (b). Error rate distribution of the raw reads (c) and clean reads (d). Base content of the raw reads (e) and clean reads (f).
Gene annotation
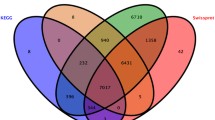
All genes, including the newly assembled fragments, were functional annotated against several databases (NR, Swiss-Prot, and evolutionary genealogy of genes: Non-supervised Orthologous Groups) using DIAMOND v0.8.37.99. For Pfam annotations, HMMER (version 3.1b2) was used with an E-value < 10−5. For Gene Ontology (GO) annotations, the Blast2GO pipeline was used, with an E-value < 10−5 35. Pathway analysis was performed against the KEGG database with KOBAS v2.1.1 (E-value < 10−5). A total of 81.58% genes were annotated in six databases (Table 1).
Gene expression analysis
Gene expression analysis was conducted based on the sequenced reads using RNA-Seq with the Expectation-Maximization (RSEM, v1.3.1) method36. Global gene expression was quantified as transcripts per million reads (TPM) in the four biological replicates. The average TPM value after normalization represented the corresponding quantitative gene expression level at each developmental stage37. The average TPM values for each stage were also used in a principal component analysis (PCA). A log10 transformation was performed to reduce the TPM values for gene expression. The PCA was carried out using an RSEM pipeline and was shown as a line plot. We measured differential gene expression between developmental stages using DESeq. 2 v1.10.138. Genes with an adjusted P-value < 0.05 and a log2 expression ratio > 2 between any two stages were identified as differentially expressed. We compared gene expression levels between stages to identify changes in dynamic gene expression among the four developmental stages (egg, larva, pupa, and adult). We also compared gene expression levels during the development among three instar larval stages; three pupal stages; and three adult stages. Gene expression was compared between the two sexes in adults.
Data Records
The raw transcriptome data were uploaded to the NCBI SRA database39. Gene information and database annotations (e.g. NR, Swiss-Prot, GO, and KEGG pathway) were uploaded to figshare40. The quantitative gene expression of all genes were evaluated by TPM reads and deposited in NCBI Gene Expression Omnibus (GEO) database with an accession number of GSE13948837.
Technical Validation
Quality control
The raw reads were quality controlled using SeqPrep and Sickle software for all samples (Fig. 2a,b). The distribution of the error rate of each sample was evaluated (Fig. 2c,d), then the bases content was calculated (Fig. 2e,f). After removing the low quality bases, we generated the clean reads. The clean reads accounted for up to 94.64% of the raw reads on average. The error rate of each reads was determined by the Phred score, and the mean error rate was 0.026% (range: 0.024–0.029%). The average Q20 (Phred score >20) was 97.54% (Online-only Table 1). The distribution of A/T/G/C was thereafter inspected to analyze the GC content, which showed a low proportion (Fig. 2e,f). These results indicated that sequencing quality was high. Clean reads were separately aligned to the reference genome using HISAT233.
Transcriptome quality assessment
The saturation of sequencing was also analyzed using RSeQC 2.3.6 package, and the data showed a high saturation of each sample sequencing (Fig. 3a). Most of the reads were mapped to the coding sequence region (Fig. 3b). The alignment of the superreads to the reference genome resulted in a total of 41,530 transcripts, and 34.03% were longer than 2700 bp, and 75.96% were longer than 900 bp (Fig. 3c). Analyses of the structures of the transcripts suggested that 11,506 transcripts were potential novel transcript isoforms (Fig. 3d), and 22,068 transcripts were completely matched to the exons.

Sequencing results. An example of the sequencing saturation (a), and the mapping percentage of the reads against the genome (b). The length distribution of the assembled transcripts (c). The type of the assembled transcripts (d).
Gene annotation
All the transcripts were annotated to 18,241 genes, including 13,760 known genes (22,159 transcripts), and 4481 novel genes (19,371 transcripts) (Table 1). All genes were functionally annotated to six databases. The percent of the annotated new assembled genes was 45.28%, which was lower than the reference gene of 93.76%. Finally, we functionally annotated 14,931 genes (81.85%): 14,891 in NR; 10,216 in Swiss-Prot; 13,283 in COG; 11,298 in Pfam; 6911 in GO; and 6653 in KEGG (Table 1).
Gene expression analysis
In this study, four biological replicates were sequenced for gene expression analysis. The correlation coefficient was calculated with Pearson correlation, and the results showed a good correlation between the four biological replicates of the different samples (Fig. 4a). The TPM distribution density of all samples were calculated, and the results also showed a good repeatability and dynamic changes among developmental stages (Fig. 4b). Gene expression levels across all samples were analyzed by PCA; the PCA diagnostic screen plot suggested that the first nine components explain more than 90% of the variance, with the first two explaining 52.13% (Fig. 4c). The graph of the first two principal components (PC1 and PC2) showed that the samples were consistently clustered by developmental stage, indicating good repeatability within the biological replicates and good separation among the developmental stages (Fig. 4d). The database annotations and expression profiles for each gene were performed37,40.

Global gene expression patterns during the 13 life stages. (a) Biological replicate sample correlation analysis. (b) Gene expression boxplot of samples during different stages, plotted as log10-transformed transcripts per million reads (TPM) values (log10(TPM + 1)). (c) The line plot of the variance explained by the first 20 principal components. (d) The scatter plot of PC1 versus PC2 in the principal component analyses (PCA), the gray circle represents the eggs, the green circle represents the larvae, the red circle represents pupae, the yellow circle represents adult females, and the blue circle represents the adult males, respectively.
Differential gene expression between developmental stages was analyzed using DESeq. 238, with an adjusted P-value < 0.05 and a log2 expression ratio > 2. The absolute TPM value > 0.1 was considered as a reliable expression. We analyzed gene expression levels during the four developmental stages, and found that 11,698 of the expressed genes were expressed during metamorphic stages, while 5458 genes were expressed in one and/or more stages specifically (Fig. 5a-1). In these stages, a total of 15017, 12952, 15400, 14417 and 15484 genes were expressed during each stage (Fig. 5a-2). During the larval stage, 18.75% (2785) genes were differentially expressed (Fig. 5b-1). Similarly, 18.73% (3093) genes were differently expressed during pupal development (Fig. 5b-2). In the adult stages, 13.36% and 10.09% genes were differently expressed during female and male adult development, respectively (Fig. 5b-3,b-4).

Gene expression during the various developmental stages. (a) Veen plot of gene expression during the four metamorphic stages (egg, larva, pupa, female and male adult), and total genes in each stage. (b) Veen plot of gene expression within larva (b-1), pupa (b-2), female (b-3) and male adult (b-4) stage, respectively. (c) Venn plot of the differentially expressed genes in each of two developmental stages. Each ellipse circle represents the differentially expressed genes between two connection stages. (d) An example of gene100 expression during the whole 13 stages.
In addition, these transcriptomes might be useful for gene expression analysis among stages. That is, 93.83% of the genes were differentially expressed among stages: 920 genes were differentially expressed between egg and 7-d-old larva; 298 genes were differentially expressed between 7-d-old larva and 5-d-old pupa; 233 genes were differentially expressed between 5-d-old pupa and 5-d-old female; and 157 genes were differentially expressed between 5-d-old pupa and 5-d-old male. However, 630 genes were differentially expressed between all pairs of adjacent stages (Fig. 5c). These data provide a comprehensive comparison of global gene expression among developmental stages. Most importantly, all the gene expression during the 13 stages can be calculated by average the TPM value (Fig. 5d).
References
Dhillon, M. K., Singh, R., Naresh, J. S. & Sharma, H. C. The melon fruit fly, Bactrocera cucurbitae: a review of its biology and management. J. Insect Sci. 5, 40 (2005).
Virgilio, M., Delatte, H., Backeljau, T. & De Meyer, M. Macrogeographic population structuring in the cosmopolitan agricultural pest Bactrocera cucurbitae (Diptera: Tephritidae). Mol. Ecol. 19, 2713–2724 (2010).
McQuate, G. T., Liquido, N. J. & Nakamichi, K. A. A. Annotated world bibliography of host plants of the melon fly, Bactrocera cucurbitae (Coquillett) (Diptera: Tephritidae). Insecta Mundi 527, 1–340 (2017).
Vargas, R. I., Piñero, J. C. & Leblanc, L. An overview of pest species of Bactrocera fruit flies (Diptera: Tephritidae) and the integration of biopesticides with other biological approaches for their management with a focus on the Pacific region. Insects 6, 297–318 (2015).
Sim, S. B. & Geib, S. M. A chromosome-scale assembly of the Bactrocera cucurbitae genome provides insight to the genetic basis of white pupae. G3-Genes Genom. Genet. 7, 1927–1940 (2017).
Zhang, Q. L. et al. Characterization of ladybird Henosepilachna vigintioctopunctata transcriptomes across various life stages. Sci. Data 5, 180093 (2018).
Morandin, C., Pulliainen, U., Bos, N. & Schultner, E. De novo transcriptome assembly and its annotation for the black ant Formica fusca at the larval stage. Sci. Data 5, 180282 (2018).
Tian, C. B. et al. Comparative transcriptome analysis of three Bactrocera dorsalis (Hendel) organs to identify functional genes in male accessory glands and ejaculatory duct. Fla. Entomol. 100, 42–51 (2017).
Wei, D. et al. Comparative analysis of differential gene expression profiling of sex-bias fat body of Bactrocera dorsalis (Diptera: Tephritidae) identifying a new vitellogenin gene. Ann. Entomol. Soc. Am. 111, 43–54 (2018).
Ding, B. Y. et al. Silencing of two insulin receptor genes disrupts nymph-adult transition of alate brown citrus aphid. Int. J. Mol. Sci. 18, 357 (2017).
Shang, F. et al. Differential expression of genes in the alate and apterous morphs of the brown citrus aphid, Toxoptera citricida. Sci. Rep. 6, 32099 (2016).
Meccariello, A. et al. Maleness-on-the-Y (MoY) orchestrates male sex determination in major agricultural fruit fly pests. Science 365, 1457–1460 (2019).
Shen, G. M. et al. Transcriptome analysis of the oriental fruit fly (Bactrocera dorsalis). Plos One 6, e29127 (2011).
Chen, E. H. et al. RNA-seq analysis of gene expression changes during pupariation in Bactrocera dorsalis (Hendel) (Diptera: Tephritidae). BMC Genomics 19, 693 (2018).
Salvemini, M. et al. De Novo Assembly and Transcriptome Analysis of the Mediterranean Fruit Fly Ceratitis capitata Early Embryos. Plos One 9, e114191 (2014).
Wei, D. et al. Gene expression profiling of ovary identified eggshell proteins regulated by 20-hydroxyecdysone in Bactrocera dorsalis. Comp. Biochem. Physiol. D 30, 206–216 (2019).
Wei, D. et al. Transcriptome profiling of the testis reveals genes involved in spermatogenesis and marker discovery in the oriental fruit fly, Bactrocera dorsalis. Insect Mol. Biol. 24, 41–57 (2015).
Zheng, W., Luo, D., Wu, F., Wang, J. & Zhang, H. RNA sequencing to characterize transcriptional changes of sexual maturation and mating in the female oriental fruit fly Bactrocera dorsalis. BMC Genomics 17, 194 (2016).
Gomulski, L. M. et al. Transcriptome profiling of sexual maturation and mating in the Mediterranean fruit fly, Ceratitis capitata. PLoS ONE 7, e30857 (2012).
Gu, Z. Y. et al. Differentially expressed genes in the fat body of Bombyx mori in response to phoxim insecticide. Pest. Biochem. Physiol. 117, 47–53 (2015).
Lin, J. H. et al. Gene expression profiling provides insights into the immune mechanism of Plutella xylostella midgut to microbial infection. Gene 647, 21–30 (2018).
Sparks, M. E., Blackburn, M. B., Kuhar, D. & Gundersen-Rindal, D. E. Transcriptome of the Lymantria dispar (gypsy moth) larval midgut in response to infection by Bacillus thuringiensis. Plos One 8, e61190 (2013).
Sim, S. B., Calla, B., Hall, B., DeRego, T. & Geib, S. M. Reconstructing a comprehensive transcriptome assembly of a white-pupal translocated strain of the pest fruit fly Bactrocera cucurbitae. Gigascience 4, 14 (2015).
Elfekih, S., Chen, C. Y., Hsu, J. C., Belcaid, M. & Haymer, D. Identification and preliminary characterization of chemosensory perception-associated proteins in the melon fly Bactrocera cucurbitae using RNA-seq. Sci. Rep. 6, 19112 (2016).
Chen, X. F. et al. Genome-wide identification and expression profiling of odorant-binding proteins in the oriental fruit fly, Bactrocera dorsalis. Comp. Biochem. Physiol. D 31, 100605 (2019).
Niwa, R. & Niwa, Y. S. Enzymes for ecdysteroid biosynthesis: their biological functions in insects and beyond. Biosci. Biotech. Biochem. 78, 1283–1292 (2014).
Kayukawa, T., Jouraku, A., Ito, Y. & Shinoda, T. Molecular mechanism underlying juvenile hormone-mediated repression of precocious larval–adult metamorphosis. Proc. Natl. Acad. Sci. USA 114, 1057–1062 (2017).
Liu, H. W. et al. A clip domain serine protease involved in moulting in the silkworm, Bombyx mori: cloning, characterization, expression patterns and functional analysis. Insect Mol. Biol. 26, 507–521 (2017).
Papanicolaou, A. et al. The whole genome sequence of the Mediterranean fruit fly, Ceratitis capitata (Wiedemann), reveals insights into the biology and adaptive evolution of a highly invasive pest species. Genome Biol. 17, 31 (2016).
Calla, B., Hall, B., Hou, S. B. & Geib, S. M. A genomic perspective to assessing quality of mass-reared SIT flies used in Mediterranean fruit fly (Ceratitis capitata) eradication in California. BMC Genomics 15, 98 (2014).
Wang, J. J. et al. Toxicities and synergistic effects of several insecticides against the oriental fruit fly (Diptera: Tephritidae). J. Econ. Entomol. 106, 970–978 (2013).
Chen, C., Khaleel, S. S., Huang, H. & Wu, C. H. Software for pre-processing Illumina next-generation sequencing short read sequences. Source Code Biol. Med. 9, 8 (2014).
Daehwan, K., Ben, L. & Salzberg, S. L. HISAT: a fast spliced aligner with low memory requirements. Nat. Methods 12, 357–360 (2015).
Mihaela, P. et al. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 33, 290–295 (2015).
Conesa, A. et al. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21, 3674–3676 (2005).
Li, B. & Dewey, C. N. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12, 323 (2011).
Gene Expression Omnibus. https://identifiers.org/geo:GSE139488 (2019).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq. 2. Genome Biol. 15, 550 (2014).
NCBI Sequence Read Archive. https://identifiers.org/ncbi/insdc.sra:SRP220120 (2019).
Wei, D. et al. Genome-wide gene expression profiling of the melon fly, Zeugodacus cucurbitae, during thirteen life stages. figshare, https://doi.org/10.6084/m9.figshare.9841040 (2019).
Acknowledgements
We appreciate Dr. Fei Pan from Institute of Plant Protection, Hainan Academy of Agricultural Sciences, Hainan, China, for help collect insects. This study was supported in part by the Foundation Project of Southwest University (SWU019033), the Chongqing Research Program of Basic Research and Frontier Technology (CSTC2016jcyjA0019), and the 111 Project (B18044).
Author information
Authors and Affiliations
Contributions
D.W. and J.J.W. conceived and designed the project; H.Q.X., D.C. and S.Y.Z. contributed to the materials and samples; D.W. and W.J.L. analyzed the data; D.W., G.S. and J.J.W. wrote the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Online-only Table
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
The Creative Commons Public Domain Dedication waiver http://creativecommons.org/publicdomain/zero/1.0/ applies to the metadata files associated with this article.
About this article

Cite this article
Wei, D., Xu, HQ., Chen, D. et al. Genome-wide gene expression profiling of the melon fly, Zeugodacus cucurbitae, during thirteen life stages. Sci Data 7, 45 (2020). https://doi.org/10.1038/s41597-020-0387-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-020-0387-9
- Springer Nature Limited