

Abstract
A multitude of sequencing-based and microscopy technologies provide the means to unravel the relationship between the three-dimensional organization of genomes and key regulatory processes of genome function. Here, we develop a multimodal data integration approach to produce populations of single-cell genome structures that are highly predictive for nuclear locations of genes and nuclear bodies, local chromatin compaction and spatial segregation of functionally related chromatin. We demonstrate that multimodal data integration can compensate for systematic errors in some of the data and can greatly increase accuracy and coverage of genome structure models. We also show that alternative combinations of different orthogonal data sources can converge to models with similar predictive power. Moreover, our study reveals the key contributions of low-frequency (‘rare’) interchromosomal contacts to accurately predicting the global nuclear architecture, including the positioning of genes and chromosomes. Overall, our results highlight the benefits of multimodal data integration for genome structure analysis, available through the Integrative Genome Modeling software package.
Similar content being viewed by others


Main
The spatial organization of eukaryotic genomes plays crucial roles in regulation of transcription, replication and cell differentiation, while malfunctions in chromatin structure is linked to disease, including cancer and premature aging disorders1,2. Advances in chromosome conformation capture (3C)-based3,4,5,6,7,8,9,10 and ligation-free methods11,12,13 and, most recently, live-cell and super-resolution microscopy14,15,16,17,18, have shed light onto key elements of genome structure organization, including the genome-wide detection of chromatin loops19,20, topologically associating domains (TADs)21 that modulate long-range promoter–enhancer interactions12,22 as well as the segregation of chromatin into nuclear compartments8,10,23,24,25,26. Each technology probes different aspects of genome architecture at different resolutions1,27,28,29.
These complementary methods provide a renewed opportunity to generate quantitative, highly predictive structural models of the entire nuclear organization30. Embedding data into three-dimensional (3D) structures is beneficial for a variety of reasons. First, all data itself originate from (often a large population of) 3D structures; so, reverse engineering that data and relating it back to an ensemble of representative 3D structures appears to be the natural way for integrating data from complementary methods via an appropriate representation of experimental errors and uncertainties. Second, generating structures consistent with multimodal data from heterogeneous and independent sources allows cross-validation of orthogonal data itself. Finally, 3D structures give access to features that are not immediately visible in the original input dataset, which can be compared with experimental data tailored to assess model predictivity. Yet, embedding data into 3D structures is a challenging task: not only is there no established protocol for data interpretation and modeling, but genome structures are dynamic in nature and can substantially vary between individual cells. A probabilistic description is thus needed surpassing traditional structural modeling that limits to a single equilibrium structure, or a small number of metastable structures.
There are several data-driven and mechanistic modeling strategies, which differ in the functional interpretation of data and sampling strategies, for generating an ensemble of 3D genome structures statistically consistent with it23,25,26,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50. These 3D structures are then examined to derive structure–function correlations and make quantitative predictions about structural features of genomic regions, study their cell-to-cell variabilities and link these to functional observations. Most strategies have relied primarily on Hi-C data, which is abundant and straightforward to interpret in terms of chromatin contacts. However, data from a single experimental method cannot possibly capture all aspects of the spatial genome organization. Integrating data from a wide range of technologies, each with complementary strengths and limitations, will likely increase accuracy and coverage of genome structure models. Several methods were adapted to combine Hi-C with one other data source14,37,39,49,51,52; nevertheless, developing hybrid methods that can systematically integrate data from many different technologies to generate structural maps of entire diploid genomes remains a major challenge.
Here we present a population-based deconvolution method that provides a probabilistic framework for comprehensive and multimodal data integration. Our approach30,36,44 de-multiplexes ensemble data into a population of 3D structures, each governed by a unique pseudo-energy function, representing a subset of the data, hence explicitly factoring in the heterogeneity of structural features across different cells. The method produces highly predictive models of the folded states of complete diploid genomes, which are statistically consistent with all input data, and is therefore distinct from resampling methods32,34,41,45,46.
Our generalized framework generates fully diploid genome models from integration of four orthogonal data types: ensemble Hi-C10, lamin B1 DamID24,53,54, large-scale HIPMap 3D fluorescence in situ hybridization (FISH) imaging55,56 and data from single-cell split-pool recognition of interactions by tag extension (SPRITE) experiments11. Such models are capable of successfully predicting with good accuracy orthogonal experimental data from a variety of other genomics-based and super-resolution imaging experiments, such as data from SON TSA-seq experiments57 and DNA-MERFISH imaging17. Specifically, our structures predict with good accuracy gene distances to nuclear speckles, gene distances to the nuclear lamina and therefore allow an in-depth analysis of the nuclear microenvironment of genes at a genome-wide scale.
We further demonstrate that integration of all data modalities produces structures of maximal accuracy and show that different combinations of data types can lead to structures of comparable accuracy. For a given available data type, we can therefore propose which additional data types would maximize the prediction accuracy of the resulting structures. Also, our results highlight that relatively low-frequency interchromosomal contacts are essential to correctly predict whole-genome structure organizations: indeed, a modified Hi-C dataset with artificially underrepresented interchromosomal contacts severely fails at reproducing the correct global genome architecture. However, integrating additional data sources from other experiments can compensate for these biases and generate structure populations with still high predictivity accuracy. Our method is potentially applicable to other cell types and organisms, with different combinations of data as described here.
Our work represents the effort at integrating orthogonal data types from Hi-C, lamina DamID, 3D HIPMap FISH and DNA SPRITE experiments to produce highly predictive genome structure populations, which ultimately showcases the benefits of multimodal data integration in the context of whole-genome modeling. Due to its modular architecture, the method we propose can be easily adapted to incorporate other data types in the modeling pipeline, as we strive for even more realistic and predictive structures to dissect the genome structure–function relationship.
Results
Multimodal data-driven population modeling as an optimization problem
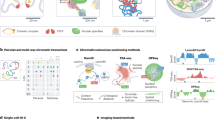
We expand our previous genome modeling framework36,37,44 and introduce a generalized formulation for the integration of a variety of orthogonal data to generate a population of full genome structures that simultaneously recapitulate all the data. Our method incorporates data types that relate to single genomic regions, such as lamin B1 DamID or radial 3D HIPMap FISH, to two genomic regions, such as Hi-C or pairwise 3D HIPMap FISH and several genomic regions, such as single-cell SPRITE experiments (Fig. 1). Our method incorporates both ensemble and single-cell data by deconvoluting ensemble data into a population of distinct single-cell genome structures, which cumulatively recapitulate all input information. Our model is defined as a population of S diploid genome structures \(X=\left\{ {\boldsymbol{X}}_1, {\boldsymbol{X}}_2, \ldots, {\boldsymbol{X}}_S \right\}\), where each structure Xs is represented by a set of 3D vectors representing the coordinates of all diploid chromatin regions. Given a collection of input data \({{{\mathcal{D}}}}_k\) from K different data sources, \({\frak{D}} = \left\{ {{{{\mathcal{D}}}}_k|k = 1, \ldots ,K} \right\}\), we aim to estimate the structure population \({{{\hat{\boldsymbol X}}}}\) such that the likelihood \(P({\frak{D}}|{{{\boldsymbol{X}}}})\) is maximized. Because most experiments, such as Hi-C and lamina DamID, provide data that are averaged over a large population of cells, and often produce unphased data, they do not reveal which contacts coexist in which structure of the population or between which homologous chromosome copies. To represent this missing information at single-cell and diploid levels, we introduce data indicator tensors \({{{\mathcal{D}}}}_k^ \ast\) for each of the data sources \({\frak{D}}^ \ast = \left\{ {{{{\mathcal{D}}}}_k^ \ast |k = 1, \ldots ,K} \right\}\) as latent variables that augment all missing information in \({{{\mathcal{D}}}}_k\) (Methods and Supplementary Table 1). Thus, the latent variables \({\frak{D}}^ \ast\) are a detailed expansion of \({\frak{D}}\) at the diploid and single-structure representation. To determine a population of genome structures consistent with all experimental data, we therefore formulate a so-called hard expectation–maximization (EM) problem, where we jointly optimize all genome structure coordinates X and all latent variables.

Top, schematic of the data-driven modeling approach. Information provided by orthogonal data modalities (Hi-C, lamina DamID, radial and pairwise HIPMap 3D FISH and DNA SPRITE) is used as input to the Integrative Genome Modeling (IGM) platform to generate a population of S = 1,000 diploid genome structures. Structures can be used to predict locations of nuclear bodies and compartments (nuclear speckles and lamina compartment), which can serve as reference points to describe locations of genes and the genome architecture. Bottom, the predicted genome structure population gives access to a large number of structural features (left), which collectively describe the nuclear microenvironment of genes on a genome-wide scale.
The solution of such a high-dimensional maximum likelihood problem requires extensive exploration of the space of all genome structure populations, which we achieve by using a series of optimization strategies for efficient and scalable model estimation (Methods, Supplementary Information and Extended Data Fig. 1)36,37,44. Convergence to an optimal solution \(({{{\hat{\boldsymbol X}}}},\hat {\frak{D}}^ \ast )\) is reached when the models statistically reproduce all the input data (details of the mathematical formulation of data types, likelihood P and optimization strategy are provided in the Methods and Supplementary Information). The optimized structure population X̂ is then used to determine locations of nuclear bodies in each single-cell model, which in turn serve as reference points to calculate a host of structural features. These features allow a thorough characterization of the nuclear microenvironment of each gene30 (Fig. 1).
Comprehensive data-driven genome population structures of HFFc6 cell line
To showcase our data integration platform, we generated a population of 1,000 3D diploid genome structures of prolate ellipsoidal HFFc6 fibroblast cell nuclei (Extended Data Fig. 2a) at 200,000 base-pair resolution by integrating data from in situ Hi-C58, lamin B1 DamID59, HIPMap large-scale 3D FISH imaging55 and DNA SPRITE experiments11 (see Extended Data Fig. 2b–d for details of the optimization statistics). These structures are statistically consistent with all input data: (i) genome-wide Hi-C contact probabilities (genome-wide Pearson correlation: 0.98, average intra-chromosomal Pearson correlation: 0.98, average intra-chromosomal stratum-adjusted correlation coefficient60: 0.89; Fig. 2a,b and Supplementary Table 3); (ii) chromatin contact probabilities to the nuclear envelope (NE) from lamin B1 DamID experiments (Pearson correlation of 0.93; Fig. 2c,d); (iii) pairwise distance distributions for 51 pairs of loci from 3D HIPMap experiments (Pearson correlation of 1.0 of cross-Wasserstein distances Fig. 2e,f); and (iv) chromatin colocalizations for more than 6,600 chromatin clusters from SPRITE experiments (Fig. 2g and Extended Data Fig. 2d). Agreement between input experiments and predictions from optimized structures was further validated by χ2 goodness-of-fit tests (Methods and Extended Data Fig. 3).

a, Genome-wide correlation of Hi-C contact frequencies (interchromosomal and intra-chromosomal) between experiment58 and simulation (r = 0.98). b, Comparison between experimental (upper diagonal region) and simulated (lower diagonal region) contact frequency maps for chromosome 2 (left) and zoomed-in region (right). c,d, Correlation of lamin B1 DamID-derived contact probabilities between experiment59 and model genome wide (c) (r = 0.93) and visual comparison of both signals for chromosome 2 (d). e, Correlation of cross-Wasserstein distance (WD) between experimental FISH data and predictions (r = 1.00; Methods). f, Cumulative distributions of pairwise FISH distances for the set of 51 pairs of loci measured in 3D HIPMap FISH experiments55, plotted for both model (left) and experiment (right). Colors indicate the sequence separation in the chromosome between imaged loci pairs, with darker hues indicating larger sequence separations. g, Examples of single-cell SPRITE clusters from three different structures, showing colocalization of loci in a single-cell structure. Colors distinguish chromosomes, and homologs are shown in the same color. Loci in the same SPRITE cluster are also shown enlarged; left cluster: chr2: 150,927,500, chr3: 6,265,500, chr6: 93,928,500, chr10: 11,602,500; center cluster: chr2: 4,872,500, chr5: 23,208,500, chr11: 57,966,500, chr19: 51,314,500, chr22: 42,294,500; right cluster: chr4: 42,821,500, chr5: 68,438,500, chr6: 106,123,500, chr8: 85,891,500, chr12: 99,185,500. Clusters assayed experimentally11, including those shown, are reproduced in our structures.
To evaluate the predictive value of our models, we must assess how well they predict independent experimental data, which were not used as input information. We first compared our chromosome structures with those from multiplex FISH imaging in a related IMR90 cell type17. Individual chromosome structures from DNA-MERFISH imaging17 show large structural variability, with distinctly different folding patterns between single-cell and homologous copies (Fig. 3a and Extended Data Fig. 4). We found good agreement between chromosome structures from our calculations and experiment (Methods), with several single-cell chromosome conformations found in our models with very similar distance matrix patterns. The range of conformational variability for chromosome 6 and chromosome 2 is nicely matched in our models for selected structures, as shown by the similarities for a range of distance matrices from the experiment and models (see Extended Data Fig. 4 for a more comprehensive comparison). For example, 72% of chromosome 6 structures in our models match to a structure from DNA-MERFISH experiments with an average distance matrix correlation of at least 0.5 or larger.

a, Comparison of high scoring simulated structures of chromosome 6 and structures from the DNA-MERFISH dataset17. Each structure is plotted to the right of its normalized distance matrix; each row shows the corresponding structures in DNA-MERFISH experiments (top row) and IGM HDSF models (bottom row). Modeled structures at 200-kb base-pair resolution have a higher genomic coverage than the imaged genome structures. Genomic regions imaged in the experiment are shown in the models with opaque beads and are connected by opaque links, while genomic regions not imaged in the experiment are shown with translucent beads. b, Correlation between experimental61 and predicted SON TSA-seq data. c, Correlation between predicted and experimental SAF from DNA-MERFISH imaging; d, Experimentally observed correlation between SAF and trans A/B ratio from DNA-MERFISH imaging is nicely reproduced in our genome structures with high correlation. e, Correlation between experimental LAF from DNA-MERFISH imaging with predictions from our genome structure population. All scatterplots are colored according to the local density of points, and the Pearson correlation scores are annotated. TSA-seq correlations are genome wide, and DNA-MERFISH data correlations involve the 1,041 loci studied in the experiment by Su et al.17. f, Predicted SAFs of genomic regions show significant correlations (r = 0.49, P value ~ 0) with the transcription frequency (TRF) of genes from RNA-MERFISH imaging. TRF values are plotted in deciles. Error bars show mean values and standard deviations of predicted SAFs in each TRF range; number of SAF values used in the statistics (from left to right): 704, 79, 66, 61, 41, 27, 11, 10. g, Box plots of the cell-to-cell variability of radial positions for genomic regions containing actively transcribed genes with the 10% highest (T10) and 10% lowest (B10) transcription levels. h, Average radial positions of genomic regions containing actively transcribed genes with the 10% highest (T10) and 10% lowest (B10) transcription levels. Transcription levels for both g and h were taken from total RNA-sequencing experiments62. Comparison between T10 and B10 was performed using the Mann–Whitney two-sided tests, with P values ~ 0 for both g and h; the asterisks represent statistical significance of 0.0001. The box bounds indicate the interquartile range (Q3 − Q1) divided by the median, and Tukey-style whiskers extend to a maximum of 1.5 times the interquartile range beyond the box. Q3 and Q1 are the third and first quartiles of the distributions, respectively. Outliers are represented as dots. The number of B10 and T10 genomic regions used for the box plots is 1,253 and 1,296, respectively. i, Pearson correlation between experimental speckle distance cell-to-cell variability (Methods) from DNA-MERFISH imaging with predictions from our genome structure population, (r = 0.58, P value ~ 0). Error bars show standard deviations of speckle distance variability values in our models, in each experimental value decile. Number of values used in the statistics (from left to right): 99, 97, 103, 101, 94, 105, 100, 98, 101 and 101.
Next, we predicted the locations of nuclear speckles in each single-cell structure, following a previously described procedure30 (Methods). Based on the chromatin structural features, we first identified those chromatin regions with high propensity to be associated with nuclear speckles. We then determined in each model the highly connected spatial partitions formed by these chromatin regions. As we previously discovered, the geometric centers of each partition in a model serve as excellent approximations of nuclear speckle locations30.
The locations of predicted speckles together with the folded genome models were then used to predict experimental SON TSA-seq data (Methods and Fig. 1). SON TSA-seq is an experimental mapping method that determines, on a genome-wide scale, the median distances between any chromatin region and nuclear speckles57. Predicted SON TSA-seq data from our models agree remarkably well with experimental data61 (Pearson correlation 0.83; Fig. 3b). Moreover, our models confirm the previously described relationship between a chromatin region’s experimental SON TSA-seq value and its mean distance to the nearest speckle57.
We then used the predicted speckle locations to determine a gene’s speckle association frequency (SAF), defined as the fraction of models in which a chromatin region is in spatial association to a speckle (Methods and Fig. 1). A recent super-resolution microscopy study detected the same quantity for approximately 1,000 loci by DNA-MERFISH imaging17. The SAF prediction for these loci from our models shows excellent agreement with the experiments (Pearson correlation 0.71; Fig. 3c).
Moreover, we predicted for each chromatin region the median trans A/B ratio (Methods), defined as the ratio of A and B compartment chromatin forming interchromosomal interactions with the target loci. Predicted trans A/B ratios show good agreement with those determined by DNA-MERFISH experiments (Pearson correlation 0.66) and a strong correlation with the SAF (Pearson correlation 0.92; Fig. 3d), again confirming previous findings17,30.
The lamina-associated repressive chromatin compartment is usually located at the NE; thus, we used the location of the NE as a reference point to simulate lamin B1 TSA-seq data (Methods), which measures the mean distances of genomic regions to the nuclear lamina57. Moreover, we also calculated the lamina association frequency (LAF) for each genomic region (Fig. 1), which also shows excellent agreement with the LAF determined by super-resolution DNA-MERFISH imaging17 (Pearson correlation 0.84 for LAF; Fig. 3e). We also observed an inverse correlation between LAF and SAF (Pearson −0.77), confirming previous experimental observations.
Overall, the accurate prediction of orthogonal observables assayed in independent experiments highlights the predictive power of our genome structures. We therefore can describe the nuclear microenvironment of each chromatin region by several structural features calculated from the models (Fig. 1 and Methods), namely: a chromatin region’s average radial position in the nucleus, the variability of its radial positions between single cells, the interior localization probability of a genomic region, the interchromosomal contact probability, the average local chromatin decompaction of the chromatin fiber and its variability across the population of models. Together with predicted SAF, LAF, trans A/B ratio and SON TSA-seq (Methods), we characterized each chromatin region by a total of 13 structural features, which define the structural microenvironment of each genomic region in the nucleus (Fig. 1). All structural features and chromosome structures are highly reproducible in independent replicate optimizations (Methods and Extended Data Fig. 5). For example, 80% of all structures of chromosome 6 in two replicate populations show almost identical structures with a correlation of at least 0.8 or larger between their corresponding distance matrices.
Studying the nuclear microenvironment of genomic regions (even at 200-kb resolution) provides useful information about the role of nuclear positions in gene function, information that is not otherwise easily accessible. For instance, we analyzed the link between a genomic region’s structural environment, in particular its nuclear location, with its gene expression propensity. We observed a significant correlation (Pearson 0.46, P value ~ 0) between the fraction of models a genomic region is in direct proximity to a nuclear speckle (SAF) and the fraction of single cells that show nascent mRNA transcripts for the corresponding genes in RNA-MERFISH experiments17; that is, its transcription frequency (TRF; Fig. 3f). This observation points to a favorable transcriptional microenvironment in the vicinity of nuclear speckles, and thus, confirms previous observations that point to a role of nuclear speckles in gene expression11,57.
We can then relate cell-to-cell variabilities of these features to functional properties. We observed a connection between the cell-to-cell variability of a genomic region’s nuclear position (Methods) with the expression level of genes located in these regions30. For instance, genomic regions containing the top 10% most highly transcribed genes showed substantially lower structural variability than regions containing the bottom 10% of transcribed genes (Fig. 3g; Mann–Whitney two-sided test, P value ~ 0, transcription data from RNA sequencing62). Thus, the most highly transcribed genes are located in genomic regions with the most stable nuclear structure. These regions also showed notably lower (more interior) average radial positions than genes present at low expression levels (Fig. 3h). We also found a significant correlation (Pearson 0.58, P value ~ 0) between our predicted cell-to-cell variability of a genomic region’s distance to the nearest speckle with that observed in DNA-MERFISH experiments (Fig. 3i).
Thus, structural features about nuclear locations of genomic regions can be directly linked to their functional potential in gene transcription. None of these structure-based findings would be possible through analysis of the input data alone.
Multimodal data integration improves predictive power
We next investigated how different combinations of data influence model accuracy. We generated four genome populations, each with different combinations of experimental data, and assessed their accuracy by comparing predicted SON TSA-seq data, lamina DamID data, SAF, LAF and median trans A/B ratios with those available from experiments (Methods and Fig. 4). For reference, we also assessed a population of random chromosome territories constrained within the nuclear volume.

Top, Model accuracy for five different genome structure populations generated from different combinations of experimental input datasets: random chromosome territory (rand), Hi-C only (H), Hi-C + lamina DamID data (HD), Hi-C + lamina DamID + SPRITE (HDS) and Hi-C + lamina DamID + SPRITE + FISH (HDSF). The first and second plots show genome-wide Pearson correlation coefficients between model predictions and experimental data for experimental SON TSA-seq data and lamin B1 DamID. The third to fifth plots show Pearson correlations between experimental and predicted data for LAF, SAF and trans A/B ratio for 1,041 imaged loci from DNA-MERFISH imaging experiments17. Error bars were computed as the standard deviation of the Pearson correlation across three independent population replicates (Methods). Data are presented as mean values ± standard deviation. Bottom plots show the comparison between experimental61 and predicted SON TSA-seq profiles of chromosomes 2 (top and bottom, respectively). Predicted profiles are shown for structure populations generated with setups H and HDSF (Methods).
Interestingly, models from Hi-C data alone (setup H) reproduce SON TSA-seq data and SAF already with high accuracy, while lamin B1 DamID and LAF show relatively poor performance (Fig. 4), which is likely related to the flat ellipsoidal shape of the HFF nucleus. Our previous studies using GM12878 cells, with a spherical nucleus, could predict both lamina TSA-seq and lamin B1 DamID data with higher accuracy from Hi-C data alone30. When Hi-C and Lamina DamID data (setup HD) were combined, predictions of TSA-seq, DamID data, SAF and LAF greatly improve (Fig. 4).
Combining SPRITE colocalization clusters and 3D FISH distance distributions with Hi-C and lamin B1 DamID, input information slightly improved correlation scores for TSA-seq and DamID data, even though the total number of spatial restraints from DNA SPRITE and FISH data were an order of magnitude smaller than those from Hi-C and lamina DamID (Extended Data Fig. 2d). Models HDS and HDSF recapitulated MERFISH imaging data well, recapitulated 3D FISH and SPRITE data, while also showing excellent predictability for TSA-seq and DamID data (Fig. 4 and Extended Data Fig. 6). Overall, the steady improvement of model accuracy with an increasing amount of input data highlights the benefits of multimodal over unimodal data integration in generating realistic and highly predictive structures.
Systematic assessment of comprehensive data integration using synthetic data
To perform a thorough assessment of multimodal data integration, we regarded a structural population as a ‘ground truth’ reference, from which a variety of synthetic data can be simulated (Methods and Fig. 5a). Models were then generated from different combinations of synthetic data, to facilitate the comparison of their predictive power on 3D genome architecture. Note that model assessment depends on the structural features being explored, and a ground truth allows a more comprehensive model validation based on a larger number of structural observables that are accessible. Moreover, we can simulate different input data at variable levels of information content to better assess their influence on model quality.

a, Schematic of the assessment process. Information corresponding to Hi-C, lamina DamID and radial and pairwise FISH data was simulated from a structure population that serves as a reference ground truth. Eight different genome structure populations were calculated from different combinations of synthetic data. Independent structural features were calculated from each population and compared with the ground truth reference to assess the accuracy of the models. b, Combinations of synthetic data included in the eight different input setups (columns). Gray boxes indicate the presence of a synthetic data type in the input setup. Hi-C* and Hi-C** indicate two differently perturbed Hi-C maps. In Hi-C* only, interchromosomal contact frequencies were scaled down by a factor of 2. In Hi-C** only, intra-chromosomal contact frequencies were scaled down by a factor of 2. c, Accuracy of models was estimated for each input setup using the Pearson correlations between predicted structural features and those in the ground truth reference. Structural features included SON and lamin B1 TSA-seq data, lamin B1 DamID data, the radius of gyration for chromosomes, distributions of chromatin radial positions, distributions of intra-chromosomal distances, and distributions of inter-chromosomal distances. Baseline predictions from the correct (non-perturbed) Hi-C-only simulation are indicated with a red horizontal line. d, OPRs for all setups. The OPR for setup s was calculated as follows: \(\mathrm{OPR}_s = \mathop {\sum}\nolimits_{f = 1}^7 {(9 - \mathrm{rank}_f^s)}\), where rankfs is the rank of setup s in assessment of feature f. rankfs is 1 for the top-ranking setup, and 8 for the poorest performing setup for feature f. Therefore, OPRs can range from 56 (best performance in all feature assessments) to 8 (poorest performance in all feature assessments). Error bars in b–d for each setup were estimated from three independent population replicates (Methods). Data are presented as mean values ± standard deviation.
We chose population H (Fig. 4) as the ground truth structure population, from which we generated the synthetic datasets, including genome-wide contact frequencies (that is, Hi-C data), contact frequencies between loci and the NE (that is, lamin B1 DamID data), and a randomly chosen subset of 1,000 radial and 1,000 pairwise distance distributions (that is, HIPMap 3D FISH datasets; Methods and Fig. 5a). These datasets represent idealized data sources, and were combined into seven different input data setups. Models were then generated for all data setups, each containing different combinations of synthetic data (Fig. 5b).
We quantitatively assessed model accuracy with the following structural properties (Fig. 5c): (i) the distribution of radial positions for each chromatin region, (ii) the distributions of pairwise distances between chromatin loci in cis and trans; (iii) the distribution of the radius of gyration for each chromosome; (iv) SON TSA-seq data; (v) lamin B1 TSA-seq data; and (vi) lamin B1 DamID data. We used the cross-Wasserstein distance to measure the similarity between two probability distributions (for features i–iii); quantities (iv–vi) were assessed by their Pearson correlations with the corresponding ground truth features (Methods). Finally, for each setup, an overall performance rank (OPR) was determined as the total sum of ranks for all individual feature assessments (Fig. 5d).
Models generated from simulated contact frequencies naturally reproduce with high accuracy the ground truth features. To better substantiate our assessment of data integration performance, we manipulated the simulated Hi-C data by scaling down the interchromosomal contact probabilities by a factor of two and used the resulting ‘perturbed’ contact map (labelled Hi-C*) as input for all model populations instead.
Structures generated from perturbed Hi-C* data alone (setup 2) showed poor performance with low correlations of ground truth features, except for intra-chromosomal distance distributions (Pearson correlation 0.79; Fig. 5c). We then generated another perturbed Hi-C** dataset, in which interchromosomal interactions remain untouched, while probabilities of intra-chromosomal interactions were scaled down by a factor of 2 (setup 8). Models generated using this dataset predicted with good accuracy all ground truth features related to the global nuclear architecture, such as SON TSA-seq, lamin B1 TSA-seq and lamina DamID signals (Pearson correlations > 0.98) as well as radial distributions of chromatin regions with substantially higher accuracy than setup 2 Hi-C* (Fig. 5c). In contrast, setup 8 showed slightly higher accuracy than setup 2 for chromosomal properties, such as the radius of gyration. It is noteworthy that intra-chromosomal distance distributions were still well reproduced in comparison to setup 2, which indicates that scaling down intra-chromosomal contacts has a less detrimental effect than interchromosomal contacts. These results showcase the surprisingly dramatic loss of information when trans contact probabilities are underestimated in Hi-C data, which generally have very low contact probabilities to begin with. Reducing interchromosomal interactions further will lead to the loss of information about the global genome architecture. Reducing relatively high-frequency intra-chromosomal contact probabilities will have a smaller impact, as sufficient information about intra-chromosomal chromatin interactions is still retained in the dataset.
To further assess the relevance of interchromosomal interactions, we generated four structure populations from (unperturbed) Hi-C data that included interchromosomal contacts only if their contact probability was larger than a given cutoff θinter, which is gradually decreased (Methods). Interestingly, good predictive models can only be generated when interchromosomal contacts with very low probabilities are included (Fig. 6). For instance, radial profiles are only reproduced with low residual errors if relatively ‘rare’ contact events are included, that is, probabilities corresponding to only 2 contact events per 1,000 structures (Fig. 6a). The chromatin compartmentalization score, which measures the spatial segregation between chromatin in the active A compartment from the inactive B compartment63 (Methods), also steadily increased when interchromosomal contacts with low contact probabilities were added (Fig. 6b). Thus, the large number of low-probability interchromosomal interactions, which define relatively ‘rare’ contact events per chromatin region, are essential for accurate genome structure modeling and for correct predictions of genome-wide SON TSA-seq, lamin B1 TSA-seq and lamin B1 DamID data (Fig. 6c). Overall, these results further underline the important role of trans interactions in predicting the correct global genome architecture in our models. Hi-C experimental conditions can influence fragment lengths, ligation efficiencies and thus the amount of informative interchromosomal proximity information captured by ligations. Hi-C variants, such as MicroC6, capture local short-range chromatin interactions at higher resolution, while the fraction of long-range and interchromosomal interactions is reduced. It is therefore of interest to test if additional orthogonal data sources can compensate for reduced levels of informative interchromosomal interactions.

We compared the accuracy of a structure population generated from unperturbed Hi-C data as a function of the lowest interchromosomal contact probability value included in the modeling. The probabilities are labeled as θinter (Methods). a, Mean radial positions plotted for chromatin regions in chromosome 1 from structures in the ground truth reference population (dark blue) and structures calculated from three representative setups (red) that included interchromosomal contacts with gradually decreasing contact probabilities: θinter = 0.02, 0.008 and 0.002. Characteristic radial profiles seen in the ground truth (Fig. 1) were only correctly reproduced when contacts were included with probabilities of at least 0.2%. From left to right, θintra = 0.02, 0.008 and 0.002. b, The A/B compartmentalization score for each setup, with error bars representing the standard deviation of the underlying distribution (Methods): compartmentalization increased as more low-frequency interchromosomal contacts were included in the modeling. c, The Pearson correlation value between the ground truth and simulations of the same seven structural observables discussed in Fig. 5 for θinter = 0.020, 0.015, 0.008 and 0.002. Gray boxes indicate negative correlation values. Structural quantities experienced a substantial correlation increase when low-probability contacts were included, indicating that overall model predictivity increases dramatically. Error bars for each setup were estimated from three independent replicate calculations (Methods); data in b and c are presented as mean values ± standard deviation.
Combining lamin B1 DamID as well as radial and pairwise distance distributions from 3D FISH experiments with the biased Hi-C* data (setup 7) produced models with high predictive power and similar accuracy for all structural features as models generated with unmodified original Hi-C data (Fig. 5c). The OPR increased monotonically with increasing amounts of added data (setups 3–7; Fig. 5d). Therefore, orthogonal data modalities appear to compensate for systematic errors affecting one of the data types (here, underrepresentation of interchromosomal contacts; Extended Data Fig. 7).
The steady improvement in model accuracy with increasing data is not only due to those features being directly restrained by the added data (which is only a small portion of all degrees of freedom), but also due to cooperative effects acting on the entire genome: each newly added data modality makes already included data more informative. This is due to the specific nature of our iterative optimization process, which reduces data ambiguity by selecting the best of a set of alternative restraints assignments, based on the current genome structures at a given iteration (Methods and Supplementary Information). For instance, if newly added information about a gene’s radial position restricts its nuclear locations, it will also make certain non-native chromatin contacts less likely, which in turn will lower the change for that gene to be wrongly selected in non-native Hi-C contact-restraint assignments. An analogy is a crossword puzzle, where gradually filling in interconnected words reduces the ambiguity of missing word solutions. Adding a data modality to our modeling process reduces, in a similar way, the ambiguity of restraints assignments of all other data types, thus making these data more informative.
Our simulations showed that adding FISH radial distributions for 1,000 loci (setup 2 to setup 3) improved prediction accuracy of radial distributions for all genes (not only those being actively restrained), as well as genome-wide SON and lamin B1 TSA-seq signals, and even interchromosomal gene distance distributions, although the radial FISH data did not contain any bivariate information (Fig. 5c).
Models generated from Hi-C* and simulated DamID data (setup 5) outperformed models from Hi-C* data and FISH radial distributions of 1,000 loci (setup 3). However, adding information for 1,000 pairwise FISH distance distributions (setup 4) produced models as accurate as those in setup 5.
The information equivalence of datasets depends naturally on the amount of data. For instance, using radial distributions of all chromatin loci would render lamina DamID data redundant. We therefore assessed (Hi-C* + radial FISH data) class models that contain increasing numbers of FISH probes. Our results confirm that, at a critical number of probes, models from Hi-C* and radial FISH data become more informative than those from Hi-C* and lamina DamID data (setup 5; Extended Data Fig. 8). Of course, these observations are made in an idealized case, and only serve as a conceptual point. The true information content of data depends on systematic errors in the experimental data, such as potential distortions due to cell fixations and other treatments in FISH experiments, as well as the base-pair resolution of the chromatin fiber representation. Also, radial positions (instead of distance to the nuclear lamina) may be an inadequate description for highly irregular nuclear shapes that vary in size. In future, actual microcopy 3D images, instead of positional metadata, should be used in the modeling process to overcome some of these issues.
Discussion
We introduced a robust pipeline for multimodal data integration to determine 3D structures of whole diploid genomes. These structures revealed a wealth of information about the structural organization of genomes over multiple length scales, along with dynamic variabilities of structural features between individual cells. Collectively these features define the nuclear microenvironment of genes on a genome-wide scale, which can be directly linked to their functional potential in gene transcription and subnuclear compartmentalization43. Our method therefore provides a useful analytical tool for comparative genome structure analysis, which could link changes in a gene’s structural organization between different cell types (or during developmental processes) with underlying functional changes. Moreover, the structures generated by our method also predict a host of orthogonal experimental data, including SON TSA-seq data, speckle and lamina association frequencies and trans A/B ratios as determined by DNA-MERFISH experiments, and reproduce chromosomal structures from super-resolution imaging experiments. These predictions could serve as first approximations to data otherwise only available through experiments with considerable added effort.
We tested the proficiency of our approach by studying the diploid genome structures of human HFFc6 cells by integrating data from Hi-C, lamin B1 DamID, 3D HIPMap FISH and SPRITE experiments. We systematically assessed the accuracy of models generated from different combinations and amount of data types. Model accuracy steadily improves with increasing amounts of data and is maximal when data integration is multimodal, indicating that single data sources might not fully capture all information about a genome’s structural organization. Moreover, orthogonal data sources can compensate for systematic biases and missing information in some data types. For instance, a biased Hi-C dataset with artificially reduced chromatin interaction frequencies shows substantially lowered accuracy. However, combining this biased dataset with additional information from lamina DamID and 3D FISH experiments recovers structures with almost identical accuracy to those generated by the unbiased Hi-C data. The improvement of performance can partly be explained by cooperative effects. Adding a complementary data type to the input set can reduce ambiguity in other data, thus making already included data more informative.
Also, different combinations of orthogonal data sources can produce models with similar levels of high accuracy and thus share similar information content. For instance, the combination of Hi-C with lamina DamID data can produce similarly accurate structures than a combination of data from Hi-C and 3D FISH experiments, given that a critical number of FISH probes is considered. Therefore, the method does not rely on a specific combination of data to produce models with high predictive values.
Interestingly, our work also underlines the essential role of low-probability interchromosomal interactions for accurate data-driven predictions of genome organizations. The multitude of relatively ‘rare’ contact events are crucial for accurate predictions of radial gene positions and overall chromatin compartmentalization. It is not sufficient to consider only the most frequent interactions in the modeling process. However, if datasets are compromised by a lack of sufficient information about trans interactions, additional orthogonal data sources can compensate for a reduced level of information.
In future, our approach will be expanded to incorporate 3D imaging data into the modeling process also, which will consider variations in nuclear shapes between individual cells and exclude volumes for some nuclear bodies. We expect that these additions will further improve the quality of models. Due to its modular organization, our software platform is readily suited for incorporating new volumetric microscopy data
In summary, here we showed that our method provides a useful tool for multimodal data integration to produce genome structure models with high predictability. Our software implementation is publicly available, widely applicable to other cell types and can be tailored to include new experimental data types.
Methods
Our population-based modeling approach uses a probabilistic framework to generate a large number of 3D genome structures (that is, the structure population) statistically consistent with all input data (that is, Hi-C, lamin B1 DamID, 3D FISH and SPRITE). Structures are generated by a deconvolution of ensemble data (Hi-C, lamin DamID and 3D FISH) and incorporation of single-cell data (SPRITE) into a population of individual diploid genome structures that represent the most likely approximation of the true population of genome structures, given all the available data. The structure optimization problem is formulated as a maximum likelihood estimation problem using an iterative optimization scheme.
Genome representation
Chromosomes are segmented into genomic regions of 200-kb DNA sequence length, each represented by chromatin domains with spherical volume. Each chromatin domain is defined by an excluded volume with a sphere radius r0 = 118 nm, which guarantees a 40% volume occupancy of the diploid genome in the nucleus. In a diploid genome, each autosome genomic region has two homologous chromatin domain copies. Overall, the diploid genome is represented by a total of N = 29,838 chromatin domains. The nuclear shape is modeled as a prolate ellipsoid of semiaxes (a, b, c) = (7,840 nm; 6,470 nm; 2,450 nm); Extended Data Fig. 2a). The semiaxes’ lengths are based on the estimates from Seaman et al.64.
Our model, the structure population, is defined as a set of S diploid genome structures X = {X1,…,XS}; a genome structure XS is a set of 3D vectors representing the center coordinates of each chromatin domain \({{{\boldsymbol{X}}}}_s = \{ {{{\vec{\boldsymbol x}}}}_{is}:{{{\vec{\boldsymbol x}}}}_{is} \in {\Bbb R}^3,i = 1,2, \ldots ,N\}\), with N as the total number of all chromatin domains in the diploid genome. The variable H indicates the total number of genomic regions, that is, the number of domains when homologous copies are not distinguished.
Note that capital letter indices, such as I and J, relate to domains without distinguishing between two homologous copies, while lowercase indices i, i’ and j, j’ distinguish between the two copies, when applicable (sex chromosomes only come in one copy).
Data source representation
We integrate data from four experimental methods, namely in situ Hi-C58 and lamin B1 DamID59, high-throughput HIPMap 3D FISH55 and SPRITE11.
Data types are categorized into three classes depending on the number of genomic loci involved. For instance, data that inform on the coordinates of only a single genomic locus will be univariate, such as the radial distance of a locus from radial FISH data or a normal distance to the nuclear lamina from lamina DamID data. Bivariate data inform on pairs of genomic loci, for instance, distances between pairs of loci from 3D FISH experiments or contacts between pairs of loci from Hi-C experiments. Multivariate data define relationships between more than two loci, for example, knowledge about colocalization of a set of loci in single cells from SPRITE experiments.
Most experiments, such as Hi-C and Lamina DamID, provide data that are averaged over a large population of cells, and so they cannot reveal which contacts coexist in which single-cell structure. Moreover, unphased data cannot discriminate between homologous chromosome copies. To represent the missing information at single-cell level and to distinguish homologous chromatin domain copies, we introduce indicator tensors \({\frak{D}}^ \ast = \left\{ {{{{\mathcal{D}}}}_k^ \ast |k = 1, \ldots ,K} \right\} = \{ {{{\boldsymbol{B}}}},{{{\boldsymbol{V}}}},{{{\boldsymbol{F}}}},{{{\boldsymbol{W}}}},{{{\boldsymbol{R}}}}\}\) as latent variables that augment missing information in data variables \({\frak{D}} = \left\{ {{{{\mathcal{D}}}}_k|k = 1, \ldots ,K} \right\} = \{ {{{\boldsymbol{U}}}},{{{\boldsymbol{E}}}},{{{\boldsymbol{M}}}},{{{\boldsymbol{A}}}},{{{\boldsymbol{T}}}}\}\), respectively (Supplementary Table 1).
Chromosome conformation capture
Hi-C data are expressed as a contact probability matrix A = (aIJ)H×H where 0 ≤ aIJ ≤ 1 is the contact probability between the genomic regions I and J44. The contact probability matrix A is incomplete and does not contain the detailed information about which of the homologous domain copies (i and i′ for genomic region I, and j and j' for J) are in contact, nor does it provide information about structures of the population in which a contact is present. To complement every cell’s contact information, we introduce the contact indicator tensor W = (wijs)N×N×S, which is a latent binary-valued third-order tensor specifying the contacts between chromatin domains i and j for each homologous copy in each structure of the population. wijs = 1 indicates that a contact between chromatin loci i and j is present in structure s, while wijs = 0 indicates that such a contact is not present. W is a detailed expansion of A at the diploid representation and single-cell level with a dependence relationship X → W → A.
Lamina DamID
Lamina DamID data are expressed by the tensor E = (eI)H, where 0 ≤ eI ≤ 1 is the probability that genomic region I is in contact with the lamina at the NE, which is derived from lamin B1 DamID data, following a similar notation as used by Li et al.37.
To complement information about homologous domains in single structures, we introduce the binary-valued latent tensor V = (vis)N×S, which indicates whether the i-th chromatin domain is in contact with nuclear lamina in the s-th structure (vis = 1) or not (vis = 0). V is a detailed expansion of E at the diploid representation and single-cell level with a dependence relationship X → V → E.
3D FISH HIPMap
Data from 3D FISH HIPMap experiments are divided into two sets of data: (i) univariate data about the radial positions of genomic loci, and (ii) bivariate data providing information about the distributions of distances between pairs of genomic loci. Large-scale FISH data provide the probability distributions of pairwise distances between genomic loci and probability distributions of radial positions of genomic loci in the nucleus. Probability distributions of both radial and pairwise distances are discretized into Q bins, which equally span the nuclear dimension. For convenience, we can assume bins are disjoint and that any distance can be assigned to only one bin.
3D FISH radial positions
We express radial 3D FISH data with the tensor U = (uIq)H×Q, with H as the number of genomic regions and Q as the total number of distance bins. uIq is the probability that the radial position of genomic locus I falls into the range defined by \({{{\mathcal{B}}}}_q = \left[ {d_q,d_{q + 1}} \right)\), with dq as the lower bound and dq+1 as the upper bound for radial positions in bin q.
To complement missing information about single-cell structures and homologous domain copies, we introduce the binary-valued latent tensor B = (biqs)N×Q×S, which indicates whether the i-th chromatin domain in structure s has a radial position in the range defined by bin \({{{\mathcal{B}}}}_q = \left[ {d_q,d_{q + 1}} \right)(b_{iqs} = 1)\) or not (biqs = 0). B is a detailed expansion of U at the diploid representation and single-cell level with a dependence relationship X → B → U.
3D FISH distance distributions
We express 3D FISH pairwise distance data by the tensor M = (mIJq)H×H×Q, where mIJq is the probability that genomic loci I and J have a distance in the range defined by bin \({{{\mathcal{B}}}}_q = [d_q,d_{q + 1})\). The binary-valued tensor F = (fijqs)N×N×Q×S complements the missing information about homologous domain copies and single cells and thus indicates whether the spatial distance between the i-th and j-th chromatin domains in structure s falls in the range of \({{{\mathcal{B}}}}_q = \left[ {d_q,d_{q + 1}} \right)\) (fijqs = 1) or not (fijqs = 0). F is a detailed expansion of M at the diploid representation and single-cell level with a dependence relationship X → F → M.
SPRITE
The SPRITE data provide information about the number and identity of genomic regions colocalized in a single-cell structure. We expressed these SPRITE clusters by a collection of tensors {Tn} = \(\left(t_{I_1, \ldots , I_n} \right)_{H^n}\), where n is the number of genomic regions in a SPRITE cluster. Each tensor entry \(t_{I_1, \ldots, I_n}\), derived from single-cell SPRITE data is the probability of genomic regions I1,…,In to be colocalized in a single structure of the population \(t_{I_1, \ldots, I_n} = 1\) or not \(t_{I_1, \ldots, I_n} = 0\). All clusters of n regions are described by the multidimensional tensor Tn, and we will use the notation Cn to indicate any of those clusters n genomic loci. Summing all the clusters of any size is indicated then by the notation \(\mathop {\sum}\nolimits_n {{\sum} {C_n} }\).
The latent indicator tensor Rn = \(\left( r_{i_1, \ldots , i_n,s}\right)_{N^n \times S}\), where \(r_{i_1, \ldots , i_n,s}\) distinguishes homologous domain copies, complements the information by indicating whether chromatin domains (different copies are distinguished) {i1,…,in} are colocalized in structure s \(r_{i_1, \ldots , i_n,s} = 1\) or not \(r_{i_1, \ldots , i_n,s} = 0\). Rn is a detailed expansion of Tn at the diploid representation and single-cell level with a dependence relationship X → Rn → Tn
In the following, we will collectively indicate the family of Tn and Rn tensors with T and R, respectively, as T = {Tn} and R = {Rn}.
Probabilistic formulation of maximum likelihood problem
We introduced a set of data variables \(\left\{ {{{{\mathcal{D}}}}_k|k = 1, \ldots 5} \right\} = \{ {{{\boldsymbol{U}}}},{{{\boldsymbol{E}}}},{{{\boldsymbol{M}}}},{{{\boldsymbol{A}}}},{{{\boldsymbol{T}}}}\}\) and a set of indicator tensors \(\left\{ {{{{\mathcal{D}}}}_k^ \ast |k = 1, \ldots ,5} \right\} = \{ {{{\boldsymbol{B}}}},{{{\boldsymbol{V}}}},{{{\boldsymbol{F}}}},{{{\boldsymbol{W}}}},{{{\boldsymbol{R}}}}\}\) as latent variables that augment missing information in data variables to distinguish homologous chromatin domain copies and in single cells. Given \(\left\{ {{{{\mathcal{D}}}}_k} \right\}\), we aimed to estimate the structure population model X such that the likelihood \(P\left( {\left\{ {{{{\mathcal{D}}}}_k} \right\},\left\{ {{{{\mathcal{D}}}}_k^ \ast } \right\}|{{{\boldsymbol{X}}}}} \right) = P\left( {{{{\boldsymbol{U}}}},{{{\boldsymbol{E}}}},{{{\boldsymbol{M}}}},{{{\boldsymbol{A}}}},{{{\boldsymbol{T}}}},{{{\boldsymbol{B}}}},{{{\boldsymbol{V}}}},{{{\boldsymbol{F}}}},{{{\boldsymbol{W}}}},{{{\boldsymbol{R}}}}|{{{\boldsymbol{X}}}}} \right)\) is maximized. The statistical dependence relationship between data sources and latent variables in an optimized structure population is \({{{\boldsymbol{X}}}} \to {{{\mathcal{D}}}}_k^ \ast \to {{{\mathcal{D}}}}_k,\forall k\), because \(\left\{ {{{{\mathcal{D}}}}_k^ \ast } \right\}\) is a detailed expansion of \(\left\{ {{{{\mathcal{D}}}}_k} \right\}\) at the diploid and single-structure representation of the data and X is the structure population consistent with \(\left\{ {{{{\mathcal{D}}}}_k^ \ast } \right\}\). Therefore, the likelihood \(P\left( {\left\{ {{{{\mathcal{D}}}}_k} \right\},\left\{ {{{{\mathcal{D}}}}_k^ \ast } \right\}|{{{\boldsymbol{X}}}}} \right)\) can be expanded to \(P\left( {\left. {\left\{ {{{{\mathcal{D}}}}_k} \right\}|\left\{ {{{{\mathcal{D}}}}_k^ \ast } \right\},{{{\boldsymbol{X}}}}} \right)P\left( {\left\{ {{{{\mathcal{D}}}}_k^ \ast } \right\}|{{{\boldsymbol{X}}}}} \right.} \right)\) and therefore
We assumed, as a first approximation, that \(P\left( {\left. {\left\{ {{{{\mathcal{D}}}}_k} \right\}|\left\{ {{{{\mathcal{D}}}}_k^ \ast } \right\},{{{\boldsymbol{X}}}}} \right)P\left( {\left\{ {{{{\mathcal{D}}}}_k^ \ast } \right\}|{{{\boldsymbol{X}}}}} \right.} \right) = \mathop {\prod}\limits_k P \left( {{{{\mathcal{D}}}}_k|{{{\mathcal{D}}}}_k^ \ast ,{{{\boldsymbol{X}}}}} \right) \cdot \mathop {\prod}\limits_k P ({{{\mathcal{D}}}}_k^ \ast |{{{\boldsymbol{X}}}})\) with k as the data source index, and \({{{\mathcal{D}}}}_k\) and \({{{\mathcal{D}}}}_k^ \ast\) as the data source k (Supplementary Table 1) and its associated latent variable, respectively. Subsequently, the conditional probability function is given according to equation (1):
We aimed to maximize the conditional probability function equation (1): namely, we wanted to find the optimal structures and the optimal latent variables that satisfy:
and thus
In addition to the five data sources from four experimental methods (Supplementary Table 1), we also included a set of spatial constraints based on additional information about the genome organization. These data were included in the form of general spatial constraints acting on N chromatin domains: (i) a nuclear volume confinement restraint that forces all chromatin domains to be inside the nuclear volume, (ii) excluded volume restraints that prevent ‘hard-core’ overlap between any two chromatin domains and (iii) a polymer chain connectivity restraint between chromatin domain neighbors in a chromosome, which guarantees the structural integrity of the chromosomal chains. Additional information about these restraints is available in the Supplementary Information.
In summary, the maximum likelihood problem is formally expressed by equation (2):
Optimization procedure
We adapted our previously developed iterative optimization procedure to solve this maximum likelihood estimation problem for determining a population of genome structures consistent with all data modalities36,37,44. Because there is no closed-form solution to this optimization problem (equation (2)), we developed a variant of the EM method to iteratively optimize local approximations of the log likelihood function37,44,65. We use an iterative solver to alternately optimize the latent variables and model parameters in a sequence of so-called modeling (M) and assignment (A) steps until joint convergence was reached.
-
Initialization step: an initial model estimate X0 is needed to start the first iteration. X0 is generated by using random chromatin domain positions that satisfy the three spatial constraints in equation (2), that is, nuclear volume, excluded volume and chain connectivity. Chromatin regions are randomly placed in a bounding sphere proportional to its chromosome territory size and randomly placed within the nucleus followed by a short optimization to eliminate excluded volume steric clashes in the structures.
Each iteration consists of two steps:
-
(1) Assignment step (A-step): given the current estimated population of genome structures X(t), which resulted from the previous A/M optimization iteration at step t, the optimal latent variables Bt + 1, Vt + 1, Ft + 1, Wt + 1, Rt + 1 are determined by solving the following log likelihood. We use an efficient heuristic strategy to estimate all latent variables (Supplementary Information).
$$\begin{array}{l}{{{\boldsymbol{B}}}}^{t + 1},{{{\boldsymbol{V}}}}^{t + 1},{{{\boldsymbol{F}}}}^{t + 1},{{{\boldsymbol{W}}}}^{t + 1},{{{\boldsymbol{R}}}}^{t + 1} = \arg max_{{{{\boldsymbol{B}}}},{{{\boldsymbol{V}}}},{{{\boldsymbol{F}}}},{{{\boldsymbol{W}}}},{{{\boldsymbol{R}}}}}\\ \log \left[ \begin{array}{l}P\left( {{{{\boldsymbol{U}}}}|{{{\boldsymbol{B}}}},{{{\boldsymbol{X}}}}^t} \right)P\left( {\boldsymbol{E}|{{{\boldsymbol{V}}}},{{{\boldsymbol{X}}}}^t} \right)P\left( {{{{\boldsymbol{M}}}}|{{{\boldsymbol{F}}}},{{{\boldsymbol{X}}}}^t} \right)P\left( {{{{\boldsymbol{A}}}}|{{{\boldsymbol{W}}}},{{{\boldsymbol{X}}}}^{{{\boldsymbol{t}}}}} \right)\\ P\left( {{{{\boldsymbol{T}}}}|{{{\boldsymbol{R}}}},{{{\boldsymbol{X}}}}^t} \right)P\left( {{{{\boldsymbol{B}}}},{{{\boldsymbol{V}}}},{{{\boldsymbol{F}}}},{{{\boldsymbol{W}}}},{{{\boldsymbol{R}}}}|{{{\boldsymbol{X}}}}^t} \right)\end{array} \right]\end{array}$$ -
(2) Modeling step (M-step): given the current latent variables Bt + 1,Vt + 1,Ft + 1,Wt + 1,Rt + 1, determined in the A-step, find the genome structure population Xt + 1 that maximizes the log likelihood of all data. A new structure population Xt + 1 is generated in which data assignments in latent variables will be physically present in the structure population X. Optimization is performed in an efficient parallel platform (Supplementary Information).
$${{{\boldsymbol{X}}}}^{t + 1} = \arg \mathop {{\max }}\limits_{{{\mathbf{x}}}} \log \left[ \begin{array}{l}P\left( {{{{\boldsymbol{U}}}}|{{{\boldsymbol{B}}}}^{t + 1},{{{\boldsymbol{X}}}}} \right)P\left( {\boldsymbol{E}|{{{\boldsymbol{V}}}}^{t + 1},{{{\boldsymbol{X}}}}} \right)P\left( {{{{\boldsymbol{M}}}}|{{{\boldsymbol{F}}}}^{t + 1},{{{\boldsymbol{X}}}}} \right)P\left( {{{{\boldsymbol{A}}}}|{{{\boldsymbol{W}}}}^{t + 1},{{{\boldsymbol{X}}}}} \right)\\ P\left( {{{{\boldsymbol{T}}}}|{{{\boldsymbol{R}}}}^{t + 1},{{{\boldsymbol{X}}}}} \right)P\left( {{{{\boldsymbol{B}}}}^{t + 1},{{{\boldsymbol{V}}}}^{t + 1},{{{\boldsymbol{F}}}}^{t + 1},{{{\boldsymbol{W}}}}^{t + 1},{{{\boldsymbol{R}}}}^{t + 1}|{{{\boldsymbol{X}}}}} \right)\end{array} \right]$$ -
Iterate A/M steps until convergence is reached (see Supplementary Information for convergence criteria). This iterative procedure ensures that all data allocations are re-evaluated using the current structure population.
Stepwise optimization strategy
We used a stepwise optimization strategy to gradually increase the optimization hardness (Extended Data Fig. 1). An initial model that already fits a portion of the data \(\left\{ {{{{\mathcal{D}}}}_k} \right\}\) can guide a more efficient search for the optimum latent variables \(\left\{ {{{{\mathcal{D}}}}_k^\prime } \right\}\) than a random structure population. Thus, gradually fitting an increasing number of data points starting from the highest to the lowest data probabilities (that is, domain contacts and domain distances from Hi-C and DamID data), or starting from largest to lowest distance tolerances (for SPRITE and 3D FISH data; Supplementary Information) will effectively guide the search of the optimal solution. In the initial step, we first calculated a structure population \({\boldsymbol{X}}^{{\mathrm{step}}_{1}}\) that integrates only data with the highest probabilities (for Hi-C and DamID data) and performed several rounds of iterative A/M optimizations until convergence is reached. At each following step, we added further data batches with gradually lower probabilities (for Hi-C and lamina DamID), and decreasing tolerances (for SPRITE and FISH data), and performed iterative rounds of A/M optimizations each time until full convergence for all data was reached (that is, all data are reproduced in the models; Extended Data Fig. 2b,c).
How the data are added to the optimization at each step and at what accuracy is controlled by a sequence of nonzero threshold values, and each data type is associated with its own sequence.
-
θ1≥…≥θfinal indicates the list of gradually decreasing Hi-C probability thresholds, such that the k-th step incorporates only those chromatin contacts in \({{{\boldsymbol{A}}}}_{\theta _k}\) with higher probability than aIJ≥θk, thus \({\boldsymbol{A}}_{\theta_k}=[{\boldsymbol{A}} \ge \uptheta_k]\).
-
λ1≥…≥λfinal indicates the list of gradually decreasing DamID contact probability thresholds, such that the k-th step incorporates those chromatin–NE contacts in \(\mathbf{E}_{\lambda _k}\) with higher probabilities than eI ≥ λk, thus \({\mathbf{E}}_{\lambda_{k}} = {\mathbf{E}}\left[{\mathbf{E}} \ge \lambda_{k}\right]\).
-
t1≥…≥tfinal indicates the list of gradually decreasing FISH distance thresholds, such that the k-th step in the optimization enforces distance values with a tolerance tk. All FISH distances are incorporated from the first optimization steps on, but their tolerances are gradually reduced with the number of optimization steps.
-
ρ1≤…≤ρfinal indicates the SPRITE thresholds, such that the k-th step enforces clusters with a volume density ρk. The volume density is related to the cluster radius, as detailed in the (Supplementary Information). All SPRITE clusters are incorporated from the beginning of the optimization, while their effective co-location density is gradually increased with each optimization step (from ρ1 to ρfinal).
We used a nonzero final bound for each data type (that is, θfinal, λfinal, tfinal, ρfinal > 0) to reduce the chances of including experimental noise in the calculations (that is, data errors are expected to have very low probabilities). To reach convergence, multiple A/M iterations are typically required at a given optimization step, which is defined by a given combination of threshold values (Extended Data Fig. 2b,c). Only if the optimization in a given step is fully converged will the optimization proceed to the next step. All data sources are integrated simultaneously.
The IGM software, as introduced here, automatically performs the sequence of A/M iterations until full convergence is reached and a genome structure population is calculated that recapitulates all the input data (at a given tolerance; Extended Data Fig. 1).
Convergence
The optimization progress is monitored by tracking the agreement between model and target distances. As detailed in the Supplementary Information, each energy term introduced in the M-step to model the effect of genomic data is associated with a residual error η that monitors whether the corresponding target distance is satisfied or not: η > 0.05 indicates a discrepancy between target and model distances larger than 5%, and is considered a violation. A round of A/M iterations (for a given combination of threshold values) is successful when the cumulative fraction of all violations (from all data types) is smaller than 0.01%. Only then does the optimization move to the next step, and optimization thresholds are lowered and more data are added. Extended Data Fig. 2d shows the histogram of residual errors in population HDSF for the different data categories used as input (polymer and volume, Hi-C, lamina DamID, SPRITE and FISH).
IGM software
The IGM requires one input file for each data type and a configuration file, which lists all parameters controlling the pipeline, including nuclear shape, genome segmentation/base-pair resolution, nuclear radius, semiaxes and MD time step. The software automatically performs a preliminary statistical analysis of genome structures, including a report of the model quality using the correlation between prediction and experiments, and radial features such as the radial positions of individual chromatin domains in the nucleus.
We refer the interested reader to the documentation for implementation details. Here, we would like to discuss the design guidelines that were cornerstones to the development: flexibility, modularity and user-friendliness.
As for flexibility, the software is able to handle different types of genomes confined to either spherical or ellipsoidal nuclei and can use any combination of ensemble Hi-C, lamin B1 DamID, 3D FISH and SPRITE data points as input. Due to IGM’s modularity, the different parts of the code communicate in such a way that any data type can be added with minimal changes, as long as the data can be cast into an energy term, thus allowing for any data customization that users may require. Parallel computing can be deployed on different schedulers in a straightforward manner. Simulation and optimization setups can be adjusted by editing a text file, which lists all the configuration parameters.
A Python wrapper is available for interfacing the different building blocks and keeping track of the optimization status.
The optimization progress is monitored by a log file that prints all the details, from current iteration violation score to the specific values of thresholds associated with it.
The IGM optimization for a population of 1,000 whole diploid genome structures at 200-kb resolution using ensemble Hi-C, lamin B1 DamID, 3D FISH HIPMap and SPRITE data takes about 10–15 h of computing time, using a controller core with 4 GB of RAM communicating with 250 2-GB-RAM engine processors. The optimized coordinates after each iteration, that is, Xt, are saved in separate files, each ~350 Mb in size. The complete package (and its documentation) is available at https://github.com/alberlab/igm/. In particular, we refer the reader to the README.md file (https://github.com/alberlab/igm/blob/master/README.md/), which also guides the reader through installing and running the platform on a simple demo.
Simulating structural observables from a population of genome structures
The same notation and variables are used here as in the description above (‘Data source representation’ and ‘Probabilistic formulation of maximum likelihood problem’) and in the Supplementary Information. \({{{\vec{\boldsymbol x}}}}_{is}=(x_{is},y_{is},z_{is})\) denotes the 3D coordinates of locus i in structure s, i and i' indicate the two copies of genomic region I.
Genomic data used as input to IGM
Ensemble Hi-C
The Hi-C indicator tensor W = (wijs) is computed as
\(R_i^{ex}\) being the excluded volume locus radius.
The simulated A = (aIJ) matrix is computed as
where CN(I) indicates the number of homologous copies associated with locus I.
Lamina DamID
The lamina DamID indicator tensor V = (vis) is computed as
where (a, b, c) are the nuclear semiaxes, r0 is the domain radius in the model, and cr is the contact range scalar (Supplementary Information). The simulated E = (eI) matrix is then computed as
Radial distance distributions (radial 3D HIPMap)
We extract the ordered radial distance distribution of region I from the S structures in the population. Assuming I has two copies, we have the list of distances
We isolate the S maximal and S minimal distances, each defining a ‘maximal’ and ‘minimal’ distance distribution. We obtain the two distributions
The collection of Z − distance distributions for different chromatin regions are cast into the U data variables (Supplementary Information) by binning the distances into appropriate \({{{\mathcal{B}}}}_q = \left[ {d_q,d_{q + 1}} \right)\) bins. In particular, if we use those distance distributions as input to an IGM calculation on a population also containing S structures (Fig. 5 and Extended Data Fig. 8), we use a straightforward approach whereby each distance in the distribution is the center of a distance bin \({{{\mathcal{B}}}}_q\) (Supplementary Information).
Pairwise distance distributions (pairwise 3D HIPMap)
We extract the ordered pairwise distance distribution of genomic pair I and J from the S structures in the population. Assuming I and J both have two copies, we have the list of distances
We isolate the S maximal and S minimal distances, each defining a ‘maximal’ and ‘minimal’ distance distribution. We obtain the two distributions
The collection of Z − distance distributions for different pairs of chromatin regions are cast into the M data variable (Supplementary Information) by binning the distances into appropriate \({{{\mathcal{B}}}}_q = \left[ {d_q,d_{q + 1}} \right)\) bins. In particular, if we use those distance distributions as input to an IGM calculation on a population also containing S structures (Fig. 5), we use a straightforward approach whereby each distance in the distribution is the center of a distance bin \({{{\mathcal{B}}}}_q\) (Supplementary Information).
Single-cell SPRITE clusters
For a given SPRITE cluster {I1,…,In}, we followed the first step of the assignment procedure (Supplementary Information; SPRITE) and determined the optimal diploid representation \(\tilde C_n\) for each structure; we computed the SPRITE residual error for all structures: if a structure has no violations, then the cluster is present in that structure, and \(t_{I_1, \ldots, I_n} = 1\); If no structure has zero violations, the cluster is not present in the population, that is, \(t_{I_1, \ldots, I_n} = 0\) (Fig. 2g).
Other structural features
A more detailed description of the following structural features is provided in ref. 30.
Distance of a locus to the nuclear center and to the lamina
The normalized radial distance of a locus i of coordinates (xis, yis, zis) to the nuclear center of an ellipsoidal nucleus (in population structure s) is computed as
that is, locus coordinates are scaled by the corresponding semiaxes. \(\left\| {{{{\vec{\boldsymbol x}}}}_i} \right\|_2 = 0\) . 1, indicates that the region is located at the geometric center (nuclear lamina).
The normal distance to an ellipsoidal surface cannot be computed exactly, so we use the radial approximation for the distance to the lamina (NE)
Radius of gyration
The radius of gyration of a chromatin segment comprising C loci \({{{\mathcal{C}}}} = (i_1,i_2, \ldots ,i_C)\) in genome structure s is computed as
where xjs are the coordinates of the j-th locus in the segment, and \({{{\boldsymbol{x}}}}_{{{\mathcal{C}}}}^{\mathrm{CM}}\) is the segment center of mass in structure s. The chromosomal radius of gyration is easily computed by replacing a chromatin segment with a whole chromosome.
Compartmentalization score
For the HFFc6 cell type, each locus is assigned to either A or B compartments using the ensemble Hi-C and the procedure used in ref. 8. For each structure, the compartmentalization score is computed as defined in ref. 63:
where NAA, NAB and NBB are the number of A–A, A–B and B–B contacts in the structure respectively. The A/B assignment for HFFc6 structures was downloaded from the 4DN portal58 under identifier 4DNFINQZ5JHV.
Average radial position
The mean radial position of a locus I in an autosome is \(\overline {r_I} = \mathop {\sum}\nolimits_{s = 1}^S {\frac{{r_{is} + r_{i\prime s}}}{{2S}}}\), with i, i′ as the two homologous copies. S is the total number of structures in the population30.
Chromatin decompaction
The local compaction of the chromatin fiber at the location of a given locus is estimated by the radius of gyration for a 1-Mb region centered at the locus (that is, comprising +500 kb upstream and 500 kb downstream of the given locus). To estimate the radius of gyration values along an entire chromosome, we use a sliding-window approach over all chromatin regions in a chromosome, as described in ref. 30.
Cell-to-cell variability of structural features30
Cell-to-cell variability, δ, of any structural feature for a chromatin region, i, in chromosome c, is calculated as
where σc,i is the standard deviation of the feature value of region i across the population and \(\overline {\sigma _c}\) is the mean standard deviation of the feature value calculated from all regions within the same chromosome, c. Positive δi values (δi > 0) result from high cell-to-cell variability of the feature (for example, radial position), whereas negative values (δI < 0) indicate low variability.
Interchromosomal interaction probability
For each chromatin region I, its interchromosomal interaction probability (ICP) is calculated as
across the full population, where \(n_{\mathrm{intra}}^s\) and \(n_{\mathrm{inter}}^s\) are the number of cis and trans contacts in structure s, respectively.
Interior chromatin localization
For a given 200-kb region, the interior localization frequency (ILF) is calculated as
where n[rI ≤ 0.5] is the number of structures where either copy of the region I has a radial position lower than 0.5, for example, in the nuclear interior.
SON TSA-seq
We followed a procedure described in ref. 30. We first identified chromatin expected to have high speckle association: we selected 5% of chromatin regions with the lowest average radial positions and generated chromatin interaction networks (CINs)66 for the selected group of chromatin regions in each structure of the population. A CIN was calculated for the selected chromatin in each model as follows: Each vertex represents a 200-kb chromatin region. An edge between two vertices i, j is drawn if the corresponding chromatin regions are in physical contact in the model, if the spatial distance dij ≤ 4r0. Approximate speckle locations are then identified as the geometric center of the resulting spatial partitions identified by Markov clustering67 of the CINs.
To predict TSA-seq signals from our models, we use
where S is the number of models, L is the number of approximate speckle locations in structure s, \(\left\| {{{{\vec{\boldsymbol x}}}}_{is} - {{{\vec{\boldsymbol x}}}}_{ls}} \right\|_2\) is the distance between the region i and the predicted nuclear body location l (in structure s), and R0 = 4 is the estimated decay constant in the TSA-seq experiment57. The normalized TSA-seq signal for region i then becomes:
where \(\overline {sig}\) is the mean signal calculated from all regions in the genome. The predicted signal is averaged over copies for regions that have more than one copy in the genome.
Lamin B1 TSA-seq
We followed the procedure described in ref. 30. For lamin locations, we first identified regions with the highest 15% radial positions in each structure, determined spatial partitions of these regions and used centers of these spatial partitions as approximate locations of lamina-associated domains. Lamina TSA-seq signal was then calculated from these center locations using the decay function described in ‘SON TSA-seq’.
Speckle and lamina association frequencies30
For a given 200-kb chromatin region I, the SAF is calculated as
where S is the number of structures in the population; \(n_{d_i < d_t}\) and \(n_{d_{i\prime } < d_t}\) are the number of structures, in which region i and its homologous copy i′ have a distance to a predicted speckle smaller than the association threshold, dt (if the chromatin region is from a sex chromosome, there is only one copy and i′ = i). The dt value is set to 1,000 nm. Distances to the speckles are computed using the predicted speckle partitions via Markov clustering.
For a given 200-kb chromatin region I, the LAF is calculated as
where S is the number of structures in the population; nri>0.85 and nri'>0.85 are the number of structures, in which region i and its homologous copy i′ have a radial position larger than 0.85 (if the chromatin region is from a sex chromosome, there is only one copy and i′ = i). Both for SAF and LAF, we tried different distance thresholds, and the selected thresholds resulted in the best correlations with experimental data. The following experimental threshold distances were used for comparison with the experimental data from Su et al.17: SAF of 500 nm and LAF of 750 nm.
Median trans A/B ratio17,30
For each chromatin region i, we defined the trans neighborhood {j} if the center-to-center distances of other regions from other chromosomes to i are smaller than 500 nm, which can be expressed as a set; \(Ne_i^t = \{ j:\mathrm{chrom}_i \ne \mathrm{chrom}_j,d_{ij} < 500\,\mathrm{nm}\}\). The trans A/B ratio is then calculated as
where \(n_A^t\) and \(n_B^t\) are the number of trans A and B regions in the set Nei for haploid region i. The median of the trans A/B ratios for a region is then calculated from all the trans A/B ratios of the homologous copies of the region observed in all the structures of the population. The values are then rescaled to have values between 0 and 1.
Comparison of simulated structures with imaged single cells
Preprocessing of the DNA-MERFISH dataset17
We collected both homologous chromosome copies from each of the 3,029 single cells that contained at least 80% assigned imaged loci and where all chromosomes are imaged. There were 935 loci for 3,029 different single cells for the high-resolution chromosome 2 dataset and 1,041 loci for 4,555 different single cells for the low-resolution whole-genome-imaged dataset. If a locus is unidentified in an image, we used linear interpolation to approximate its coordinates within the image. For low-resolution chromosome 6 data, we filtered out those structures containing at least 75% of assigned loci.
Preprocessing of the IGS dataset68
We collected both copies from each single cell for the target chromosomes. Because the number of imaged loci varies per chromosome, we considered only chromosome structures with a coverage of at least ten genomic regions in a single cell to allow meaningful comparisons. At the end of the pipeline, there were 82 imaged single cells for chromosome 2 and 52 for chromosome 6.
Calculation and comparison of distance matrices
Chromosome structures were extracted from the images and imaged loci mapped to genomic bins at 200-k base-pair resolution. To compare structures from models and microscopy images, we only considered loci in the models that had been imaged in experiments.
We computed the distance matrix for each structure s as
where n is the number of loci in the chromosome at 200-kb resolution and coordinates are from either one of the simulated or the imaged chromosomal structures.
The matching score between any two structures is the Pearson correlation coefficient between the corresponding minimum–maximum normalized (flattened) distance matrices. To search for matching structures, we iterated over all possible structure pairs, and identified for each structure in one set its best match in the other by selecting the one with the largest correlation score.
Data analysis
Correlations
Unless otherwise specified, Pearson correlation was used to compare a given quantity across different populations. All Pearson correlation values are associated with a P value < 10−8 and we indicated that with ~0. The chromosomal stratum-adjusted correlation coefficients in Supplementary Table 3 were computed following the procedure detailed by Yang et al.60, using a smoothing parameter h = 0 and an upper-bound resolution of 50 Mb.
Goodness-of-fit test
We performed a chi-squared goodness-of-fit test on all four input data types (that is, Hi-C, lamin B1 DamID, 3D HIPMap FISH and single-cell SPRITE) of the HDSF population of structures. The test null hypothesis is that both the input data (from the experiment) and the output data (simulated from the structure population) are drawn from the same underlying distribution. We used a standard confidence value α = 0.05 for assessing the test results. For Hi-C and lamin B1 DamID data, the modeled and experimental cumulative distributions of probability of locus–locus contacts of a locus with another or the NE were compared, respectively. For 3D HIPMap data, the modeled and experimental cumulative pairwise distance distributions were compared. As for single-cell SPRITE data, we assigned a value of 1 or 0 to any of the 6,617 SPRITE clusters from the experiment that were or were not present in any of the structures of the population, by quantifying the SPRITE residual errors (Methods and Supplementary Information). The resulting distribution of binary values was then compared with the experimental distribution, which only contained values of 1. Large P values associated with the test statistics indicate that the initial null hypothesis can be rejected with great confidence; thus, it is reasonable to assume that input and output come from the same distribution (Extended Data Fig. 3).
Error bars
Error bars in Figs. 4, 5c,d and 6c and Extended Data Fig. 8b,c were computed by generating three independent population replicates for each modeling setup. Each replicate started from different random starting conditions. Any two replicates differ in the initial coordinate initialization \({{{\boldsymbol{X}}}}_i^0 \ne {{{\boldsymbol{X}}}}_j^0\), and undergo the same optimization procedure. Different random seeds were used each time to generate initial random chromosome positions within the nuclear volume. The average and standard deviation of the statistics from the three replicates are plotted in the figures.
Cross-Wasserstein distance
Let Q and P denote the cumulative probability distributions of distributions q and p of variable y, then the Wasserstein distance (WD)
is customarily used to estimate the amount of work required to transform one distribution into the other; ‘work’ measured as the amount of distribution weight to be moved, multiplied by the distance it has to be moved. We used the ordinary Wasserstein distance to compare two distributions within the same population.
When comparing probability distributions between two different genome populations or between one population and a set of experimental data, we used the notion of cross- (‘all versus all’) Wasserstein distance: we computed the set of all Wasserstein distance values for applicable distribution pairs within the same populations (cross-WD) and then computed a simple correlation between the two sets (score). Let us assume we want to compare the set of distance distributions of n pairs C = {(i1,j1),⋯,(in,jn)} between population 1 and population 2 (either one could be an experimental distribution), then we will compute
which is the correlation between two sets of n(n − 1)/2 Wasserstein distance values. For a given haploid pair I−J, the four diploid pair distributions were concatenated, \(p_{IJ} = p_{ij} \cup p_{ij\prime } \cup p_{i\prime j} \cup p_{i\prime j\prime }\). We use cross-Wasserstein distance to compare distance distributions in Fig. 2e, to compare radial, cis and trans pairwise distance distributions, and chromosomal radius of gyration in Figs. 5c and 6c and Extended Data Fig. 8b.
Data analysis
The codes used in our work are based on standard, publicly available software packages. Pre- and post-processing data and the generation of figures were performed using the Anaconda (v4.10) packages Matplotlib v3.4, Scikit Learn v1.0, Scipy v1.5 and NetworkX v2.3. Figures were then assembled using Adobe Illustrator. Chimera (v1.13)69 was used for visualization of the 3D structures generated.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
The following datasets were used to generate or validate the structures: ensemble Hi-C (4DN portal; accession code 4DNES2R6PUEK), lamin B1 DamID (4DN portal; accession code 4DNESXZ4FW4T), 3D HIPMap FISH (4DN portal; https://data.4dnucleome.org/publications/80007b23-7748-4492-9e49-c38400acbe60), single-cell SPRITE (4DN portal identifier: 4DNESJYGTI8S, private), SON TSA-seq (4DN portal; 4DNES85R9TIB), transcription data (ENCODE; accession code ENCSR735JKB). Super-resolution single-cell imaging data are available at the referenced papers. The pre-processed experimental inputs of different data sources (Hi-C, lamin B1 DamID, 3D HIPMap FISH and single-cell SPRITE) for the HFF cell line and the simulated HDSF population are available at https://doi.org/10.5281/zenodo.6540731. Other data (including configuration files and synthetic data input files) are available upon request. The configuration files and pre-processed data input files are sufficient to reproduce the structure populations with the IGM software.
Code availability
The IGM platform is available at www.github.com/alberlab/igm/. This includes, but is not limited to, the source code, a README file detailing code installation and execution, accompanying documentation, and a demo that uses a reduced data input for users to familiarize with the input, expected outputs and execution steps.
References
Misteli, T. The self-organizing genome: principles of genome architecture and function. Cell 183, 28–45 (2020).
Misteli, T. Higher-order genome organization in human disease. Cold Spring Harb. Perspect. Biol. 2, a000794 (2010).
Dekker, J. et al. The 4D nucleome project. Nature 549, 219–226 (2017).
Fang, R. et al. Mapping of long-range chromatin interactions by proximity ligation-assisted ChIP–seq. Cell Res. 26, 1345–1348 (2016).
Fullwood, M. J. et al. An oestrogen-receptor-α-bound human chromatin interactome. Nature 462, 58–64 (2009).
Hsieh, T.-H. S. et al. Mapping nucleosome resolution chromosome folding in yeast by Micro-C. Cell 162, 108–119 (2015).
Li, X. et al. Long-read ChIA-PET for base-pair resolution mapping of haplotype-specific chromatin interactions. Nat. Protoc. 12, 899–915 (2017).
Lieberman-Aiden, E. et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 326, 289–293 (2009).
Mumbach, M. R. et al. HiChIP: efficient and sensitive analysis of protein-directed genome architecture. Nat. Methods 13, 919–922 (2016).
Rao, S. S. P. et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 159, 1665–1680 (2014).
Quinodoz, S. A. et al. Higher-order inter-chromosomal hubs shape 3D genome organization in the nucleus. Cell 174, 744–757 (2018).
Beagrie, R. A. et al. Complex multi-enhancer contacts captured by genome architecture mapping. Nature 543, 519–524 (2017).
Zheng, M. et al. Multiplex chromatin interactions with single-molecule precision. Nature 566, 558–562 (2019).
Nir, G. et al. Walking along chromosomes with super-resolution imaging, contact maps and integrative modeling. PLoS Genet. 14, e1007872 (2018).
Bintu, B. et al. Super-resolution chromatin tracing reveals domains and cooperative interactions in single cells. Science 362, eaau1783 (2018).
Wang, S. et al. Spatial organization of chromatin domains and compartments in single chromosomes. Science 353, 598–602 (2016).
Su, J.-H., Zheng, P., Kinrot, S. S., Bintu, B. & Zhuang, X. Genome-scale imaging of the 3D organization and transcriptional activity of chromatin. Cell 182, 1641–1659 (2020).
Takei, Y. et al. Integrated spatial genomics reveals global architecture of single nuclei. Nature 590, 344–350 (2021).
Fudenberg, G. et al. Formation of chromosomal domains by loop extrusion. Cell Rep. 15, 2038–2049 (2016).
Sanborn, A. L. et al. Chromatin extrusion explains key features of loop and domain formation in wild-type and engineered genomes. Proc. Natl Acad. Sci. USA 112, E6456–E6465 (2015).
Dixon, J. R. et al. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 485, 376–380 (2012).
Schoenfelder, S. & Fraser, P. Long-range enhancer–promoter contacts in gene expression control. Nat. Rev. Genet. 20, 437–455 (2019).
Falk, M. et al. Heterochromatin drives compartmentalization of inverted and conventional nuclei. Nature 570, 395–399 (2019).
Guelen, L. et al. Domain organization of human chromosomes revealed by mapping of nuclear lamina interactions. Nature 453, 948–951 (2008).
Mirny, L. A., Imakaev, M. & Abdennur, N. Two major mechanisms of chromosome organization. Curr. Opin. Cell Biol. 58, 142–152 (2019).
Nuebler, J., Fudenberg, G., Imakaev, M., Abdennur, N. & Mirny, L. A. Chromatin organization by an interplay of loop extrusion and compartmental segregation. Proc. Natl Acad. Sci. USA 115, E6697–E6706 (2018).
Kempfer, R. & Pombo, A. Methods for mapping 3D chromosome architecture. Nat. Rev. Genet. 21, 207–226 (2020).
McCord, R. P., Kaplan, N. & Giorgetti, L. Chromosome conformation capture and beyond: toward an integrative view of chromosome structure and function. Mol. Cell 77, 688–708 (2020).
Sparks, T. M., Harabula, I. & Pombo, A. Evolving methodologies and concepts in 4D nucleome research. Curr. Opin. Cell Biol. 64, 105–111 (2020).
Yildirim, A. et al. Population-based structure modeling reveals key roles of nuclear microenvironment in gene functions. Preprint at bioRxiv https://doi.org/10.1101/2021.07.11.451976 (2022).
Barbieri, M. et al. Complexity of chromatin folding is captured by the strings and binders switch model. Proc. Natl Acad. Sci. USA 109, 16173–16178 (2012).
Baù, D. et al. The three-dimensional folding of the α-globin gene domain reveals formation of chromatin globules. Nat. Struct. Mol. Biol. 18, 107–114 (2011).
Bianco, S. et al. Computational approaches from polymer physics to investigate chromatin folding. Curr. Opin. Cell Biol. 64, 10–17 (2020).
Di Stefano, M., Nützmann, H.-W., Marti-Renom, M. A. & Jost, D. Polymer modelling unveils the roles of heterochromatin and nucleolar organizing regions in shaping 3D genome organization in Arabidopsis thaliana. Nucleic Acids Res. 49, 1840–1858 (2021).
Giorgetti, L. et al. Predictive polymer modeling reveals coupled fluctuations in chromosome conformation and transcription. Cell 157, 950–963 (2014).
Hua, N. et al. Producing genome structure populations with the dynamic and automated PGS software. Nat. Protoc. 13, 915–926 (2018).
Li, Q. et al. The three-dimensional genome organization of Drosophila melanogaster through data integration. Genome Biol. 18, 145 (2017).
Nagano, T. et al. Single-cell Hi-C reveals cell-to-cell variability in chromosome structure. Nature 502, 59–64 (2013).
Paulsen, J. et al. Chrom3D: three-dimensional genome modeling from Hi-C and nuclear lamin-genome contacts. Genome Biol. 18, 21 (2017).
Rosenthal, M. et al. Bayesian estimation of three-dimensional chromosomal structure from single-cell Hi-C data. J. Comput. Biol. 26, 1191–1202 (2019).
Serra, F. et al. Automatic analysis and 3D-modelling of Hi-C data using TADbit reveals structural features of the fly chromatin colors. PLoS Comput. Biol. 13, e1005665 (2017).
Stevens, T. J. et al. 3D structure of individual mammalian genomes studied by single-cell Hi-C. Nature 544, 59–64 (2017).
Tan, L., Xing, D., Chang, C. H., Li, H. & Xie, X. S. Three-dimensional genome structures of single diploid human cells. Science 361, 924–928 (2018).
Tjong, H. et al. Population-based 3D genome structure analysis reveals driving forces in spatial genome organization. Proc. Natl Acad. Sci. USA 113, E1663–E1672 (2016).
Trieu, T. & Cheng, J. Large-scale reconstruction of 3D structures of human chromosomes from chromosomal contact data. Nucleic Acids Res. 42, e52 (2014).
Umbarger, M. A. et al. The three-dimensional architecture of a bacterial genome and its alteration by genetic perturbation. Mol. Cell 44, 252–264 (2011).
Yildirim, A., Boninsegna, L., Zhan, Y. & Alber, F. Uncovering the principles of genome folding by 3D chromatin modeling. Cold Spring Harb. Perspect. Biol. 14, a039693 (2021).
Zhang, B. & Wolynes, P. G. Prediction of chromosome conformations with maximum entropy principle. Biophys. J. 108, 537a (2015).
Zhu, G. et al. Reconstructing spatial organizations of chromosomes through manifold learning. Nucleic Acids Res. 46, e50 (2018).
Boninsegna, L., Yildirim, A., Zhan, Y. & Alber, F. Integrative approaches in genome structure analysis. Structure 30, 24–36 (2022).
Abbas, A. et al. Integrating Hi-C and FISH data for modeling of the 3D organization of chromosomes. Nat. Commun. 10, 2049 (2019).
Girelli, G. et al. GPSeq reveals the radial organization of chromatin in the cell nucleus. Nat. Biotechnol. 38, 1184–1193 (2020).
Kind, J. et al. Genome-wide maps of nuclear lamina interactions in single human cells. Cell 163, 134–147 (2015).
van Steensel, B. & Belmont, A. S. Lamina-associated domains: links with chromosome architecture, heterochromatin and gene repression. Cell 169, 780–791 (2017).
Finn, E. H. et al. Extensive heterogeneity and intrinsic variation in spatial genome organization. Cell 176, 1502–1515 (2019).
Shachar, S., Pegoraro, G. & Misteli, T. HIPMap: a high-throughput imaging method for mapping spatial gene positions. Cold Spring Harb. Symp. Quant. Biol. 80, 73–81 (2015).
Chen, Y. et al. Mapping 3D genome organization relative to nuclear compartments using TSA-seq as a cytological ruler. J. Cell Biol. 217, 4025–4048 (2018).
Krietenstein, N. et al. Ultrastructural details of mammalian chromosome architecture. Mol. Cell 78, 554–565 (2020).
Wang, Y. et al. SPIN reveals genome-wide landscape of nuclear compartmentalization. Genome Biol. 22, 36 (2021).
Yang, T. et al. HiCRep: assessing the reproducibility of Hi-C data using a stratum-adjusted correlation coefficient. Genome Res. 27, 1939–1949 (2017).
Zhang, L. et al. TSA-seq reveals a largely conserved genome organization relative to nuclear speckles with small position changes tightly correlated with gene expression changes. Genome Res. 31, 251–264 (2021).
Dunham, I. et al. An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74 (2012).
Nagano, T. et al. Cell-cycle dynamics of chromosomal organization at single-cell resolution. Nature 547, 61–67 (2017).
Seaman, L., Meixner, W., Snyder, J. & Rajapakse, I. Periodicity of nuclear morphology in human fibroblasts. Nucleus 6, 408–416 (2015).
Kalhor, R., Tjong, H., Jayathilaka, N., Alber, F. & Chen, L. Genome architectures revealed by tethered chromosome conformation capture and population-based modeling. Nat. Biotechnol. 30, 90–98 (2012).
Hagberg, A., Swart, P. & S. D. Chult. Exploring network structure, dynamics, and function using NetworkX. https://www.osti.gov/biblio/960616-exploring-network-structure-dynamics-function-using-networkx (2008).
Enright, A. J., Van Dongen, S. & Ouzounis, C. A. An efficient algorithm for large-scale detection of protein families. Nucleic Acids Res. 30, 1575–1584 (2002).
Payne, A. C. et al. In situ genome sequencing resolves DNA sequence and structure in intact biological samples. Science 371, eaay3446 (2021).
Pettersen, E. F. et al. UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 (2004).
Acknowledgements
This work was supported by the National Institutes of Health (NIH; grants U54DK107981 and UM1HG011593 to F.A.), and an NSF CAREER grant (1150287 to F.A.). We thank the laboratories of J. Dekker (University of Massachusetts Medical School), B. Van Steensel (Netherlands Cancer Institute), T. Misteli (NIH) and A. Belmont (University of Illinois Urbana-Champaign) for kindly providing the experimental data (in situ Hi-C, lamina DamID, 3D HIPMap FISH, DNA SPRITE and SON TSA-seq) used for generating and validating our genome models. We thank W. Li for proofreading the section about the probability functions.
Author information
Authors and Affiliations
Contributions
L.B. and F.A. designed research. L.B., A.Y. and Y.Z. performed all calculations and data analysis. L.B., A.Y. and F.A. interpreted results and data analysis with input from X.J.Z. G.P., L.B. and A.Y. wrote software and documentation. S.A.Q. and M.G. contributed new data sources. E.H.F. provided data and help in data interpretation. L.B., A.Y. and F.A. wrote the manuscript with input from X.J.Z. All authors approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Methods thanks Ming Hu and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editor: Lin Tang, in collaboration with the Nature Methods team.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Flowchart of the Stepwise Iterative Optimization pipeline.
Ensemble Hi-C, lamina DamID, 3D HIPMap FISH and SPRITE data are used as input to the Stepwise Iterative Optimization protocol which underlies the Integrated Genome Modeling platform. A randomly initialized diploid genome population with chromosome territories X0 is first thermally relaxed subject to envelope and polymer restraints only (not shown). Then, genomic data are gradually added and structures are optimized via a sequence of iterative A/M optimization steps. Optimization hardness is gradually increased by adding batches of data and reducing the tolerance, as visually indicated (see also Methods). For example, at the end of i-th A/M step, all contacts with probability larger than θi (that is, all matrix entries specified by \({{{\boldsymbol{A}}}}_{\theta _{{{i}}}}\)), all lamina contacts with probability larger than \(\lambda _{{{i}}}\) (that is, all entries \({{{\boldsymbol{E}}}}_{\lambda _{{{i}}}}\)), all 3D HIPMap FISH distances with a tolerance equal to \({{{\boldsymbol{t}}}}_{{{i}}}\) (that is, \({{{\boldsymbol{U}}}}_{{{{t}}}_i}\)and \({{{\boldsymbol{M}}}}_{{{{t}}}_i}\)) and all SPRITE clusters with volume density ρi (that is \(\mathbf{T}_{\rho _i}\)) are included (see Methods). Multiple sequential A/M iterations may be needed for a given set of optimization thresholds in order to generate an intermediate population \({{{\hat{\boldsymbol X}}}}^{({{{i}}})}\) which successfully incorporates all the data restraints that have been added up to that point. At the end of the pipeline, all data up to the final threshold values are included, and, after additional iterations lead to convergence (all data is satisfied), the optimized population \({{{\hat{\boldsymbol X}}}}^{({{{final}}})}\) is returned, together with the final violation statistics (see also Extended Data Fig. 2).
Extended Data Fig. 2 Optimization statistics for HFFc6 all-data genome model.
(A) Top and side view of one full genome structure from the optimized HDSF population, with the ellipsoidal nuclear lamina axes annotated (in nm): the same color is used for homologous chromosomes. (B) Fraction of violations plotted as a function of A/M iterations during the HDSF population optimization: jumps in the curve (iterations 6 and 11) indicate the gradual addition of more data batches (that is data added at optimization thresholds (Methods)). All data are added by iteration 12, but additional iterations are run to ensure robust convergence with a violation fraction < 10−5. (C) Optimization thresholds (θi,λi,ti and ρi−1), which control the rate and size of data batches being added, shown as a function of the number of A/M iterations: a red vertical line indicates the iteration when all data points are added to the modeling. Final values are non-zero, which reproduces typical experimental setups where finite precision is only available. \(\theta _{final} = \theta _{final}^{intra} = 0.008\) (Hi-C probability), λfinal = 0.3 (lamina DamID probability), tfinal = 25nm (FISH distance tolerance), ρfinal = 0.005nm−3 (SPRITE volume density), see also Methods and Extended Data Fig. 1. (D) Final violation statistics broken down into the different restraint categories; each panel shows the normalized histogram of residual errors (η > 0.05, see Supplementary Information) associated with violations in a given data category. No bars are showing in the SPRITE panel because all applied SPRITE restraints are satisfied, and none is violated. The accompanying table details the number of applied restraints and the number of violations: over 99.999% of polymer restraints, over 99.999% of Hi-C restraints, 99.98% of FISH restraints, and 100% of both SPRITE and lamina DamID restraints are satisfied in the optimized population. The number of FISH and SPRITE restraints is orders of magnitude smaller than polymer, Hi-C and DamID restraints.
Extended Data Fig. 3 χ2 goodness-of-fit test between the predicted data from IGM HDSF populations and the input data from experiments.
Each panel compares the cumulative probability distributions from experiments (blue) and simulation (red). For Hi-C (A) and laminB1 DamID data (B), the cumulative distributions of probability of contacts of a locus with another locus (Hi-C) or the nuclear envelope (DamID) are compared. (C) To demonstrate the good agreement between 3D HIPMap data from experiment and models, we show an example for a distribution of pairwise distances between loci 2.4 Mb and 273.5 Mb for chromosome 1. All the other distance distributions are also accurately reproduced with p-values ~1.0. (D) As for single cell SPRITE data, we assign a value of 1 or 0 to any of the 6617 SPRITE clusters from experiment that are or are not present in any of the structures of the population, by quantifying the SPRITE residual errors (Methods and Supporting Information). The resulting distribution of binary values is then compared with the experimental distribution, which only contain values of 1. The large p-values indicate that the null hypothesis can be accepted (confidence level α = 0.05) and that input and output are in fact drawn from the identical underlying probability distribution.
Extended Data Fig. 4 Validating chromosome structures from HDSF population with single cell structures from imaging experiments.
(AB) Comparison of distance matrices of single cell chromosome 6 (A) and chromosome 2 (B) structures from simulated models and DNA-MERFISH imaging data17. Models reproduce a variety of folding patterns observed in experiment very efficiently. Numbers above the distance matrix indicate Pearson correlation between simulated and experimental distance matrices. (CD) Comparison of distance matrices of single cell chromosome 6 (C) and chromosome 2 (D) structures from simulated models and fibroblast in situ genome sequencing (IGS) imaged single cells68. Models reproduce a variety of folding patterns observed in experiment very efficiently. Numbers above the distance matrix indicate Pearson correlation between simulated and experimental distance matrices.
Extended Data Fig. 5 Reproducibility across IGM replicates.
Reproducibility of 15 structural features in independent HDSF replicate calculations starting from different random starting configurations, see Methods. These features also include the reproducibility of cell-to-cell variability of several features from two independent population replicates. The high Pearson’s correlation values in each panel validate the robust reproducibility of all features (ICP = interchromosomal contact probability, SAF = speckle association frequency, LAF = lamina association frequency).
Extended Data Fig. 6 Prediction of experimental SPRITE and FISH data in HFFc6 H, HD, HDS, HDSF populations.
(Top panels) SPRITE11 cumulative residual (left) and fraction of violated SPRITE restraints (right) for each of the data-driven populations discussed in Fig. 4. Lamina DamID restraints tend to stretch the genome towards the lamina, whereas SPRITE restraints squeeze the targeted loci close to one another: an optimal balance is only found when both data modalities are simultaneously integrated, for example, populations HDS and HDSF. (Bottom) FISH cumulative residual (left) and cross WD score (right). The cumulative residual is defined as the sum of the residual errors η for all violations; the cross WD score is the Pearson correlation between two cross WD sets (see Methods and Supporting Information). FISH distributions55 are gradually better predicted with increasing amount of data and most efficiently recapitulated in population HSDF only, as suggested by a cross WD score of 0.999 and the smallest cumulative residual.
Extended Data Fig. 7 Relevance of low frequency inter-chromosomal contacts.
(Unperturbed) Hi-C, lamina DamID and 1000 radial and 1000 pairwise FISH distance distributions extracted from the ground truth (Fig. 5) are used to generate a population of structures. The predicted radial profiles for chromosome 1 are compared with the underlying ground truth at different stages of the optimization process. Specifically, lamina DamID and FISH data have been all added up to the final thresholds λfinal and tfinal, and low frequency inter chromosomal contacts added up to probability θinter = 0.02 (left) and θinter = 0.008 (right). Radial profiles are better reproduced in multi-modal Hi-C + lamina DamID + FISH models at θinter = 0.02 than they are in Hi-C only models with the same setup (Fig. 6A), and then refined by lowering the contact probability θinter. This provides alternative evidence that independent data sources can account for missing information; here, inter chromosomal contacts with probability smaller than 0.008. (θinter = 0.02, 0.008).
Extended Data Fig. 8 Comparing information content of lamina DamID data against increasingly larger radial distance distribution FISH data sets.
Additional Hi-C* and radial FISH only populations (3a, 3b and 3c) are analyzed and compared with previous Hi-C*-radial FISH population 3 and Hi-C*-DamID only population 5 from Fig. 5. (A) The four populations with FISH data differ in the number of radial distributions used in the input (500, 1,000, 5,000 and 10,000). (B) The seven quantities from Fig. 5C are predicted for each population and compared with the ground truth. (C) The overall performance rank for these five populations indicates that a sufficiently large sample of radial distance distributions can match and outperform the information provided by lamina DamID data. Error bars for each setup were estimated from three independent population replicates (see Methods); data in panels (B) and (C) are presented as mean values +/− standard deviation.
Supplementary information
Supplementary Information
Supplementary Discussion and Supplementary Tables 1–3
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Boninsegna, L., Yildirim, A., Polles, G. et al. Integrative genome modeling platform reveals essentiality of rare contact events in 3D genome organizations. Nat Methods 19, 938–949 (2022). https://doi.org/10.1038/s41592-022-01527-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41592-022-01527-x
- Springer Nature America, Inc.
This article is cited by
-
Computational methods for analysing multiscale 3D genome organization
Nature Reviews Genetics (2024)
-
Evaluating the role of the nuclear microenvironment in gene function by population-based modeling
Nature Structural & Molecular Biology (2023)
-
SnapFISH: a computational pipeline to identify chromatin loops from multiplexed DNA FISH data
Nature Communications (2023)
-
Studying the impact of the nuclear topography on gene function
Nature Structural & Molecular Biology (2023)
-
Multiscale genome organization symposium — annual biophysical society meeting 2023
Biophysical Reviews (2023)