
Abstract
Estrogen Receptor alpha (ERα) is a ligand-inducible transcription factor that mediates estrogen signaling in hormone-responsive cells, where it controls key cellular functions by assembling in gene-regulatory multiprotein complexes. For this reason, interaction proteomics has been shown to represent a useful tool to investigate the molecular mechanisms underlying ERα action in target cells. RNAs have emerged as bridging molecules, involved in both assembly and activity of transcription regulatory protein complexes. By applying Tandem Affinity Purification (TAP) coupled to mass spectrometry (MS) before and after RNase digestion in vitro, we generated a dataset of nuclear ERα molecular partners whose association with the receptor involves RNAs. These data provide a useful resource to elucidate the combined role of nuclear RNAs and the proteins identified here in ERα signaling to the genome in breast cancer and other cell types.
Design Type(s) | parallel group design • protein and RNA interaction identification objective • cell type comparison design |
Measurement Type(s) | RNA-Protein Interaction • protein-protein interaction detection |
Technology Type(s) | mass spectrometry |
Factor Type(s) | experimental condition • biological replicate |
Sample Characteristic(s) | MCF7 cell |
Machine-accessible metadata file describing the reported data (ISA-Tab format)
Similar content being viewed by others


Background & Summary
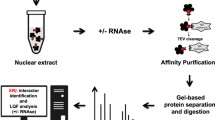
The Estrogen Receptor alpha (ERα), a ligand-inducible transcription factor encoded by the ESR1 gene, is a key mediator of the estrogen signaling in hormone-responsive breast cancer (BC)1. This receptor subtype exerts a direct control on gene transcription machinery by binding chromatin cistromes2, where it assembles in large functional multiprotein complexes. These complexes comprise several molecular partners endowed with different functions, including co-regulators3,4,5 and epigenetic modulators6,7,8 that drive gene expression changes underlying BC development and progression9. Thus, the identification and characterization of the multiprotein complexes involved in the mechanism of action of ERα are crucial steps to understand the molecular bases of its signaling. Systematic application of interaction proteomics by coupling complexes purification with mass spectrometry analyses, represent an optimal experimental approach to gain such information. In the context of regulatory network assembly, it has emerged that RNAs of different nature may play a critical role as hinge of multiprotein complexes. Yang and colleagues10 revealed the importance of long non-coding RNAs in mediating androgen receptor function, acting as bridging molecules in the formation of functional protein complexes of this nuclear hormone receptor. On the other side, it is known that ERα has the ability to bind RNAs and that some of those are present within the receptor-mediated transcriptional complexes11. Thus, we assessed here the involvement of RNAs in mediating ERα-driven nuclear protein network assembly. To this aim, ERα-bound native proteins were purified from BC cell nuclei, before and after in vitro RNase treatment, and subjected to protein identification and quantification (Fig. 1a). In detail, Tandem Affinity Purification (TAP)12,13,14 followed by mass spectrometry was performed in parallel from Ct-ERα (MCF7 cells stably expressing ERα tagged with the TAP moiety at the C-terminus) and parental cells (CTRL: MCF7 parental line). This resulted in the initial identification of 1423 ERα protein partners15. Indeed, to avoid the inclusion of false positives and strengthen the specificity of our data, we defined ERα molecular partners only those proteins that were completely absent in the CTRLs purifications performed in parallel, according to the analytical steps detailed in the methods section.

Characterization of ERα interactome. (a) Summary of the experimental workflow applied to generate the protein datasets. Ct-ERα: MCF7 cells stably expressing TAP-tag at the ERα C-terminal; CTRL: wt MCF7 cells. (b) Classification of ERα molecular partners; asterisks indicate statistically enriched molecule types (p < 0.01 hypergeometric test). (c) Functional enrichment analysis by IPA of ERα-associated proteins (B-H: Benjamini-Hochberg corrected p-value). (d) Venn diagram showing the overlap between ERα interactors identified here by Tandem affinity purification (TAP) and a dataset previously generated through Chromatin immunoprecipitation followed by mass spectrometry (ChIP-MS), described by Nassa et al.8.
The interactome identified, comprising among other enzymes, transcription regulators and kinases (Fig. 1b) (p-value < 0.05, hypergeometric test), defines molecular functions known to be involved in ERα cellular functions, such as cell growth and proliferation, gene expression, cell cycle and cell death and survival (Fig. 1c) (p-value < 0.05, Fisher’s test, Benjamini-Hochberg correction). The identified proteins include several known ERα molecular partners12, including a sizable fraction (32%, 196) recently shown to be part of chromatin-associated multiprotein complexes important for ERα activity in BC8 (Fig. 1d). Then, to understand whether and how RNA moieties can mediate the assembly or composition of these complexes, nuclear extracts were pre-treated by RNase digestion before TAP and MS analysis using the same experimental approach detailed in Fig. 1a. To assess the effectiveness of the enzymatic treatment, RNA was purified from an aliquot of each nuclear extract before and after RNase and analyzed by microfluidic electrophoresis as previously described14,16. In parallel, aliquots of each sample were kept from each TAP passage and analyzed by western blotting for the presence and integrity of ERα as detailed in the technical validation section. Molecular partners associated with the receptor even after RNase treatment were identified according to the same procedure described above, by comparing RNase+ and RNase− datasets, after filtering nonspecific interactors retrieved also in control purifications.
The dataset obtained results in 1296 proteins. By comparing ERα interactomes under the two experimental conditions, it turned out that they shared about 90% of the interactors (Fig. 2a). A quantitative approach was then applied, by using Perseus17, to identify proteins whose concentration was significantly reduced by RNase digestion, compared to untreated samples. The quantitative analyses were performed considering the 1222 ERα partners identified and quantified in both untreated (RNase−) and treated (RNase+) samples. Statistical analyses revealed that the concentration of about 35% of these proteins was significantly modulated by RNA depletion (q-value ≤ 0,05) (Volcano plot in Fig. 2a and in figshare15), indicating that RNAs are likely to mediate their association with the receptor in BC nuclei. ERα molecular partners affected by RNase treatment, meaning the interacting proteins whose concentration in the purified samples was affected by RNase pre-treatment (reported in the Volcano plot in light blue) comprise several enzymes, such as, transcription regulators and kinases (Fig. 2b) (p-value < 0.05, hypergeometric test). These factors are involved in key estrogen receptor functions in BC, including gene expression and protein synthesis regulation as shown in Fig. 2c. Complex analyses via g:Profiler18 was then used to gather information concerning ERα multiprotein complexes assembly15.

Analysis of ERα interactome changes upon RNase treatment. (a) Venn diagram showing the overlap between ERα molecular partners identified by Tandem affinity purification (TAP) before and after RNase treatment and Volcano plot summarizing quantitative changes of ERα-associated overlapping proteins upon treatment with RNase. Dotted line (threshold) represents the cut-off (q-value ≤ 0.05). (b) Classification of ERα molecular partners; asterisks indicate statistically significant molecule types (p < 0.01 hypergeometric test). (c) Functional enrichment analysis by IPA of ERα-associated proteins (B-H: Benjamini-Hochberg corrected p-value). (d) Network representation of the ESR1-TRAP/Mediator coactivator-complex; thickness of links (lines) among nodes (proteins) is proportional to the strength of the physical interaction. Information about co-expression, physical interactions and strength derive from GeneMANIA.
Considering the functions of the 1222 ERα partners, it turned out that they assemble in multiple complexes15, such as for example the Mediator complex, known to have a key role in estrogen receptor-mediated gene transcription, and the ESR1-TRAP/Mediator coactivator-complex (where ESR1 indicates the gene coding for the ERα protein). The latter, as shown as an example in Fig. 2d, includes several proteins whose association with the receptor was decreased by RNase treatment.
In conclusion, the RNA-dependent nuclear interactome reported here will be useful to investigate in greater detail the molecular mechanisms underlying ERα actions in BC cells, characterizing the RNA(s) involved and other key nodes of this regulatory network, toward identification of druggable targets against breast and other cancers where ERα plays a pivotal role.
Methods
ERα nuclear complexes purification
MCF7 cells stably expressing ERα fused with the TAP-tag at the C-terminus (Ct-ERα), to allow proper protein complexes purification, were generated as earlier detailed13,19. Ct-ERα and wt (CTRL) MCF7 cells (ATCC HTB-22), exponentially growing, were harvested by scraping in cold PBS and lysed as previously described19 in order to obtain nuclear extracts as reported by Giurato and co-workers14. To this aim, cell pellets were resuspended in 3 volumes of hypotonic buffer (20 mM HEPES pH 7.4, 5 mM NaF, 10 µM sodium molybdate, 0.1 mM EDTA, 1 mM PMSF and 1X protease inhibitors cocktail (Sigma Aldrich) and incubated on ice for 15 minutes. Cytosolic fraction was discarded after adding 0.5% Triton X-100 and spinning for 30 sec at 15000 × g at 4 °C. Nuclear pellets were then resuspended in 1 volume of nuclear lysis buffer (20 mM HEPES pH 7.4, 25% glycerol, 420 mM NaCl, 1.5 mM MgCl2, 0.2 mM EDTA, 1 mM PMSF and 1X protease inhibitors cocktail (Sigma Aldrich), incubated for 30 minutes at 4 °C under gentle shaking and centrifuged for 30 min at 15000 × g at 4 °C. Supernatants were then collected, diluted 1:3 with nuclear lysis buffer without NaCl, to restore the physiological saline concentration, and quantified.
For TAP procedure, IgG-Sepharose beads (GE Healthcare), pre-treated according to the manufacturer’s instructions and equilibrated in TEV buffer (50 mM Tris-HCl pH 8.0, 0.5 mM EDTA, 0.1% Triton X-100, 150 mM NaCl, 1 mM DTT), were added to nuclear protein extracts and incubated for 3 hours at 4 °C with gentle rotation, as described earlier12,20. 100 µg/ml RNaseA were added to the samples before binding, as already reported14,16 (see Table 1). After incubation, unbound proteins were discarded following centrifugation and the beads were thoroughly washed with 100xVol of IPP150 buffer (20 mM HEPES pH 7.5, 8% glycerol, 150 mM NaCl, 0.5 mM MgCl2, 0.1 mM EDTA, 0.1% Triton X-100) and equilibrated in 30xVol of TEV Buffer in Poly-Prep Chromatography columns (0.8 × 4 cm, Bio-Rad) at 4 °C. Then, 4xBeads Vol of Cleavage Buffer (TEV Buffer containing 1U/μl beads of TEV protease, Invitrogen) were added and two subsequent cleavage reactions were carried out for 2 hours and 30 minutes respectively at 16 °C with gentle shaking. The eluates were then collected, after sedimentation of beads still binding uncut and non-specific proteins. The same procedure was performed in parallel from Ct-ERα and wt MCF7 cells that served as negative control to identify nonspecifically isolated proteins to be discarded.
Nano LC-MS/MS and data analysis
For mass spectrometry analyses, three biological replicates of TEV-eluted samples from control MCF-7 and from Ct-ERα cells before and after RNase treatment were analyzed. Protein extracts were precipitated with 10% TCA in acetone solution and and the proteins were reduced, alkylated and in-solution digested with trypsin (Promega) with the ProteaseMAX™ Surfactant (Promega) protocol according to manufacturers’ instructions. The resulting peptides were desalted and concentrated before mass spectrometry by the STAGE-TIP method using a C18 resin disk (3 M Empore). The peptides were eluted with 0.1% TFA/50% ACN, dried and solubilized in 7 μL 0.1% FA for mass spectrometry analysis. Each peptide mixture was analyzed on an Easy nLC1000 nano-LC system connected to a quadrupole Orbitrap mass spectrometer (QExactive, ThermoElectron, Bremen, Germany) equipped with a nanoelectrospray ion source (EasySpray/Thermo). For the liquid chromatography separation of the peptides an EasySpray column capillary of 25 cm bed length was employed. The flow rate was 300 nL/min, and the peptides were eluted with a 2–30% gradient of solvent B in 60 minutes. Solvent A was aqueous 0.1% formic acid and solvent B 100% acetonitrile/0.1% formic acid. The data-dependent acquisition automatically switched between MS and MS/MS mode. Survey full scan MS spectra were acquired from a mass-to-charge ratio (m/z) of 400 to 1,200 with the resolution R = 70,000 at m/z 200 after accumulation to a target of 3,000,000 ions in the C-trap. For MS/MS, the ten most abundant multiple-charged ions were selected for fragmentation in the high-energy collision dissociation (HCD) cell at a target value of 100,000 charges or maximum acquisition time of 100 ms. The MS/MS scans were collected at a resolution of 17,500. Target ions already selected for MS/MS were dynamically excluded for 30 seconds. The resulting MS raw files of control MCF-7 cells were submitted for protein identification using Proteome Discoverer (ver 2.1) software with the Mascot (ver 2.6.1) search engine. The search criteria for Mascot searches were: trypsin digestion with two missed cleavage allowed, Carbamidomethyl (C) as fixed modification and Acetyl (N-term), Gln- > pyro-Glu (N-term Q), Oxidation (M) as variable modifications. The parent mass tolerance was 10 ppm and MS/MS tolerance 0.1 Da. Database searches were done against the UniProt Human database (September 2018) and known contaminants provided by MaxQuant. All of the reported protein identifications were statistically significant (p < 0.05) in Mascot, and further filtered in ProteomeDiscoverer to report only high and medium protein FDR confidence identifications.
For the experiment performed in presence or absence of RNase the resulting MS raw files were submitted to the MaxQuant software (version 1.6.2.10)21 for protein identification and quantitation using the Andromeda search engine. MaxQuant search was done against the UniProt Human database (September 2018). Carbamidomethyl (C) was set as a fixed modification and protein N-acetylation and methionine oxidation were set as variable modifications. First search peptide tolerance of 20 ppm and main search error 4.5 ppm were used. Trypsin without proline restriction enzyme option was used, with two allowed miscleavages. The minimal unique + razor peptides number was set to 1, and the allowed FDR was 0.01 (1%) for peptide and protein identification.
Further analysis of the LC-MS/MS data followed two steps. Firstly, only proteins identified in the Ct-ERα samples (before and after RNase treatment) and not present in parental MCF7 cells (CTRL) were considered. In details, proteins were considered specifically associated with Ct-ERα before RNase treatment if they were present in at least two out of three biological replicates, but not after RNase treatment. The same approach was followed to consider proteins specifically associated with Ct-ERα after RNase. As commonly identified proteins we reported those present in at least two out of three replicate samples, in both experimental conditions tested. Moreover, known contaminants provided by MaxQuant and potential contaminants identified only in the Ct-ERα samples (e.g. Keratins, Immunoglobulins) were discarded and excluded from further analysis15. Statistical analysis was performed using Perseus software (version 1.6.1.3). To this aim, data (INTENSITY) was log10 transformed, filtered to include only proteins identified and quantified in at least two of the three replicates in at least one experimental group. Missing values were imputed with values representing a normal distribution with default settings in Perseus 1.6.1.317. To find statistically significant differences between the two groups (RNase vs Sample) a T-test with a permutation-based approach was applied, with an FDR cut-off of 0.05. The lists of proteins whose association with the receptor changed after depletion of RNA have been deposited in figshare15. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium22 via the PRIDE23 partner repository with the dataset identifier PXD01263024 for interaction and quantitative proteomics datasets comprising CTRLs and Ct-ERα samples before and after RNase treatment. The protein interactions from this publication have been submitted to the IMEx (http://wwww.imexconsortium.org) consortium through IntAct25 and assigned the identifier IM-2695426.
Functional annotation analyses
Functional annotation analysis was performed using Ingenuity Pathway Analysis (IPA, QIAGEN, Redwood City, www.qiagen.com/ingenuity) setting the following parameters:
-
Reference set: Ingenuity Knowledge Base
-
Relationship to include: Direct and Indirect
-
Filter: Consider only molecules and/or relationship where (species:Human) AND (confidence: Experimentally Observed)
Only functions showing a Benjamini Hochberg (B-H) pvalue ≤ 0.05 were considered.
Molecular type information was retrieved from IPA and the pvalue associated to over-enrichment of each molecular category was computed using hypergeometric test. The information for protein categories was retrieved from UniProt.
Over-representation analysis on protein complexes was performed using g:Profiler18, in order to gather information concerning ERα multiprotein complexes assembly, setting the following parameters:
-
Organism: “Homo Sapiens”
-
All results: yes
-
Statistical domain scope: “Only annotated genes”
-
Significance threshold: “g:SCS threshold”
-
User threshold: “0.05”
-
Numeric ID treated as: “ENTREZGENE_ACC”
-
Data sources: “CORUM”
Only complexes showing an adjusted-pvalue ≤ 0.05 were considered. Network was created with Cytoscape v. 3.7.127, using the GeneMANIA28 application and considering only “Physical Interaction” and “Co-expression”. Physical interactions, co-expression, and strength information derive from GeneMANIA internal database.
Data Records
Data records are available to be downloaded from Figshare archive, in which data were deposited including uncropped Western blots of the TAP procedures15.
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the following dataset identifier: PXD012630 for interaction and quantitative proteomics datasets comprising CTRLs and Ct-ERα samples before and after RNase treatment24.
The Protein-Protein interactions have been submitted to the IMEx (www.imexconsortium.org) consortium through IntAct25 and assigned the identifier IM-2695426.
Technical Validation
To ensure quality and robustness of the data presented here, our datasets were generated from three, independent biological replicates by using cell cultures processed independently from authenticated and Mycoplasma-free MCF7. This cell line represents a widely used model of Luminal A BC that have been demonstrated to have fundamentally altered the course of breast cancer research and have contributed to the improvement of patient’s outcomes29, suggesting that it is unlikely that the results obtained here are all specific only of the cell line used.
Each TAP biological replicate was performed independently and both controls and samples were analyzed in parallel. The effect of the RNase treatment and efficiency of the affinity purification procedure were rigorously assessed as described previously14,16 and reported in Fig. 3a,b respectively. Correlation between the biological triplicate purifications (about 90%) was verified by analyzing log10 intensities with R version 3.5.2 (see code availability 7), applying the function cor from R-package sats v3.5.2 with Pearson correlation. Additionally, principal component analysis (PCA) highlighted the differences between the two treatments (−/+ RNase treatment) and the minimal variation among biological replicates of each group. Lastly, to exclude that the observed proteome changes are due to a reduction of receptor concentration in the crude nuclear extracts as a consequence of RNase treatment, ERα (ESR1) median intensities of the three biological replicates before and after RNase treatment were compared (Fig. 3c). This and other laboratories investigated previously ERα interactome in breast cancer cells using different approaches and under different experimental conditions8,12,13,30,31.

Quality controls of the experimental procedure. (a) Electrophoretic analysis of RNA extracted from nuclear extracts (starting material) before and after RNase treatment. (b) Representative Western Blot (one of the three biological replicates of the study) of the different steps of the Tandem Affinity Purification protocol in wt MCF-7 (CTRL, up) and Ct-ERα cells before (middle) and after (down) RNase treatment. The presence of the indicated proteins has been evaluated in different fractions as described: lanes 1 and 2, Crude nuclear extracts before and after IgG-Sepharose binding respectively; Lanes 3, IgG-Sepharose-bound proteins before TEV elution; Lanes 4 (TEV elution), proteins eluted from IgG-Sepharose. (c) Bar plot showing protein levels (%) of ERα and some of its interactors (+vs − RNase). Unchanged proteins are reported in gray, decreased proteins in blue.
The dataset described here includes, also, a sizeable fraction of previously reported ERα interactors from BioGRID database (about 30%). In addition, it reveals that some of the best known key mediators of estrogen signaling to the genome, such as NCOA332 and DDX533, remain associated with ERα after RNase treatment. Others, representing about 10% (according to the ESR1/ERα BioGRID known interactions) of the 201 proteins whose concentration resulted to be modulate by RNase treatment, and including KDM1A34, MED135, SAFB236, SMARCA2/45, TRIM2537, EZH238, MPG39 and RBM340, decrease strongly upon RNA removal, suggesting that their interaction with the receptor might be mediated by RNAs41 (Fig. 3c).
Code Availability
The following software and versions were used for quality control and data analysis:
1. For protein identification from raw MS data, Proteome Discoverer software (Thermo) version 2.1 was used: https://tools.thermofisher.com/ with the Mascot 2.6.1 search engine.
2. For quantitative data analysis, MaxQuant software version 1.6.2.10 with the Andromeda search engine was used: http://www.coxdocs.org/docu.php?id=maxquant:start
3. For statistical proteomics analysis, Perseus software version 1.6.1.3 was used: (http://www.perseus-framework.org)
4. The UniProt human database was used (September 2018) as database for protein searches: https://www.uniprot.org/
5. Functional analysis was performed with IPA software version 2.4: http://www.qiagenbioinformatics.com/products/ingenuity-pathway-analysis
6. Complexes analysis was performed using g:Profiler: https://biit.cs.ut.ee/gprofiler/gost
7. Statistical analyses were performed using R 3.5.2: www.r-project.org
8. The R script and the data used to create volcano plot are available at the Github repository https://github.com/LabMedMolGE/VolcanoPlot.
References
Chen, G. G., Zeng, Q. & Tse, G. M. Estrogen and its receptors in cancer. Med Res Rev 28, 954–974 (2008).
Cicatiello, L. et al. Estrogen receptor alpha controls a gene network in luminal-like breast cancer cells comprising multiple transcription factors and microRNAs. Am J Pathol 176, 2113–2130 (2010).
Lonard, D. M. & O’Malley, B. W. Nuclear receptor coregulators: modulators of pathology and therapeutic targets. Nat Rev Endocrinol 8, 598–604 (2012).
Green, K. A. & Carroll, J. S. Oestrogen-receptor-mediated transcription and the influence of co-factors and chromatin state. Nat Rev Cancer 7, 713–722 (2007).
Metivier, R. et al. Estrogen receptor-alpha directs ordered, cyclical, and combinatorial recruitment of cofactors on a natural target promoter. Cell 115, 751–763 (2003).
Toska, E. et al. PI3K pathway regulates ER-dependent transcription in breast cancer through the epigenetic regulator KMT2D. Science 355, 1324–1330 (2017).
Feng, Q. et al. An epigenomic approach to therapy for tamoxifen-resistant breast cancer. Cell Res 24, 809–819 (2014).
Nassa, G. et al. Inhibition of histone methyltransferase DOT1L silences ERalpha gene and blocks proliferation of antiestrogen-resistant breast cancer cells. Sci Adv 5, eaav5590 (2019).
Ross-Innes, C. S. et al. Differential oestrogen receptor binding is associated with clinical outcome in breast cancer. Nature 481, 389–393 (2012).
Yang, L. et al. lncRNA-dependent mechanisms of androgen-receptor-regulated gene activation programs. Nature 500, 598–602 (2013).
Lanz, R. B. et al. A steroid receptor coactivator, SRA, functions as an RNA and is present in an SRC-1 complex. Cell 97, 17–27 (1999).
Tarallo, R. et al. Identification of proteins associated with ligand-activated estrogen receptor alpha in human breast cancer cell nuclei by tandem affinity purification and nano LC-MS/MS. Proteomics 11, 172–179 (2011).
Cirillo, F. et al. Molecular mechanisms of selective estrogen receptor modulator activity in human breast cancer cells: identification of novel nuclear cofactors of antiestrogen-ERalpha complexes by interaction proteomics. J Proteome Res 12, 421–431 (2013).
Giurato, G. et al. Quantitative mapping of RNA-mediated nuclear estrogen receptor beta interactome in human breast cancer cells. Sci Data 5, 180031 (2018).
Nassa, G. et al. The RNA-mediated estrogen receptor α interactome of hormone-dependent human breast cancer cell nuclei. figshare. https://doi.org/10.6084/m9.figshare.c.4398032 (2019).
Tarallo, R. et al. The nuclear receptor ERβ engages AGO2 in regulation of gene transcription, RNA splicing and RISC loading. Genome Biology 18, 189 (2017).
Tyanova, S. et al. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat Methods 13, 731–740 (2016).
Reimand, J. et al. g:Profiler-a web server for functional interpretation of gene lists (2016 update). Nucleic Acids Res 44, W83–89 (2016).
Ambrosino, C. et al. Identification of a hormone-regulated dynamic nuclear actin network associated with estrogen receptor alpha in human breast cancer cell nuclei. Mol Cell Proteomics 9, 1352–1367 (2010).
Nassa, G. et al. A large set of estrogen receptor beta-interacting proteins identified by tandem affinity purification in hormone-responsive human breast cancer cell nuclei. Proteomics 11, 159–165 (2011).
Tyanova, S., Temu, T. & Cox, J. The MaxQuant computational platform for mass spectrometry-based shotgun proteomics. Nat Protoc 11, 2301–2319 (2016).
Deutsch, E. W. et al. The ProteomeXchange consortium in 2017: supporting the cultural change in proteomics public data deposition. Nucleic Acids Res 45, D1100–D1106 (2017).
Perez-Riverol, Y. et al. The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Res 47, D442–D450 (2019).
Nyman, T. A. Estrogen Receptor alpha (ERα) interactome before and after RNAse treatment in human breast cancer cells nuclei. PRIDE, https://identifiers.org/pride.project:PXD012630 (2019).
Orchard, S. et al. The MIntAct project–IntAct as a common curation platform for 11 molecular interaction databases. Nucleic Acids Res 42, D358–363 (2014).
Nassa, G. The RNA-mediated estrogen receptor α interactome of hormone-dependent human breast cancer cell nuclei. IntAct, https://identifiers.org/imex:IM-26954 (2019).
Shannon, P. et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 13, 2498–2504 (2003).
Warde-Farley, D. et al. The GeneMANIA prediction server: biological network integration for gene prioritization and predicting gene function. Nucleic Acids Res 38, W214–220 (2010).
Lee, A. V., Oesterreich, S. & Davidson, N. E. MCF-7 cells–changing the course of breast cancer research and care for 45 years. J Natl Cancer Inst 107, djv073 (2015).
Mohammed, H. et al. Endogenous purification reveals GREB1 as a key estrogen receptor regulatory factor. Cell Rep 3, 342–349 (2013).
Papachristou, E. K. et al. A quantitative mass spectrometry-based approach to monitor the dynamics of endogenous chromatin-associated protein complexes. Nat Commun 9, 2311 (2018).
Yi, P. et al. Structure of a biologically active estrogen receptor-coactivator complex on DNA. Mol Cell 57, 1047–1058 (2015).
Samaan, S. et al. The Ddx5 and Ddx17 RNA helicases are cornerstones in the complex regulatory array of steroid hormone-signaling pathways. Nucleic Acids Res 42, 2197–2207 (2014).
Hu, Q. et al. Enhancing nuclear receptor-induced transcription requires nuclear motor and LSD1-dependent gene networking in interchromatin granules. Proc Natl Acad Sci USA 105, 19199–19204 (2008).
Nagalingam, A. et al. Med1 plays a critical role in the development of tamoxifen resistance. Carcinogenesis 33, 918–930 (2012).
Townson, S. M. et al. SAFB2, a new scaffold attachment factor homolog and estrogen receptor corepressor. J Biol Chem 278, 20059–20068 (2003).
Walsh, L. A. et al. An Integrated Systems Biology Approach Identifies TRIM25 as a Key Determinant of Breast Cancer Metastasis. Cell Rep 20, 1623–1640 (2017).
Wu, Y. et al. Tamoxifen Resistance in Breast Cancer Is Regulated by the EZH2-ERalpha-GREB1 Transcriptional Axis. Cancer Res 78, 671–684 (2018).
Likhite, V. S., Cass, E. I., Anderson, S. D., Yates, J. R. & Nardulli, A. M. Interaction of estrogen receptor alpha with 3-methyladenine DNA glycosylase modulates transcription and DNA repair. J Biol Chem 279, 16875–16882 (2004).
Jogi, A. et al. Nuclear expression of the RNA-binding protein RBM3 is associated with an improved clinical outcome in breast cancer. Mod Pathol 22, 1564–1574 (2009).
Tsai, M. C. et al. Long noncoding RNA as modular scaffold of histone modification complexes. Science 329, 689–693 (2010).
Acknowledgements
Work supported by: Italian Association for Cancer Research (Grant IG-17426), Italian Ministries of Education, University and Research (Flagship Project InterOmics), University of Salerno (Fondi FARB 2017), Regione Campania, Progetto GENOMAeSALUTE (POR Campania FESR 2014/2020, azione 1.5; CUP: B41C17000080007) and Genomix4Life. We also acknowledge ELIXIR-IIB (www.elixir-italy.org), the Italian Node of the European ELIXIR infrastructure (www.elixir-europe.org), for the computational power support provided. G.P. and V.G. are PhD Students of the Research Doctorates in Biomedical Sciences and Technologies of the University Roma Tre and in Molecular Oncology, Immunology and Experimental Development of Innovative Therapies of the University of Catanzaro ‘Magna Graecia’, respectively.
Author information
Authors and Affiliations
Contributions
All authors participated in conception and design of the study. G.N., A.S., V.G., G.P., J.L. and F.R. performed in vitro experimental work, T.N. and G.N. carried out the proteomics analyses, G.G. performed the statistical and bioinformatics analyses, G.N., G.G., R.T. and A.W. coordinated and finalized figure preparation, manuscript drafting and revision. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
ISA-Tab metadata file
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
The Creative Commons Public Domain Dedication waiver http://creativecommons.org/publicdomain/zero/1.0/ applies to the metadata files associated with this article.
About this article

Cite this article
Nassa, G., Giurato, G., Salvati, A. et al. The RNA-mediated estrogen receptor α interactome of hormone-dependent human breast cancer cell nuclei. Sci Data 6, 173 (2019). https://doi.org/10.1038/s41597-019-0179-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-019-0179-2
- Springer Nature Limited
This article is cited by
-
Integrative analysis of transcriptomic and metabolomic profiles reveals enhanced arginine metabolism in androgen-independent prostate cancer cells
BMC Cancer (2023)
-
Combinatorial targeting of a chromatin complex comprising Dot1L, menin and the tyrosine kinase BAZ1B reveals a new therapeutic vulnerability of endocrine therapy-resistant breast cancer
Breast Cancer Research (2022)
-
PLK1 inhibition exhibits strong anti-tumoral activity in CCND1-driven breast cancer metastases with acquired palbociclib resistance
Nature Communications (2020)