

Abstract
The combined effect of multiple mutations on protein function is hard to predict; thus, the ability to functionally assess a vast number of protein sequence variants would be practically useful for protein engineering. Here we present a high-throughput platform that enables scalable assembly and parallel characterization of barcoded protein variants with combinatorial modifications. We demonstrate this platform, which we name CombiSEAL, by systematically characterizing a library of 948 combination mutants of the widely used Streptococcus pyogenes Cas9 (SpCas9) nuclease to optimize its genome-editing activity in human cells. The ease with which the editing activities of the pool of SpCas9 variants can be assessed at multiple on- and off-target sites accelerates the identification of optimized variants and facilitates the study of mutational epistasis. We successfully identify Opti-SpCas9, which possesses enhanced editing specificity without sacrificing potency and broad targeting range. This platform is broadly applicable for engineering proteins through combinatorial modifications en masse.





Similar content being viewed by others


Data availability
Source data for the count matrices determined for SpCas9 variants on the basis of pooled characterization that are shown in Fig. 3 are provided with the online version of this paper. GUIDE-seq data are available from the European Nucleotide Archive under accession PRJEB32521.
Code availability
The custom scripts for data analysis are available at https://github.com/AWHKU/BC-analyzer.
Change history
23 July 2019
An amendment to this paper has been published and can be accessed via a link at the top of the paper.
References
Bornscheuer, U. T. et al. Engineering the third wave of biocatalysis. Nature 485, 185–194 (2012).
Weinreich, D. M., Delaney, N. F., Depristo, M. A. & Hartl, D. L. Darwinian evolution can follow only very few mutational paths to fitter proteins. Science 312, 111–114 (2006).
Slaymaker, I. M. et al. Rationally engineered Cas9 nucleases with improved specificity. Science 351, 84–88 (2016).
Kleinstiver, B. P. et al. High-fidelity CRISPR–Cas9 nucleases with no detectable genome-wide off-target effects. Nature 529, 490–495 (2016).
Chen, J. S. et al. Enhanced proofreading governs CRISPR–Cas9 targeting accuracy. Nature 550, 407–410 (2017).
Casini, A. et al. A highly specific SpCas9 variant is identified by in vivo screening in yeast. Nat. Biotechnol. 36, 265–271 (2018).
Hu, J. H. et al. Evolved Cas9 variants with broad PAM compatibility and high DNA specificity. Nature 556, 57–63 (2018).
Packer, M. S. & Liu, D. R. Methods for the directed evolution of proteins. Nat. Rev. Genet. 16, 379–394 (2015).
Romero, P. A. & Arnold, F. H. Exploring protein fitness landscapes by directed evolution. Nat. Rev. Mol. Cell Biol. 10, 866–876 (2009).
Gasperini, M., Starita, L. & Shendure, J. The power of multiplexed functional analysis of genetic variants. Nat. Protoc. 11, 1782–1787 (2016).
Fowler, D. M. & Fields, S. Deep mutational scanning: a new style of protein science. Nat. Methods 11, 801–807 (2014).
Ma, S., Saaem, I. & Tian, J. Error correction in gene synthesis technology. Trends Biotechnol. 30, 147–154 (2012).
Kosuri, S. & Church, G. M. Large-scale de novo DNA synthesis: technologies and applications. Nat. Methods 11, 499–507 (2014).
Engler, C., Kandzia, R. & Marillonnet, S. A one pot, one step, precision cloning method with high throughput capability. PLoS ONE 3, e3647 (2008).
Gibson, D. G. et al. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods 6, 343–345 (2009).
Trudeau, D. L., Smith, M. A. & Arnold, F. H. Innovation by homologous recombination. Curr. Opin. Chem. Biol. 17, 902–909 (2013).
Wong, A. S., Choi, G. C., Cheng, A. A., Purcell, O. & Lu, T. K. Massively parallel high-order combinatorial genetics in human cells. Nat. Biotechnol. 33, 952–961 (2015).
Wong, A. S. et al. Multiplexed barcoded CRISPR–Cas9 screening enabled by CombiGEM. Proc. Natl Acad. Sci. USA 113, 2544–2549 (2016).
Cheng, A. A., Ding, H. & Lu, T. K. Enhanced killing of antibiotic-resistant bacteria enabled by massively parallel combinatorial genetics. Proc. Natl Acad. Sci. USA 111, 12462–12467 (2014).
Doudna, J. A. & Charpentier, E. The new frontier of genome engineering with CRISPR–Cas9. Science 346, 1258096 (2014).
Hsu, P. D., Lander, E. S. & Zhang, F. Development and applications of CRISPR–Cas9 for genome engineering. Cell 157, 1262–1278 (2014).
Mali, P., Esvelt, K. M. & Church, G. M. Cas9 as a versatile tool for engineering biology. Nat. Methods 10, 957–963 (2013).
Barrangou, R. & Horvath, P. A decade of discovery: CRISPR functions and applications. Nat. Microbiol. 2, 17092 (2017).
Kim, S., Bae, T., Hwang, J. & Kim, J. S. Rescue of high-specificity Cas9 variants using sgRNAs with matched 5′ nucleotides. Genome Biol. 18, 218 (2017).
Kulcsar, P. I. et al. Crossing enhanced and high fidelity SpCas9 nucleases to optimize specificity and cleavage. Genome Biol. 18, 190 (2017).
Zhang, D. et al. Perfectly matched 20-nucleotide guide RNA sequences enable robust genome editing using high-fidelity SpCas9 nucleases. Genome Biol. 18, 191 (2017).
Kato-Inui, T., Takahashi, G., Hsu, S. & Miyaoka, Y. Clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated protein 9 with improved proof-reading enhances homology-directed repair. Nucleic Acids Res. 46, 4677–4688 (2018).
Sternberg, S. H., LaFrance, B., Kaplan, M. & Doudna, J. A. Conformational control of DNA target cleavage by CRISPR–Cas9. Nature 527, 110–113 (2015).
Singh, D. et al. Mechanisms of improved specificity of engineered Cas9s revealed by single-molecule FRET analysis. Nat. Struct. Mol. Biol. 25, 347–354 (2018).
Fu, Y. et al. High-frequency off-target mutagenesis induced by CRISPR–Cas nucleases in human cells. Nat. Biotechnol. 31, 822–826 (2013).
Lee, J. K. et al. Directed evolution of CRISPR–Cas9 to increase its specificity. Nat. Commun. 9, 3048 (2018).
Haeussler, M. et al. Evaluation of off-target and on-target scoring algorithms and integration into the guide RNA selection tool CRISPOR. Genome Biol. 17, 148 (2016).
Fu, Y., Sander, J. D., Reyon, D., Cascio, V. M. & Joung, J. K. Improving CRISPR–Cas nuclease specificity using truncated guide RNAs. Nat. Biotechnol. 32, 279–284 (2014).
Vakulskas, C. A. et al. A high-fidelity Cas9 mutant delivered as a ribonucleoprotein complex enables efficient gene editing in human hematopoietic stem and progenitor cells. Nat. Med. 24, 1216–1224 (2018).
Ran, F. A. et al. In vivo genome editing using Staphylococcus aureus Cas9. Nature 520, 186–191 (2015).
Zetsche, B. et al. Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR–Cas system. Cell 163, 759–771 (2015).
Komor, A. C., Kim, Y. B., Packer, M. S., Zuris, J. A. & Liu, D. R. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 533, 420–424 (2016).
Nishida, K. et al. Targeted nucleotide editing using hybrid prokaryotic and vertebrate adaptive immune systems. Science 353, aaf8729 (2016).
Gaudelli, N. M. et al. Programmable base editing of A*T to G*C in genomic DNA without DNA cleavage. Nature 551, 464–471 (2017).
Li, X. et al. Base editing with a Cpf1–cytidine deaminase fusion. Nat. Biotechnol. 36, 324–327 (2018).
Honma, K. et al. RPN2 gene confers docetaxel resistance in breast cancer. Nat. Med. 14, 939–948 (2008).
Kampmann, M., Bassik, M. C. & Weissman, J. S. Functional genomics platform for pooled screening and generation of mammalian genetic interaction maps. Nat. Protoc. 9, 1825–1847 (2014).
Olson, C. A., Wu, N. C. & Sun, R. A comprehensive biophysical description of pairwise epistasis throughout an entire protein domain. Curr. Biol. 24, 2643–2651 (2014).
Aakre, C. D. et al. Evolving new protein–protein interaction specificity through promiscuous intermediates. Cell 163, 594–606 (2015).
Guschin, D. Y. et al. A rapid and general assay for monitoring endogenous gene modification. Methods Mol. Biol. 649, 247–256 (2010).
Tsai, S. Q. et al. GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR–Cas nucleases. Nat. Biotechnol. 33, 187–197 (2015).
Tsai, S. Q., Topkar, V. V., Joung, J. K. & Aryee, M. J. Open-source guideseq software for analysis of GUIDE-seq data. Nat. Biotechnol. 34, 483 (2016).
Acknowledgements
We thank members of the Wong lab for helpful discussions, and Z. Dong, L. Qin, N. Shirgaonkar and L. Pardeshi from the Genomics, Bioinformatics and Single Cell Analysis Core of the Faculty of Health Sciences at the University of Macau for their technical support. We thank J. Chan for support at the High Performance Computing Cluster (HPCC) of ICTO of the University of Macau. We thank T. Ochiya for OVCAR8-ADR cells. We thank the Faculty Core Facility at the LKS Faculty of Medicine of The University of Hong Kong for providing and maintaining the equipment needed for flow cytometry analysis and cell sorting. This work was supported by The University of Hong Kong start-up and internal funds, the Croucher Foundation Start-up Allowance and the Hong Kong Research Grants Council (ECS-27105716, GRF-17104619 and TRS-T12-710/16-R) (to A.S.L.W.); the Swedish Research Council (2016-02830) and the National Natural Science Foundation of China (81672098) (to Z.Z.); and the Science and Technology Development Fund of Macau S.A.R. (FDCT 085/2014/A2), the Research Services and Knowledge Transfer Office of the University of Macau (MYRG2016-00211-FHS and MYRG2018-00017-FHS), and the Start-up fund from the Faculty of Health Sciences, University of Macau (to K.H.W. and K.T.).
Author information
Authors and Affiliations
Contributions
G.C.G.C. and A.S.L.W. conceived the work. G.C.G.C., P.Z., C.T.L.Y., B.K.C.C., F.X., D.T. and A.S.L.W. designed and performed the experiments and interpreted and analyzed the data. G.C.G.C., C.T.L.Y., K.T., K.H.W. and A.S.L.W. performed computational analyses on next-generation sequencing data for CombiSEAL experiments. G.C.G.C., P.Z., A.S.L.W., S.B., H.Y.C. and Z.Z. performed GUIDE-seq experiments and analyzed the data. G.C.G.C. and A.S.L.W. wrote the paper.
Corresponding author
Ethics declarations
Competing interests
A.S.L.W. and G.C.G.C. have filed a patent application that is based on this work.
Additional information
Peer review information: Lei Tang and Nicole Rusk were the primary editors on this article and managed its editorial process and peer review in collaboration with the rest of the editorial team.
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Integrated supplementary information
Supplementary Figure 1
Examples of strategies for characterizing combinatorial mutations on a protein sequence.
Supplementary Figure 2 Strategy for seamless assembly of the barcoded combination mutant library pool.
a, To create barcoded DNA parts in storage vectors, genetic inserts were generated by PCR or synthesis, and cloned in the storage vectors harboring a random barcode (pAWp61 and pAWp62; digested with EcoRI and BamHI) with Gibson assembly reactions. BsaI digestion was performed to generate the barcoded DNA parts (that is, P1, P2,…, P(n)). BbsI sites and a primer-binding site for barcode sequencing were introduced in between the insert and the barcode for pAWp61 and pAWp62, respectively. b, To create the barcoded combination mutant library, the pooled DNA parts and destination assembly vectors were digested with BsaI and BbsI, respectively. A one-pot ligation created a pooled vector library, which was further iteratively digested and ligated with the subsequent pool of DNA parts to generate higher-order combination mutants. The barcoded inserts were linked with compatible overhangs that are originated from the protein-coding sequence after digestion with type IIS restriction enzymes (that is, BsaI and BbsI), thereby no fusion scar is formed in the ligation reactions. All barcodes were localized into a contiguous stretch of DNA. The final combination mutant library was encoded in lentiviruses and delivered into targeted human cells. The integrated barcodes representing each combination were amplified from the genomic DNA within the pooled cell populations in an unbiased fashion and quantified using high-throughput sequencing to identify shifts in representation under different experimental conditions. c, High correlations between barcode representations (normalized barcode counts obtained from a single set of experiment) within the plasmid and infected OVCAR8-ADR cell pools indicate efficient lentiviral delivery of the library into human cells. High reproducibility for barcode representations between two biological replicates in OVCAR8-ADR cells infected with the library. R is the Pearson’s r.
Supplementary Figure 3 Fluorescence-activated cell sorting of SpCas9 library-infected human cells harboring on- and off- target reporters.
OVCAR8-ADR reporter cell lines that express RFP and GFP genes driven by UBC and CMV promoters, respectively, and a tandem U6 promoter-driven expression cassette of gRNA targeting the RFP site (RFPsg5 or RFPsg8) were either uninfected or infected with the SpCas9 library. RFPsg5-ON and RFPsg8-ON lines harbor sites that match completely with the gRNA sequence, while RFPsg5-OFF5-2 and RFPsg8-OFF5 lines contain synonymous mutations on the RFP and are mismatched to the gRNA. Cells were sorted under flow cytometry into bins each encompassing ~5% of the population with low RFP fluorescence. These experiments were repeated independently twice with similar results.
Supplementary Figure 4 Positive correlation between enrichment score determined from the pooled screen and individual validation data.
The normalized log2(E) for each SpCas9 combination mutant is the mean score determined from the pooled screens in two biological replicates, and the normalized RFP disruption value is the mean cell percentage with depleted RFP level when compared to WT determined from three biological replicates. R is the Pearson’s r.
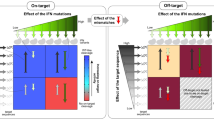
Supplementary Figure 5 Heatmaps depicting editing efficiency for the on- and off- target sites.
Editing efficiency was measured by the log-transformed enrichment ratio (log2(E)) determined for each SpCas9 combination mutant. Enriched and depleted mutants have >0 and <0, respectively. To aid visualization, amino acid residues that are predicted to make contacts with the target DNA strand or located at the linker region connecting SpCas9’s HNH and RuvC domains are grouped on the y-axis, while those predicted to make contacts with the non-target DNA strand are presented on the x-axis. The combinations for those with no enrichment are indicated in gray.
Supplementary Figure 6 Frequency of N20-NGG and G-N19-NGG sites in the reference human genome.
A custom Python code was used to find the occurrence of N20-NGG and G-N19-NGG sites in both strands of the reference human genome hg19, as an estimate of the targeting ranges of Opti-SpCas9 and other engineered SpCas9 variants including eSpCas9(1.1), SpCas9-HF1, HypaCas9, and evoCas9, respectively. N20-NGG sites are about 4.3 times more frequent than G-N19-NGG sites in the human genome.
Supplementary Figure 7 Summary of T7 endonuclease I (T7E1) assay results for DNA mismatch cleavage in OVCAR8-ADR cells.
Cells were infected with an SpCas9 variant and the indicated gRNA, and genomic DNA were collected for T7E1 assay after 11 to 16 days post-infection. Indel quantification for the infected samples is displayed as a bar graph.
Supplementary Figure 8 Expression of SpCas9 variants in OVCAR8-ADR cells.
Cells were infected with lentiviruses encoding WT SpCas9, Opti-SpCas9, eSpCas9(1.1), HypaCas9, SpCas9-HF1, Sniper-Cas9, evoCas9, xCas9, or OptiHF-SpCas9. Protein lysates were extracted for Western blot analysis, and immunoblotted with anti-SpCas9 antibodies. Beta-actin was used as loading control. We failed to detect the expression of SpCas9-HF1 and xCas9 in OVCAR8-ADR cells, which could be due to their non-optimized sequence for expression in mammalian cells (ref. 24 and Nat. Biotechnol. 36, 888–893, 2018) and thus SpCas9-HF1 and xCas9 were not included in other activity assays. These experiments were repeated independently for three times with similar results.
Supplementary Figure 9 Evaluation of the editing efficiency of SpCas9 variants with gRNAs bearing or lacking an additional mismatched 5’ guanine (5’G) using GFP disruption assay.
OVCAR8-ADR cells expressing WT SpCas9, Opti-SpCas9, eSpCas9(1.1), or HypaCas9 were infected with lentiviruses encoding gRNAs carrying or lacking an additional mismatched 5’G. Editing efficiency was measured by cell percentage with depleted GFP level using flow cytometry. Values and error bars reflect the mean and s.d. of four independent biological replicates.
Supplementary Figure 10 Opti-SpCas9 exhibits reduced off-target activity when compared to wild-type SpCas9.
Assessment of SpCas9 variants for off-target editing brought by VEGFA site 3 or DNMT1 site 4 gRNA at eight endogenous loci. Percentage of indel was measured using T7E1 assay, averaged from three independent experiments. Dash indicates none detected. Specificity of WT SpCas9 and its variants with VEGFA site 3 gRNA at OFF1 loci is plotted as the ratio of on-target to off-target activity (on-target activity data was obtained from Supplementary Fig. 7).
Supplementary Figure 11 Characterization of SpCas9 variants for editing target sites harboring sequences that are perfectly matched with the gRNA’s spacer or contain mismatch(es) using GFP disruption assay.
OVCAR8-ADR cells expressing WT SpCas9, Opti-SpCas9, eSpCas9(1.1), or HypaCas9 were infected with lentiviruses encoding gRNAs carrying no or one- to four-base mismatch(es) against the target. Editing efficiency was measured by cell percentage with depleted GFP level using flow cytometry. Values and error bars reflect the mean and s.d. of three independent biological replicates.
Supplementary Figure 12 On-target editing activity of SpCas9 variants using truncated gRNAs.
a,b, OVCAR8-ADR cells expressing WT SpCas9, Opti-SpCas9, eSpCas9(1.1), or HypaCas9 were infected with lentiviruses encoding gRNAs of varied length (17 to 19 nucleotides) targeting the GFP sequence (a) and endogenous loci (b). Editing efficiency was measured by cell percentage with depleted GFP level using flow cytometry (a) and T7E1 assay (b). The list of gRNA sequences used is presented in Supplementary Table 5. For (a), values and error bars reflect the mean and s.d. of four independent biological replicates.
Supplementary information
Supplementary Information
Supplementary Figs. 1–12
Supplementary Table 1
List of SpCas9 combination mutants that were generated and tested.
Supplementary Table 2
Enrichment scores determined for SpCas9 variants on the basis of pooled characterization.
Supplementary Table 3
A table comparing the on- and off-target activities of SpCas9 variants.
Supplementary Table 4
List of constructs used in this work.
Supplementary Table 5
List of gRNA protospacer sequences used in this study.
Supplementary Table 6
List of reporter cell lines used in this work.
Supplementary Table 7
List of primers and PCR conditions used for the T7E1 assay.
Supplementary Table 8
Adaptor and primer sequences for GUIDE-seq.
Source data
Source Data
Source Data for Fig. 3a.
Rights and permissions
About this article

Cite this article
Choi, G.C.G., Zhou, P., Yuen, C.T.L. et al. Combinatorial mutagenesis en masse optimizes the genome editing activities of SpCas9. Nat Methods 16, 722–730 (2019). https://doi.org/10.1038/s41592-019-0473-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41592-019-0473-0
- Springer Nature America, Inc.
This article is cited by
-
Engineering Cas9: next generation of genomic editors
Applied Microbiology and Biotechnology (2024)
-
Quantification of evolved DNA-editing enzymes at scale with DEQSeq
Genome Biology (2023)
-
High-throughput screening of genetic and cellular drivers of syncytium formation induced by the spike protein of SARS-CoV-2
Nature Biomedical Engineering (2023)
-
A cleavage rule for selection of increased-fidelity SpCas9 variants with high efficiency and no detectable off-targets
Nature Communications (2023)
-
Machine learning-coupled combinatorial mutagenesis enables resource-efficient engineering of CRISPR-Cas9 genome editor activities
Nature Communications (2022)