

Abstract
Natural killer (NK) cells are lymphocytes of the innate immune system. A key feature of NK cells is their ability to recognize a wide range of cells in distress, particularly tumour cells and cells infected with viruses. They combine both direct effector functions against their cellular targets and participate in the generation, shaping and maintenance of a multicellular immune response. As our understanding has deepened, several therapeutic strategies focused on NK cells have been conceived and are currently in various stages of development, from preclinical investigations to clinical trials. Here we explore in detail the complexity of NK cell biology in humans and highlight the role of these cells in cancer immunity. We also analyse the harnessing of NK cell immunity through immune checkpoint inhibitors, NK cell engagers, and infusions of preactivated or genetically modified, autologous or allogeneic NK cell products.
Similar content being viewed by others

Main
2023 heralds the fiftieth anniversary of the pioneering publications that set the stage for the discovery of NK cells1,2,3. Further characterized as a unique cellular entity distinct from other known immune cells and also officially named in 19754,5, NK cells are now known to belong to the group of innate lymphoid cells (ILCs)6—a family of cells that has been recognized as such since 20087. ILCs are lymphocytes of the innate immune system that do not express the type of diversified antigen receptors found on T cells and B cells8. Among the five major ILC subsets, type 1, 2 and 3 ILCs (ILC1, ILC2 and ILC3 cells, respectively) are mostly tissue-resident cells and mirror CD4+ T helper type 1 (TH1), TH2 and TH17 cells, respectively, in terms of cytokine production, whereas NK cells that are present both in the blood and tissues can be considered to be innate counterparts of CD8+ cytotoxic T cells9. Over the past five decades, the importance and potential of NK cells has been extensively explored. What began as academic intrigue has evolved into a promising area of immunotherapy, particularly in the fight against cancer.
What are NK cells?
NK cells are effector ILCs that arise from bone marrow progenitor cells6,10,11,12,13,14. The total number of NK cells in humans has been estimated to be 2 × 1010 cells (95% confidence interval: 0.5 × 1010–6 × 1010), making up around 1% of total immune cell types in the body and 2% of total lymphocytes15. At steady state in healthy individuals, NK cells are present mainly in the liver, bone marrow and blood where they constitute around 10% of the total number of peripheral lymphocytes. Their functions are tightly regulated by a repertoire of inhibitory and activating receptors, enabling them to recognize and to directly or indirectly eliminate stressed cells while sparing normal cells. The vast majority of mature NK cells are cytolytic, and all NK cells can produce a number of cytokines, including interferon-γ (IFNγ), growth factors such as FMS-like tyrosine kinase 3 ligand (FLT-3L) and granulocyte–macrophage colony-stimulating factor (GM-CSF), and chemokines, including XCL1 and CCL56,10,11,12,13,14.
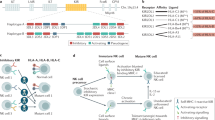
Human NK cells are usually classified on the basis of the expression of the two surface molecules CD56 (encoded by NCAM) and CD16a (encoded by FCGR3A)12. CD56brightCD16− NK cells exhibit lower cytotoxicity but produce cytokines, growth factors and chemokines. By contrast, CD56dimCD16+ NK cells are highly cytotoxic due to their expression of granzymes (GZMA, GZMB) and perforin (PRF1), and can also produce cytokines, growth factors and chemokines16. More recently, single-cell RNA-sequencing analyses have provided insights into the diversity of NK cells17,18. Unsupervised classification algorithms based on gene expression enabled the identification of three major NK cell populations (Fig. 1) and also revealed the presence of several subpopulations within them. Type 1 NK (NK1) cells, which are the most abundant in blood, correspond to CD56dimCD16+ NK cells and, besides the strong expression of CD16 (FCGR3A) and cytotoxicity effector molecules (GZMA, GZMB, PRF1), they selectively express additional genes such as SPON2, the biological function of which in NK cells remains to be elucidated. The NK2 cell population corresponds to CD56brightCD16− NK cells. These cells exhibit a characteristic transcriptional signature, including granzyme K (GZMK), a characteristic chemokine profile (XCL1, XCL2), cell surface markers (CD44, SELL, KLRC1) and strong expression of the transcription factor TCF1 (encoded by TCF7). Finally, a third major population, tentatively referred to as NK3 cells, comprises mainly CD16dim adaptive NKG2Chigh (encoded by KLRC2) NK cells, including CD57+ cells19,20,21. This subset presents memory-like properties with enhanced functional responses, in a manner resembling memory T cells, after recognition of various viral, bacterial, cytokine or hapten stimuli22,23,24,25,26. They exhibit a characteristic cytotoxic signature (GZMH), surface markers (KLRC2, CD3E) and a specific cytokine signature (IL32 and CCL5)27,28. The abundance of each of the various NK cell subsets depends on their anatomical localization and pathophysiological conditions. NK1 cells are present at higher concentrations in the bone marrow, spleen and blood, whereas NK2 cells are more prevalent in the lungs, tonsils, lymph nodes and intestines. NK2 cells show greater tissue imprinting compared with NK1 cells and share several common markers with CD8+ tissue-resident memory T cells, notably, increased expression of CD103 (encoded by ITGAE) and CD49a (in the lungs and intestines) and CXCR6 (in the bone marrow, spleen and lymph node)9,14. Recently, a human pan-cancer atlas of NK cells described tumour-associated NK cells characterized by reduced cytotoxicity and high expression of stress-associated proteins (DNAJB1 and HSP gene family), inhibitory MHC-class-I-specific receptors (members of the KIR family) and several transcription factors that control the fitness of NK cells such as NR4A1, EGR3 and KLF629.

In the peripheral blood of healthy individuals, three dominant NK cell subsets—NK1, NK2 and NK3—are discernible through single-cell transcriptomic analysis. This figure delineates the distinct transcription factors, secreted molecules and cell surface receptors that are characteristic of each subset.
Another level of complexity in the dissection of NK cell biology resides in the strong similarities with ILC1 cells30. For example, NK cells and ILC1 cells that express the activating NK cell receptor NKp46 (encoded by NCR1, also known as CD335) are dependent upon T-bet (encoded by TBX21) and secrete IFNγ 30. Moreover, NK cells can transition to ILC1-like cells in the presence of environmental cues such as transforming growth factor-β (TGFβ), and these cells have been shown to promote tumour development in some preclinical models31,32. Yet, cytotoxic ILC1 cells or ILC1-like cells have also been described to have a role in immunosurveillance of malignancies33. More studies are needed to understand the biology of NK cells as compared to ILC1 cells, as distinctive mechanisms involved in the control of NK cells and ILC1 function have already been described34. To effectively harness the antitumour properties of NK cells, it is therefore essential to distinguish the phenotype and effector functions of closely related types of ILCs, that is, circulating NK cells, tissue infiltrating NK cells, ILC1 cells and tumour-associated NK cells. In mice, bona fide NK cells and ILC1 cells can be distinguished by the mutually exclusive cell surface expression of CD49b (encoded by Itga2) and CD49a (encoded by Itga1) respectively30. Syndecan-4 is also a useful marker that enables discrimination between mouse ILC1 and NK cells35. In humans, it has been proposed that CD200R, CXCR3, CXCR6, DNAM-1 and TRAIL could distinguish ILC1 cells from NK cells36,37, but the phenotype of ILC1 cells varies according to the tissue in which these cells reside, making the identification of pan-ILC1 markers challenging38. At the transcriptomic level, a gene signature with eight cross-species and cross-organ NK-specific markers, EOMES, GZMA, IRF8, JAK1, NKG7, PLEK, PRF1 and ZEB2, has been proposed35. Importantly, these eight markers also discriminate human NK cells from the other ILC subtypes and CD4 T cells. By contrast, only three cross-species and cross-organ ILC1-specific markers, IL7R, LTB and RGS1, have been identified, but these three markers were also expressed by other ILC populations and CD4+ T cells.
Recognizing cells in distress
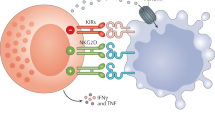
An essential feature of NK cells is their ability to distinguish between normal cells and cells in distress, and to eliminate the latter directly or indirectly6,10,11,12,13,14,39 (Table 1). NK cells recognize their targets by expressing multiple cell surface receptors6,10,11,12,13,14,39. NK cells sense the level of expression of MHC class I (MHC-I) molecules through a variety of MHC-I-specific inhibitory receptors. These include killer cell immunoglobulin-like receptors (KIRs) in humans, lectin-like Ly49 molecules in mice and CD94–NKG2A heterodimers in both species. These MHC-I receptors belong to the large family of inhibitory receptors that mediate their function through intracytoplasmic immune receptor tyrosine-based inhibitory motifs40,41. Moreover, NK cells recognize self-molecules that are induced or upregulated on the cell surface of stressed cells42,43. The prototypical example of this type of stress-induced self-recognition is the activation of NK cells triggered by cell surface receptors such as NKG2D (encoded by KLRK1), NKp46 and NKp30 (encoded by NCR3, also known as CD337), and which recognize MICA/B and ULBPs44, ecto-calreticulin45 and B7-H646, respectively, on the surface of stressed cells. The ligands for NKG2D are induced in response to the DNA-damage-response pathway, the integrated stress response, cellular hyperproliferation, activated p53 and heat-shock-induced stress43,47. Ligands for other activating receptors are also thought to be dependent on these stress-induced signalling pathways, but some of these ligands remain undefined and the precise mechanisms regulating the expression of the identified ligands are unclear. The NK cell activating receptors associate with adaptors that carry immunoreceptor tyrosine-based activation motifs40,48. With inputs of both activating and inhibitory signals, NK cells can recognize and contribute to the elimination of a variety of tumours of all histotypes49,50. NK cells can also recognize their cellular targets in the presence of antibodies and trigger antibody-dependent cell cytotoxicity (ADCC), owing to the low-affinity IgG Fc region receptor CD16a, which is mainly expressed on the surface of NK1 and NK3 cells. NK-cell-mediated ADCC has been postulated to contribute to the efficacy of therapeutic antibodies such as the anti-CD20 antibody rituximab51. After recognition, NK cells can eliminate their cellular targets through two main mechanisms: direct cytotoxicity and production of cytokines. Once activated, NK cells form an immunological synapse with the target cell, enabling the cytotoxic granules released by the NK cell to be directed towards the target cell and not to surrounding bystander cells52. Direct cytotoxicity of NK cells is also mediated by the interaction between FAS ligand expressed on NK cells and FAS expressed on the target cells. Indeed, engagement of FAS ligand by FAS leads to target cell death by apoptosis53. After the target cell is marked for death, the NK cell detaches and can move on to find another potential target. This ability to engage multiple targets sequentially is referred to as ‘serial killing’, and improves NK cell immunity. Moreover, the production of pro-inflammatory cytokines such as IFNγ and TNF by NK cells might also exert anti-proliferating, anti-angiogenic and pro-apoptotic effects on cancer cells, which could contribute to their antitumour activity54.
Why harness NK cells for cancer treatment?
Immunotherapy has undoubtedly revolutionized clinical oncology over the past decade55,56. In particular, immune checkpoint inhibitors and CAR-T cells have shown impressive responses across multiple malignancies55,56. However, only a subset of patients benefit from these treatments and unmet medical needs remain high. New immunotherapies, in particular, approaches with the potential to circumvent the ability of tumours to evade T cell-directed strategies, are warranted. Given the critical role that innate immune responses have in immunity, harnessing these responses opens new possibilities for tumour control57 but remains a challenge58. In that regard, NK cells have a unique set of antitumour properties that differ from those of T cells (Table 1). NK cells do not require antigen-specific priming and recognize a wide range of cells in distress, regardless of their embryological origin or the type of cell-stress trigger49,50. They also exhibit the notable property of controlling the development of metastases by putting metastatic cells into a dormant state through the production of IFNγ or by killing circulating tumour cells before they invade the metastatic niche59,60,61. Importantly, NK cells are not only killer cells that help to suppress tumours, but they are also involved in the generation, shaping and maintenance of adaptive immune responses by promoting, for example, the antigen-presenting function of dendritic cells, such as type 1 conventional dendritic cells at the tumour bed, through the secretion of FLT-3L, XCL1 and CCL5, and by acting directly on T cells through IFNγ62,63,64. In contrast to T cells, infusion of NK cells has been shown to be safe in patients with allogeneic grafts, as NK cells do not mediate graft-versus-host disease49,50,65,66. Finally, a very important distinguishing factor between T and NK cells lies in the increase in NK cell function when tumour cells downregulate MHC-I expression on the cell surface. Loss of MHC-I expression is a common T cell immune evasion mechanism67. By contrast, as NK cells express inhibitory MHC-I receptors, MHC-I loss contributes to the recognition and efficient elimination of tumour cells by NK cells68,69,70,71. Thus, several features of NK cell biology make their use an interesting and complementary to other modalities used in oncology, including monoclonal-antibody-based therapies, cell-based therapies or a combination of both (Fig. 2).

Two primary therapeutic strategies are being explored to enhance the antitumour efficacy of NK cells: monoclonal-antibody-based therapies (top) and cell-based therapies (bottom). The monoclonal-antibody-based NK therapies encompass the activation of NK cell antitumour immunity using immune checkpoint inhibitors (red antibodies) such as anti-LAG3, anti-NKG2A, anti-TIM-3 and anti-TIGIT monoclonal antibodies, and the augmentation of NK cell antitumour response through monoclonal-antibody-derived tools that stimulate their activating receptors, such as NK cell engagers (light green). The cell-based NK therapies use various sources of NK cell products that are injected into the patients, such as ex vivo conditioned NK cells, genetically manipulated NK cells and CAR NK cells. Activating and inhibitory NK cell receptors (purple) and their cognate ligands expressed on tumour cells are shown (dark green).
Unleashing NK cells
NK cell activity can be restrained by the engagement of checkpoint inhibitors expressed on their surface. Several inhibitory receptors such as NKG2A, lymphocyte activation gene 3 (LAG3), T cell immunoglobulin and mucin domain-containing 3 (TIM-3) and T cell immunoreceptor with Ig and immunoreceptor tyrosine-based inhibitory motif domains (TIGIT), which mediate inhibition on both NK and T cells, have been described. A number of monoclonal antibodies blocking these immune checkpoints are currently undergoing clinical evaluation. In addition to the anticipated indirect effect on T cell function therapies, both preclinical and clinical data point to their potential impact on unleashing the NK cell compartment (Fig. 2 and Table 2). Here we present studies assessing the therapeutic impact of these four inhibitory checkpoint inhibitors on NK cell functionality. However, more research is warranted to understand the effect of targeting these inhibitory cell surface receptors on NK cells and their potential contribution to therapeutic response.
NKG2A
The CD94–NKG2A heterodimer receptor is expressed by approximately half of NK cells, as well as by a subset of CD8+ T cells. Blocking NKG2A can unleash both T cell- and NK-cell-mediated antitumour immune responses72,73. One such antibody is monalizumab, a humanized IgG4 antibody that specifically blocks the interaction between human NKG2A and its cognate non-classical MHC-I molecule HLA-E72. Current development efforts for monalizumab are focused on evaluating its efficacy in various combination strategies for lung cancer.
LAG3
LAG3 is expressed on various immune cells, including NK cells, activated and exhausted T cells, B cells, regulatory T cells and dendritic cells74,75,76,77. LAG3 is a protein that shares similarities with the CD4 molecule and has the ability to interact with MHC-II molecules78,79. Moreover, LAG3 can also bind to galectin-3, fibrinogen-like protein 1 and liver sinusoidal endothelial cell lectin80,81,82,83,84. The exact mechanism of action of LAG3 is not fully understood at this time. Nevertheless, it is a promising immunotherapeutic target with more than 20 LAG3-targeting therapeutics in clinical trials. Within the class, relatlimab has been approved in combination with the anti-PD-1 antibody nivolumab for the treatment of unresectable or metastatic melanoma. Preclinical studies have shown that blocking LAG3 boosts NK cell tumour immunity85.
TIM-3
TIM-3 is an inhibitory receptor that is expressed by various immune cells, including T cells, NK cells, regulatory T cells, dendritic cells and macrophages. In healthy adults, NK1 cells and a subset of NK2 cells express TIM-386. Blockade of TIM-3 in combination with anti-PD1 suppress disease progression in MHC-I-deficient tumour-bearing mice87. In patients with metastatic melanoma, NK cells are functionally impaired compared with those from healthy individuals. This impairment was reversed by blocking TIM-388. Furthermore, TIM-3 expression is correlated with the disease stage, and blocking TIM-3 reverses the dysfunction of NK cells in patients with lung adenocarcinoma89. In multiple myeloma, both peripheral blood and bone marrow NK cells from patients express higher levels of TIM-3 compared with the control individuals90. Blockade of TIM-3 restored NK-cell-mediated killing of multiple myeloma tumour cells in vitro and significantly inhibited tumour growth in mouse models of multiple myeloma.
TIGIT
TIGIT is an inhibitory receptor expressed by T cells and NK cells. It binds to its ligands, poliovirus receptor (encoded by PVR, also known as CD155), nectin-2 and nectin-391, triggering inhibitory signalling. TIGIT competes with DNAM-1 (encoded by CD226) for binding to PVR and nectin-292. TIGIT signalling reduces NK cytotoxicity and cytokine release. TIGIT-positive NK cells have been detected in various human cancers and tumour-infiltrating lymphocytes isolated from patients with colorectal cancer. Blockade of TIGIT restored NK cell dysfunction and promoted NK-cell-mediated tumour immunity in different mouse models of cancer93.
Boosting NK cells
Immune cell engagers are bioengineered molecules designed to steer immune cells toward tumours. These engagers primarily consist of multi-armed antibodies that act as bridges between tumour cells and effector cells, facilitating the establishment of an immune synapse. Cell engagers achieve this by targeting tumour antigens and activating receptors expressed on immune effectors. NK cell engagers (NKCEs) have emerged as promising immunotherapies to redirect NK cells and activate their antitumour activity (Fig. 2).
CD16-based NKCEs
NK cells exhibit effector activity through ADCC by recognizing and killing cells opsonized with IgG1 and IgG3 antibodies through CD16a. A common polymorphism in CD16, either phenylalanine (Phe158, low affinity) or valine (Val158, high affinity), affects its affinity for the Fc portion of antibodies and can influence NK-cell-mediated ADCC94. Various strategies have been used to enhance NK-cell-mediated antitumour potential95. These include glycoengineering the Fc region of cytotoxic antibodies to improve their binding to CD16a96,97, as well as amino acid substitutions98. Another approach involves the development of CD16-engaging Fv fragments, which directly target CD16a on NK cells along with the tumour antigen. This approach bypasses the CD16 Phe/Val158 polymorphism, minimizes off-target interactions with complement or other Fc receptors, and inhibits the displacement of the Fc portion of the therapeutic monoclonal antibodies by the high concentration of circulating antibodies. AFM13, a bispecific NKCE targeting CD30 and CD16, has been evaluated as a monotherapy in a registrational phase 2 trial for CD30-positive relapsed or refractory lymphomas99. However, despite initial promising data as a monotherapy, the development of this drug is now focused on combinations, mainly with allogeneic NK cells. Other bispecific antibodies targeting EGFR (AFM24) and CD123 (AFM28) are currently undergoing phase 1/2 and phase 1 trials, respectively (Table 2). Moreover, several antibodies targeting CD16 and tumour antigens such as CD19, CD20, CD33, CD133 and EPCAM have demonstrated efficacy in preclinical studies100,101,102,103.
Trispecific natural killer engagers have been developed by incorporating an IL-15 cytokine element linking the two antibody domains104. IL-15 provides activation, proliferation and survival signals to NK cells105. GTB-3550 (anti-CD16, anti-CD33 and IL-15) showed a clinical benefit but a second-generation camelid nanobody CD33-targeting trispecific natural killer engager (GTB-3650) has been developed with improved tissue penetration106. Furthermore, GTB-7550, targeting CD19 (anti-CD16, anti-CD19 and IL-15), has shown enhanced NK cell proliferation and function against multiple B cell malignancies in vitro and is currently undergoing preclinical testing107. Note that the expression of CD16 is downregulated in the tumour microenvironment (TME) through its shedding108, which may limit the benefit of these molecules.
NKG2D-based NKCEs
NKG2D is a cell surface activating receptor expressed on all NK cells in humans and mice, as well as on the cell surface of all CD8+ T cells in humans. The chronic binding of NKG2D to its cognate ligands on the cell surface can lead to desensitization of NK cells, impairing their effector functions109. An NK cell engager targeting NKG2D and the multiple-myeloma-associated antigen CS1 has demonstrated efficacy in a preclinical mouse model of multiple myeloma110. Another bispecific NKCE has been developed to target both NKG2D and HER2, promoting cytotoxicity of NK cells in vitro111. Furthermore, trispecific NK cell engager therapies targeting HER2 (DF1001), BCMA (DF3001), CD33 (DF2001) and EGFR (DF9001) on tumour cells are currently being evaluated in phase 1/2 or phase 1 clinical trials for several haematologic and solid tumours (Table 2). Early signs of clinical activity without dose-limiting toxicities have been reported for DF1001112. Moreover, several molecules have been designed fusing an NKG2D ligand (such as MICA, ULBP1 or ULBP2) with a single-chain variable fragment targeting a tumour antigen. These molecules directed against antigens such as BCMA, CEA, CD19, CD24, CD138 or VEGFR2 have demonstrated antitumour activity in both in vitro and in vivo preclinical models113,114.
Natural cytotoxicity receptor-based NKCEs
NKCEs have been developed to target activating receptors on NK cells with a preferential expression on NK cells, such as the natural cytotoxicity receptors NKp46 and NKp30. In contrast to CD16a and NKG2D, which are expressed by myeloid cells and T cells, respectively, and of which the cell surface expression is downregulated in cancer conditions115, NKp46 exhibits stable expression in various cancers. Although it is expressed by ILC1 cells, subpopulations of ILC3 cells and discrete T cell subsets, NKp46 is the most preferentially expressed activating cell surface receptor for NK cells116,117. The trispecific antibody-based NK cell engager therapeutic (ANKET) platform consists of an antigen-binding antibody fragment that engages NKp46 on NK cells, another antibody fragment that binds to a tumour antigen and an Fc fragment that binds to Fcγ receptors, such as CD16a on NK cells115. In vitro studies have shown that NKp46-ANKET induces potent NK cell activation and promotes NK-cell-mediated lysis of tumour cells. Across a variety of haematological and solid tumour models, NKp46-ANKET has been shown to effectively increase the recruitment of NK cells to the tumour site and control tumour growth115. An NKp46-ANKET targeting CD123 (NKp46-ANKETCD123 also known as SAR′579/IPH6101) is currently being evaluated in a phase 1/2 clinical trial for the treatment of relapsed/refractory AML118 (Table 2). Moreover, NKp46-ANKET targeting of CD19 or CD20 has shown promising preclinical results in promoting tumour cell killing in models of paediatric B cell precursor acute lymphoblastic leukaemia119. A tetraspecific NKp46-ANKET has also been developed, incorporating a variant of interleukin-2 (IL-2v) that stimulates the IL-2 receptor complex without binding to IL-2Rα (CD25) to prevent regulatory T cell activation and endothelial cell binding120. This tetraspecific NKp46-ANKET, targeting CD20, has demonstrated preferential NK cell activation and proliferation compared with T cells in vitro and massive NK cell infiltration at the tumour bed in preclinical models. Additional multifunctional NK cell engagers targeting NKp46 and alternative tumour antigens have been generated. For example, CYT-303 targets NKp46 and glypican 3, which is overexpressed in hepatocellular carcinomas, and has shown antitumour activity121. Another NK cell engager, CYT-338, targets NKp46 and CD38 and have demonstrated efficacy against multiple myeloma in vitro122.
NKp30 is a type I transmembrane activating receptor that is expressed on NK cells, subsets of ILC2 cells, ILC3 cells, and CD8+ αβ and γδ T cells in humans123,124. NKp30 binds to the surface molecule B7-H6 and the nuclear factor HLA-B-associated transcript 3 (BAT3) expressed in diverse tumour cell lines125. However, in tumour contexts, the soluble form of its ligand, B7-H6, has been associated with the downregulation of NKp30 on NK cells126. This downregulation could limit the potential of NKp30 as a target for bispecific antibodies. NKp30-NKCE targeting EGFR or BCMA has shown preclinical efficacy.
NKCEs have entered the clinic quite recently and, with the exception of AFM13, all molecules are still in phase 1 (Table 2). In contrast to T cell engagers, which can be associated with adverse events such as neurotoxicity and cytokine release syndrome (an acute systemic inflammatory syndrome characterized by fever and multiple-organ dysfunction), NK cell engagers aim to provide a safer alternative, which ongoing clinical trials seem to have confirmed so far. While T cell engagers have demonstrated single-agent activity in several haematologic malignancies, both NK cell and T cell engagers face a common challenge in effectively controlling solid tumours. Of the ten NKCEs in the clinic for which the tumour targeting antigens have been disclosed, three are directed against solid tumours (Table 2).
NK cells as drug products
In addition to monoclonal-antibody-based approaches, the use of NK cells as drug products constitutes a rapidly evolving sector of oncological biotherapy. This paradigm encompasses a spectrum of clinical strategies, including both autologous and allogeneic applications, leveraging cellular progenitors from diverse biological origins such as placental tissues, umbilical cord blood, peripheral blood and induced pluripotent stem cell-derived lineages. These approaches further diverge into distinct modalities, spanning from in vitro pre-activation techniques to cutting-edge genomic editing interventions49,50,127,128. Several cancer conditions and oncological treatments, notably chemotherapy, are known to attenuate both the abundance and the operative capacity of patient’s endogenous NK cells. This depletion underscores the therapeutic rationale for adoptive NK cell transfer, a strategy to enhance the efficacy and resilience of NK cells within the TME. Allogeneic NK cell infusions have emerged as particularly advantageous49,50,127,128. Their attributes include immediate availability, robust antitumoural potency against a wide spectrum of neoplastic targets and absence of graft-versus-host disease. Many ongoing clinical trials are investigating the therapeutic efficacy of allogeneic NK cells across a variety of malignancies, underscoring both the potential and the burgeoning research interest in this domain. The favourable safety profile of allogeneic NK cell infusions has catalysed the development of ready-to-use off-the-shelf therapeutic products. However, allogeneic NK cell infusions necessitate preconditioning regimens, currently comprising agents such as fludarabine and cyclophosphamide to mitigate rejection risks49,50,127,128. Conversely, autologous NK cell therapy, which harnesses a patient’s own cellular resources, obviates the need for such conditioning treatments. Thus, autologous NK cells could have a role in clinical situations in which patients are not eligible for an allogeneic product (for example, in the context of consolidation treatment or minimal residual disease), because the latter would require harsh conditioning. Nonetheless, despite advancements in feeder-cell-free expansion protocols and the attainment of significant proliferative yields—typically 1,000- to 2,000-fold within a 2 to 3 week timeframe—the production of autologous NK cells remains time intensive129. This temporal demand represents a logistical challenge in the clinical deployment of autologous NK-cell-based interventions.
Enhancing NK cell performance through ex vivo conditioning
A crucial element of current adoptive NK cell therapy is administering large doses of robustly stimulated NK cells. Ex vivo cytokine stimulation is a widely used method for activating NK cells and facilitating their large-scale expansion. This enables their formulation and cryo-preservation, rendering them ready for infusion as a therapeutic product. Although IL-15 and IL-21 enhance NK cell proliferation and cytotoxicity, their effects vary based on dose, stimulation sequence and duration. Specifically, IL-15 enhances NK cell longevity and antitumour activity by boosting the metabolic fitness of NK cells130. IL-21 increases the NK cell ability to identify and kill tumour cells by raising activating receptor expression and promoting the production of key anti-tumour mediators such as IFNγ and TNF. Notably, sustained conditioning of NK cells with TGFβ during ex vivo expansion can render them more resilient in the TME131. Among the methods to stimulate the ex vivo expansion of NK cells, feeder cell methods using membrane-bound IL-15 (mbIL-15) and another leveraging soluble IL-12, IL-15 and IL-18 are notable. The latter produces NK cells in a cytokine-induced memory-like (CIML) state, reminiscent of viral reactivation in T cell memory132. Indeed, memory capacities of NK cells have been documented22 and their reactions against tumours could possibly be substantially and sustainably amplified133. Early clinical trials using these cytokine-activated NK cells appear promising both in terms of safety and efficacy134,135. Expansions using feeder cells with mbIL-21 (FC21) have also been successful, yielding a 1,000-fold increase in potent NK cells within 2 weeks. Such cells display enhanced cytotoxicity against diverse tumours136,137. The adoption of a cell-free method using particles derived from plasma membranes bearing mbIL-21 (PM21) has further expanded the potential of NK cell therapies to better enable manufacturability and safety. A key feature of these NK cell methods is that they allow for cryo-preservation to enable clinical delivery. Importantly, FC21-NK or PM21-NK cells, which result from these methods, align well with NKCEs due to their co-expression of NKp46 and CD16138. Adoption of these methodologies awaits results from clinical trials.
Enhancing NK cell function through genetic engineering
Despite their remarkable abilities, the effectiveness of NK cells in controlling tumour growth can be limited by immunosuppressive factors in the TME139,140,141. Advances in molecular engineering and gene editing will potentially help to overcome some of these limitations. A primary concern is TGFβ, which is overproduced in many cancers and dampens NK cell function. Introducing dominant-negative TGFβ receptors make NK cells resistant to these effects142. Hypoxia in the TME can hamper NK cell function139,140,141. Cells engineered to express a high-affinity CD16a receptor or IL-2 exhibit tolerance to hypoxic conditions comparable to those encountered in the TME143. Moreover, strategies to optimize the metabolism of NK cells have been shown to enhance their function. For example, deleting or reducing the cytokine-inducible SH2-containing protein (CISH) in NK cells enhances their metabolic fitness and anti-tumour response, offering another route for immunotherapy enhancement144,145. It has also been shown that MYC expression acts as a metabolic rheostat that regulates NK cell growth and effector responses146. Thus, ectopic MYC expression in NK cells could help promote cell survival and self-renewal and improve NK cell anti-tumour activity and persistence of NK cells in the TME.
The ability to target NK cells to tumours can be further enhanced by engineering them to express CARs that can recognize specific antigens on tumour cells, and trigger NK cell activation and cytotoxicity independent of the native NK cell receptors147. Several clinical trials are underway to address the efficacy of CAR NK cells against human tumours148. Promising results were reported from a first-in-human phase 1/2 clinical trial of a CAR NK designed to target CD19 for the treatment of B cell lymphoma149. In this case, the NK cells were also engineered to produce IL-15 to stimulate proliferation and persistence.
Driving durable responses through combination strategies
As alluded to earlier, NK cell therapies have achieved some success in haematologic malignancies, but their efficacy against solid tumours remains largely unexplored. As we deepen our understanding of tumour cell evasion mechanisms and develop therapies, the potential for combination strategies to magnify NK cell function becomes clear. These combinations aim to harness both innate and adaptive immunity for optimal anti-tumour activity. Current investigational practices combine NK cell therapies with cytotoxic antibodies or checkpoint inhibitors49,50,127,128. For example, allogeneic NK cells paired with the anti-PD-1 antibody pembrolizumab have demonstrated promising results in advanced biliary tract cancer, surpassing previous monotherapy outcomes150. An emerging method involves combining NK cell therapy with NKCEs, leading to both direct and NKCE-mediated tumour attacks. For example, the AFM-13 and CIML-NK cell combination is undergoing clinical evaluation and has shown promisingearly signs of activity134. Oncolytic viruses, which target and eliminate cancer cells while stimulating immunity, can further potentiate NK cell therapy151. Their combination might amplify NK cell tumour infiltration and activity. Lastly, the combination of NK cell therapies and targeted therapies such as MEK and CDK4/6 inhibitors152, BH3 mimetics153, and radiotherapy154, represent promising oppotunities for therapeutic exploration. Depending on the treatment, they can trigger immunogenic cell deaths, making tumours more recognizable to the immune system. Moreover, targeted therapies may reduce the presence of immunosuppressive cells, fostering NK cell function. Together, these multifaceted approaches could herald a new era of enhanced tumour control.
Conclusions and future directions
Beyond the potential of four immune checkpoint inhibitors to activate both T cells and NK cells (Table 2), considerable advancements are evident in NK cell therapies. As it stands, the most advanced of these therapies are in phase 2 clinical trials, featuring at least one NK cell engager and seven NK cell products, of which two are CAR NK cells (Table 2). Over 40 additional clinical trials are in phase 1, with numerous assets under exploration at the preclinical stage, signal a burgeoning interest in harnessing NK cells for cancer therapy and a shift towards clinical validation.
NK cell therapy holds promise to improve oncology treatments. Achieving this vision demands progress in understanding NK cell biology, technology enhancements, efficient manufacturing methods and clear regulatory guidelines. With a plethora of immuno-oncology options available, the challenge lies in determining the best combinations partners for NK cell therapy tailored to specific patient needs and therapeutic scenarios. The broad tumour-recognition ability of NK cells increases demand for scalable, cost-effective manufacturing solutions. Maintaining cell viability and potency post-cryopreservation is crucial. This capability enables centralized, off-the-shelf production and wider distribution to clinics.
NK cell therapies that do not cause infusion-related complications may enable potential re-dosing or the possibility to use adoptive NK cells either as an induction therapy to lead to complete response followed by a bridging therapy with well-tolerated treatments, or as maintenance therapy. To optimize pharmacokinetic persistence and anti-tumour effects, the need for conditioning regimens is a frequent topic of debate. The resolution of this debate will probably require clinical studies to explicitly test the requirement of conditioning. The use of a conditioning chemotherapeutic regimen would be optimal if it could also have synergistic antitumour effects. A key element to assess such questions will be correlative studies to track adoptive versus endogenous NK cells to enable assessment of whether the use of conditioning makes a difference in the pharmacokinetics of the adoptive NK cells. Such studies will be key for back-translational knowledge on how to design future cell therapeutics.
Augmented NK cells
To amplify the therapeutic index of NK-cell-based treatments, there is a need to promote NK cell trafficking to the tumour sites, their metabolic profile and their effector capacity (both direct cytotoxicity and soluble factor production) (Box 1). This can be realized through specific drugs or advanced NK cell products. With regard to next-generation NK cell products, techniques such as gene editing (for example, targeting CISH155 or CD38156) or pretreatment with agents like nicotinamide157 can be used. As CD38 is an ectoenzyme that consumes NAD+, inhibition of CD38 or supplementation of nicotinamide leads to NAD+ accumulation and boosts NK cell metabolic fitness and effector functions156,157. Moreover, growing evidence highlights the importance of epigenetic regulation in immune cell differentiation and function. Exhausted CD8+ T cells display unique epigenetic modifications that set them apart from functional memory CD8+ T cells. Along this line, the histone-methyltransferase enhancer of zeste homologue 2 (encoded by EZH2), is known to influence NK cell differentiation and function158, prompting exploration of how harnessing NK cell epigenetic regulation might contribute to the efficacy of NK cell therapies.
NK cell persistence
The persistence of NK cells in the host is an important consideration in immunotherapy. The long-term persistence of CAR-T cells, spanning months to years, was initially postulated to contribute to sustained responses159. However, it remains to be explored whether the peak of cell expansion can also be a determinant of efficacy. Thus, how long NK cells should reside in the host to be effective, and how the duration could be extended to enhance the clinical efficacy remain to be clarified (Box 1). Robust pharmacokinetic studies during ongoing clinical studies and associated back-translational studies will be very important to better understand the immunological persistence and how it can be further improved.
Building on NK cell hallmarks
The ability of NK cells to identify and target stressed cells offers expansive therapeutic possibilities. Indeed, their ability to detect surface molecules on cells infected by intracellular pathogens or those in other stress conditions could be used to deploy NK cell therapies beyond the realm of cancer treatment. Whether these therapies are antibody based, solely cell based or a mixture of the two, they are poised to be explored in therapeutic areas such as infection, inflammation, ageing and metabolic disorders. Along this line, the development of autologous, non-genetically-modified NK cell products in Alzheimer’s disease and of allogeneic NK cell infusions alone or in combination with rituximab in lupus nephritis have been launched. Moreover, as follicular T cells promote autoimmunity and express high levels of PD-1, CAR NK cells expressing the extracellular domain of PDL-1 have been generated and showed efficacy in preclinical settings160. Finally, it is noteworthy that the phenotypic similarities between NK and ILC1 cells and their potential functional differences have prompted investigations into whether some of the NK cell therapies, either monoclonal-antibody based or cell based, can involve ILC1. Regardless of this important clarification, the progress in the application of NK cells underscores their versatility and importance for the advancement of modern medicine.
References
Herberman, R. B., Nunn, M. E., Lavrin, D. H. & Asofsky, R. Effect of antibody to theta antigen on cell-mediated immunity induced in syngeneic mice by murine sarcoma virus. J. Natl Cancer Inst. 51, 1509–1512 (1973).
Oldham, R. K. & Herberman, R. B. Evaluation of cell-mediated cytotoxic reactivity against tumor associated antigens with 125I-iododeoxyuridine labeled target cells. J. Immunol. 111, 862–871 (1973).
Takasugi, M., Mickey, M. R. & Terasaki, P. I. Reactivity of lymphocytes from normal persons on cultured tumor cells. Cancer Res. 33, 2898–2902 (1973).
Kiessling, R., Klein, E. & Wigzell, H. “Natural” killer cells in the mouse. I. Cytotoxic cells with specificity for mouse Moloney leukemia cells. Specificity and distribution according to genotype. Eur. J. Immunol. 5, 112–117 (1975).
Kiessling, R., Klein, E., Pross, H. & Wigzell, H. “Natural” killer cells in the mouse. II. Cytotoxic cells with specificity for mouse Moloney leukemia cells. Characteristics of the killer cell. Eur. J. Immunol. 5, 117–121 (1975).
Vivier, E. et al. Innate or adaptive immunity? The example of natural killer cells. Science 331, 44–49 (2011).
Spits, H. et al. Innate lymphoid cells—a proposal for uniform nomenclature. Nat. Rev. Immunol. 13, 145–149 (2013).
Vivier, E. et al. Innate lymphoid cells: 10 years on. Cell 174, 1054–1066 (2018).
Sojka, D. K. et al. Tissue-resident natural killer (NK) cells are cell lineages distinct from thymic and conventional splenic NK cells. eLife 3, e01659 (2014).
Moretta, L. et al. Human natural killer cells: their origin, receptors and function. Eur. J. Immunol. 32, 1205–1211 (2002).
Moretta, A., Bottino, C., Mingari, M. C., Biassoni, R. & Moretta, L. What is a natural killer cell? Nat. Immunol. 3, 6–8 (2002).
Vivier, E., Tomasello, E., Baratin, M., Walzer, T. & Ugolini, S. Functions of natural killer cells. Nat. Immunol. 9, 503–510 (2008).
Morvan, M. G. & Lanier, L. L. NK cells and cancer: you can teach innate cells new tricks. Nat. Rev. Cancer 16, 7–19 (2016).
Freud, A. G., Mundy-Bosse, B. L., Yu, J. & Caligiuri, M. A. The broad spectrum of human natural killer cell diversity. Immunity 47, 820–833 (2017).
Sender, R. et al. The total mass, number, and distribution of immune cells in the human body. Proc. Natl Acad. Sci. USA 120, e2308511120 (2023).
Lanier, L. L., Le, A. M., Civin, C. I., Loken, M. R. & Phillips, J. H. The relationship of CD16 (Leu-11) and Leu-19 (NKH-1) antigen expression on human peripheral blood NK cells and cytotoxic T lymphocytes. J. Immunol. 136, 4480–4486 (1986).
Crinier, A. et al. High-dimensional single-cell analysis identifies organ-specific signatures and conserved NK cell subsets in humans and mice. Immunity 49, 971–986 (2018).
Vivier, E. et al. High-dimensional single-cell analysis of natural killer cell heterogeneity in human blood. Preprint at Research Square https://doi.org/10.21203/rs.3.rs-3870228/v1 (2024).
Hendricks, D. W. et al. Cutting edge: NKG2ChiCD57+ NK cells respond specifically to acute infection with cytomegalovirus and not Epstein-Barr virus. J. Immunol. 192, 4492–4496 (2014).
Lopez-Verges, S. et al. CD57 defines a functionally distinct population of mature NK cells in the human CD56dimCD16+ NK-cell subset. Blood 116, 3865–3874 (2010).
Lopez-Verges, S. et al. Expansion of a unique CD57+NKG2Chi natural killer cell subset during acute human cytomegalovirus infection. Proc. Natl Acad. Sci. USA 108, 14725–14732 (2011).
Sun, J. C., Beilke, J. N. & Lanier, L. L. Adaptive immune features of natural killer cells. Nature 457, 557–561 (2009). This study, along with ref. 23, describes NK cells with memory-like functions.
Cooper, M. A. et al. Cytokine-induced memory-like natural killer cells. Proc. Natl Acad. Sci. USA 106, 1915–1919 (2009).
Cooper, M. A. & Yokoyama, W. M. Memory-like responses of natural killer cells. Immunol. Rev. 235, 297–305 (2010).
Lee, J. et al. Epigenetic modification and antibody-dependent expansion of memory-like NK cells in human cytomegalovirus-infected individuals. Immunity 42, 431–442 (2015).
Schlums, H. et al. Cytomegalovirus infection drives adaptive epigenetic diversification of NK cells with altered signaling and effector function. Immunity 42, 443–456 (2015).
Rückert, T., Lareau, C. A., Mashreghi, M. F., Ludwig, L. S. & Romagnani, C. Clonal expansion and epigenetic inheritance of long-lasting NK cell memory. Nat. Immunol. 23, 1551–1563 (2022).
Brownlie, D. et al. Expansions of adaptive-like NK cells with a tissue-resident phenotype in human lung and blood. Proc. Natl Acad. Sci. USA 118, e2016580118 (2021).
Tang, F. et al. A pan-cancer single-cell panorama of human natural killer cells. Cell 186, 4235–4251 (2023).
Spits, H., Bernink, J. H. & Lanier, L. NK cells and type 1 innate lymphoid cells: partners in host defense. Nat. Immunol. 17, 758–764 (2016).
Cortez, V. S. et al. SMAD4 impedes the conversion of NK cells into ILC1-like cells by curtailing non-canonical TGF-β signaling. Nat. Immunol. 18, 995–1003 (2017). This study, along with ref. 32, describes the TGFβ-induced transition of NK cells into ILC1-like cells with a decrease antitumour functions.
Gao, Y. et al. Tumor immunoevasion by the conversion of effector NK cells into type 1 innate lymphoid cells. Nat. Immunol. 18, 1004–1015 (2017).
Kansler, E. R. et al. Cytotoxic innate lymphoid cells sense cancer cell-expressed interleukin-15 to suppress human and murine malignancies. Nat. Immunol. 23, 904–915 (2022).
Scarno, G. et al. Divergent roles for STAT4 in shaping differentiation of cytotoxic ILC1 and NK cells during gut inflammation. Proc. Natl Acad. Sci. USA 120, e2306761120 (2023).
Lopes, N. et al. Tissue-specific transcriptional profiles and heterogeneity of natural killer cells and group 1 innate lymphoid cells. Cell Rep. Med. 3, 100812 (2022).
Nagasawa, M. et al. KLRG1 and NKp46 discriminate subpopulations of human CD117+CRTH2− ILCs biased toward ILC2 or ILC3. J. Exp. Med. 216, 1762–1776 (2019).
Crinier, A. et al. Multidimensional molecular controls defining NK/ILC1 identity in cancers. Semin. Immunol. 52, 101424 (2021).
Yudanin, N. A. et al. Spatial and temporal mapping of human innate lymphoid cells reveals elements of tissue specificity. Immunity 50, 505–519 (2019).
Björkström, N. K., Strunz, B. & Ljunggren, H. G. Natural killer cells in antiviral immunity. Nat. Rev. Immunol. 22, 112–123 (2022).
Vivier, E., Nunes, J. A. & Vely, F. Natural killer cell signaling pathways. Science 306, 1517–1519 (2004).
Long, E. O. Negative signaling by inhibitory receptors: the NK cell paradigm. Immunol. Rev. 224, 70–84 (2008).
Gasser, S., Orsulic, S., Brown, E. J. & Raulet, D. H. The DNA damage pathway regulates innate immune system ligands of the NKG2D receptor. Nature 436, 1186–1190 (2005). This study, along with refs. 43–46, describes stress-induced ligands recognized by NK-cell-activating receptors.
Raulet, D. H. & Guerra, N. Oncogenic stress sensed by the immune system: role of natural killer cell receptors. Nat. Rev. Immunol. 9, 568–580 (2009).
Bauer, S. et al. Activation of NK cells and T cells by NKG2D, a receptor for stress-inducible MICA. Science 285, 727–729 (1999).
Sen Santara, S. et al. The NK cell receptor NKp46 recognizes ecto-calreticulin on ER-stressed cells. Nature 616, 348–356 (2023).
Brandt, C. S. et al. The B7 family member B7-H6 is a tumor cell ligand for the activating natural killer cell receptor NKp30 in humans. J. Exp. Med. 206, 1495–1503 (2009).
Wolf, N. K., Kissiov, D. U. & Raulet, D. H. Roles of natural killer cells in immunity to cancer, and applications to immunotherapy. Nat. Rev. Immunol. 23, 90–105 (2023).
Lanier, L. L. Up on the tightrope: natural killer cell activation and inhibition. Nat. Immunol. 9, 495–502 (2008).
Myers, J. A. & Miller, J. S. Exploring the NK cell platform for cancer immunotherapy. Nat. Rev. Clin. Oncol. 18, 85–100 (2021).
Laskowski, T. J., Biederstadt, A. & Rezvani, K. Natural killer cells in antitumour adoptive cell immunotherapy. Nat. Rev. Cancer 22, 557–575 (2022).
Hatjiharissi, E. et al. Increased natural killer cell expression of CD16, augmented binding and ADCC activity to rituximab among individuals expressing the FcγRIIIa-158 V/V and V/F polymorphism. Blood 110, 2561–2564 (2007).
Friedman, D. et al. Natural killer cell immune synapse formation and cytotoxicity are controlled by tension of the target interface. J. Cell Sci. 134, jcs258570 (2021).
Lettau, M., Paulsen, M., Schmidt, H. & Janssen, O. Insights into the molecular regulation of FasL (CD178) biology. Eur. J. Cell Biol. 90, 456–466 (2011).
Braumuller, H. et al. T-helper-1-cell cytokines drive cancer into senescence. Nature 494, 361–365 (2013).
Sharma, P. et al. Immune checkpoint therapy-current perspectives and future directions. Cell 186, 1652–1669 (2023).
Baker, D. J., Arany, Z., Baur, J. A., Epstein, J. A. & June, C. H. CAR T therapy beyond cancer: the evolution of a living drug. Nature 619, 707–715 (2023).
Demaria, O. et al. Harnessing innate immunity in cancer therapy. Nature 574, 45–56 (2019).
Cao, L. L. & Kagan, J. C. Targeting innate immune pathways for cancer immunotherapy. Immunity 56, 2206–2217 (2023).
Lopez-Soto, A., Gonzalez, S., Smyth, M. J. & Galluzzi, L. Control of metastasis by NK cells. Cancer Cell 32, 135–154 (2017). This report, along with refs. 60 and 61, describes anti-metastatic functions of NK cells.
Liu, X. et al. Immune checkpoint HLA-E:CD94-NKG2A mediates evasion of circulating tumor cells from NK cell surveillance. Cancer Cell 41, 272–287 (2023).
Correia, A. L. et al. Hepatic stellate cells suppress NK cell-sustained breast cancer dormancy. Nature 594, 566–571 (2021).
Bottcher, J. P. et al. NK cells stimulate recruitment of cDC1 into the tumor microenvironment promoting cancer immune control. Cell 172, 1022–1037 (2018). This study, along with ref. 63, describes the role of cytokines and chemokines secreted by NK cells in eliciting a multicellular antitumour immune response.
Barry, K. C. et al. A natural killer-dendritic cell axis defines checkpoint therapy-responsive tumor microenvironments. Nat. Med. 24, 1178–1191 (2018).
Kirchhammer, N. et al. NK cells with tissue-resident traits shape response to immunotherapy by inducing adaptive antitumor immunity. Sci. Transl. Med. 14, eabm9043 (2022).
Olson, J. A. et al. NK cells mediate reduction of GVHD by inhibiting activated, alloreactive T cells while retaining GVT effects. Blood 115, 4293–4301 (2010).
Simonetta, F., Alvarez, M. & Negrin, R. S. Natural killer cells in graft-versus-host-disease after allogeneic hematopoietic cell transplantation. Front. Immunol. 8, 465 (2017).
Dhatchinamoorthy, K., Colbert, J. D. & Rock, K. L. Cancer immune evasion through loss of MHC class I antigen presentation. Front. Immunol. 12, 636568 (2021).
Kärre, K., Ljunggren, H. G., Piontek, G. & Kiessling, R. Selective rejection of H-2-deficient lymphoma variants suggests alternative immune defence strategy. Nature 319, 675–678 (1986).
Moretta, A. et al. Major histocompatibility complex class I-specific receptors on human natural killer and T lymphocytes. Immunol. Rev. 155, 105–117 (1997).
Yokoyama, W. M. Natural killer cell immune responses. Immunol. Res. 32, 317–325 (2005).
Parham, P. MHC class I molecules and KIRs in human history, health and survival. Nat. Rev. Immunol. 5, 201–214 (2005).
André, P. et al. Anti-NKG2A mAb is a checkpoint inhibitor that promotes anti-tumor immunity by unleashing both T and NK cells. Cell 175, 1731–1743 (2018). This study, along with ref. 74, describes the role of monalizumab in promoting antitumour immunity.
van Montfoort, N. et al. NKG2A blockade potentiates CD8 T cell immunity induced by cancer vaccines. Cell 175, 1744–1755 (2018).
Andreae, S., Buisson, S. & Triebel, F. MHC class II signal transduction in human dendritic cells induced by a natural ligand, the LAG-3 protein (CD223). Blood 102, 2130–2137 (2003).
Baixeras, E. et al. Characterization of the lymphocyte activation gene 3-encoded protein. A new ligand for human leukocyte antigen class II antigens. J. Exp. Med. 176, 327–337 (1992).
Kisielow, M., Kisielow, J., Capoferri-Sollami, G. & Karjalainen, K. Expression of lymphocyte activation gene 3 (LAG-3) on B cells is induced by T cells. Eur. J. Immunol. 35, 2081–2088 (2005).
Merino, A. et al. Chronic stimulation drives human NK cell dysfunction and epigenetic reprograming. J. Clin. Invest. 129, 3770–3785 (2019).
Huard, B. et al. Characterization of the major histocompatibility complex class II binding site on LAG-3 protein. Proc. Natl Acad. Sci. USA 94, 5744–5749 (1997).
Workman, C. J., Rice, D. S., Dugger, K. J., Kurschner, C. & Vignali, D. A. Phenotypic analysis of the murine CD4-related glycoprotein, CD223 (LAG-3). Eur. J. Immunol. 32, 2255–2263 (2002).
Hu, S., Liu, X., Li, T., Li, Z. & Hu, F. LAG3 (CD223) and autoimmunity: emerging evidence. J. Autoimmun. 112, 102504 (2020).
Kouo, T. et al. Galectin-3 shapes antitumor immune responses by suppressing CD8+ T cells via LAG-3 and inhibiting expansion of plasmacytoid dendritic cells. Cancer Immunol. Res. 3, 412–423 (2015).
Liu, E. et al. Cord blood NK cells engineered to express IL-15 and a CD19-targeted CAR show long-term persistence and potent antitumor activity. Leukemia 32, 520–531 (2018).
Sordo-Bahamonde, C. et al. LAG-3 blockade with relatlimab (BMS-986016) restores anti-leukemic responses in chronic lymphocytic leukemia. Cancers 13, 2112 (2021).
Wang, J. et al. Fibrinogen-like protein 1 is a major immune inhibitory ligand of LAG-3. Cell 176, 334–347 (2019).
Khan, M., Arooj, S. & Wang, H. NK cell-based immune checkpoint inhibition. Front. Immunol. 11, 167 (2020).
Ndhlovu, L. C. et al. Tim-3 marks human natural killer cell maturation and suppresses cell-mediated cytotoxicity. Blood 119, 3734–3743 (2012).
Seo, H. et al. IL21 therapy combined with PD-1 and Tim-3 blockade provides enhanced NK cell antitumor activity against MHC class I-deficient tumors. Cancer Immunol. Res. 6, 685–695 (2018).
da Silva, I. P. et al. Reversal of NK-cell exhaustion in advanced melanoma by Tim-3 blockade. Cancer Immunol. Res. 2, 410–422 (2014).
Xu, L. et al. Increased Tim-3 expression in peripheral NK cells predicts a poorer prognosis and Tim-3 blockade improves NK cell-mediated cytotoxicity in human lung adenocarcinoma. Int. Immunopharmacol. 29, 635–641 (2015).
Jiang, W. et al. Tim-3 blockade elicits potent anti-multiple myeloma immunity of natural killer cells. Front. Oncol .12, 739976 (2022).
Sanchez-Correa, B. et al. DNAM-1 and the TIGIT/PVRIG/TACTILE axis: novel immune checkpoints for natural killer cell-based cancer immunotherapy. Cancers 11, 877 (2019).
Alteber, Z. et al. Therapeutic targeting of checkpoint receptors within the DNAM1 axis. Cancer Discov. 11, 1040–1051 (2021).
Zhang, Q. et al. Blockade of the checkpoint receptor TIGIT prevents NK cell exhaustion and elicits potent anti-tumor immunity. Nat. Immunol. 19, 723–732 (2018).
Temming, A. R. et al. Functional attributes of antibodies, effector cells, and target cells affecting nk cell-mediated antibody-dependent cellular cytotoxicity. J. Immunol. 203, 3126–3135 (2019).
van der Horst, H. J., Nijhof, I. S., Mutis, T. & Chamuleau, M. E. D. Fc-engineered antibodies with enhanced Fc-effector function for the treatment of B-cell malignancies. Cancers 12, 3041 (2020).
Shields, R. L. et al. Lack of fucose on human IgG1 N-linked oligosaccharide improves binding to human FcγRIII and antibody-dependent cellular toxicity. J. Biol. Chem. 277, 26733–26740 (2002).
Suzuki, E. et al. A nonfucosylated anti-HER2 antibody augments antibody-dependent cellular cytotoxicity in breast cancer patients. Clin. Cancer Res. 13, 1875–1882 (2007).
Wang, X., Mathieu, M. & Brezski, R. J. IgG Fc engineering to modulate antibody effector functions. Protein Cell 9, 63–73 (2018).
Rothe, A. et al. A phase 1 study of the bispecific anti-CD30/CD16A antibody construct AFM13 in patients with relapsed or refractory Hodgkin lymphoma. Blood 125, 4024–4031 (2015).
Wiernik, A. et al. Targeting natural killer cells to acute myeloid leukemia in vitro with a CD16 × 33 bispecific killer cell engager and ADAM17 inhibition. Clin. Cancer Res. 19, 3844–3855 (2013).
Vallera, D. A. et al. Heterodimeric bispecific single-chain variable-fragment antibodies against EpCAM and CD16 induce effective antibody-dependent cellular cytotoxicity against human carcinoma cells. Cancer Biother. Radiopharm. 28, 274–282 (2013).
Gleason, M. K. et al. Bispecific and trispecific killer cell engagers directly activate human NK cells through CD16 signaling and induce cytotoxicity and cytokine production. Mol. Cancer Ther. 11, 2674–2684 (2012).
Gleason, M. K. et al. CD16xCD33 bispecific killer cell engager (BiKE) activates NK cells against primary MDS and MDSC CD33+ targets. Blood 123, 3016–3026 (2014).
Vallera, D. A. et al. IL15 trispecific killer engagers (TriKE) make natural killer cells specific to CD33+ targets while also inducing persistence, in vivo expansion, and enhanced function. Clin. Cancer Res. 22, 3440–3450 (2016).
Schmohl, J. U. et al. Engineering of anti-CD133 trispecific molecule capable of inducing NK expansion and driving antibody-dependent cell-mediated cytotoxicity. Cancer Res. Treat. 49, 1140–1152 (2017).
Felices, M. et al. Potent cytolytic activity and specific IL15 delivery in a second-generation trispecific killer engager. Cancer Immunol. Res. 8, 1139–1149 (2020).
Miller, J. et al. P618: second-generation CD19 targeting tri-specific killer engager drives robust nk cell function against B cell malignancies. HemaSphere 6, 517–518 (2022).
Romee, R. et al. NK cell CD16 surface expression and function is regulated by a disintegrin and metalloprotease-17 (ADAM17). Blood 121, 3599–3608 (2013).
Coudert, J. D., Scarpellino, L., Gros, F., Vivier, E. & Held, W. Sustained NKG2D engagement induces cross-tolerance of multiple distinct NK cell activation pathways. Blood 111, 3571–3578 (2008).
Chan, W. K. et al. A CS1-NKG2D bispecific antibody collectively activates cytolytic immune cells against multiple myeloma. Cancer Immunol. Res. 6, 776–787 (2018).
Raynaud, A. et al. Anti-NKG2D single domain-based antibodies for the modulation of anti-tumor immune response. Oncoimmunology 10, 1854529 (2020).
Safran, H. et al. Phase 1/2 study of DF1001, a novel tri-specific, NK cell engager therapy targeting HER2, in patients with advanced solid tumors: phase 1 DF1001 monotherapy dose-escalation results. J. Clin. Oncol. 41, 2508–2508 (2023).
von Strandmann, E. P. et al. A novel bispecific protein (ULBP2-BB4) targeting the NKG2D receptor on natural killer (NK) cells and CD138 activates NK cells and has potent antitumor activity against human multiple myeloma in vitro and in vivo. Blood 107, 1955–1962 (2006).
Wang, Y. et al. BCMA-targeting bispecific antibody that simultaneously stimulates NKG2D-enhanced efficacy against multiple myeloma. J. Immunother. 43, 175–188 (2020).
Gauthier, L. et al. Multifunctional natural killer cell engagers targeting NKp46 trigger protective tumor immunity. Cell 177, 1701–1713 (2019).
Pessino, A. et al. Molecular cloning of NKp46: a novel member of the immunoglobulin superfamily involved in triggering of natural cytotoxicity. J. Exp. Med. 188, 953–960 (1998).
Walzer, T. et al. Identification, activation, and selective in vivo ablation of mouse NK cells via NKp46. Proc. Natl Acad. Sci. USA 104, 3384–3389 (2007).
Gauthier, L. et al. Control of acute myeloid leukemia by a trifunctional NKp46-CD16a-NK cell engager targeting CD123. Nat. Biotechnol. 41, 1296–1306 (2023).
Colomar-Carando, N. et al. Exploiting natural killer cell engagers to control pediatric B-cell precursor acute lymphoblastic leukemia. Cancer Immunol. Res. 10, 291–302 (2022).
Demaria, O. et al. Antitumor immunity induced by antibody-based natural killer cell engager therapeutics armed with not-alpha IL-2 variant. Cell Rep. Med. 3, 100783 (2022). This study describes the first tetraspecific NK cell engagers armed with a variant of IL-2.
Arulanandam, A. et al. 756P glypican-3 (GPC3) and NKp46 directed FLEX-NK engager antibody (CYT-303) recruits natural killer (NK) cells to tumors in a preclinical hepatocellular carcinoma (HCC) mouse model. Ann. Oncol. 33, S889 (2022).
Lin, L. et al. P842: novel multifunctional tetravalent CD38 NKp46 FLEX-NK engagers actively target and kill multiple myeloma cells. HemaSphere 6, 736–737 (2022).
Pende, D. et al. Identification and molecular characterization of NKp30, a novel triggering receptor involved in natural cytotoxicity mediated by human natural killer cells. J. Exp. Med. 190, 1505–1516 (1999).
Pinheiro, P. F., Justino, G. C. & Marques, M. M. NKp30—a prospective target for new cancer immunotherapy strategies. Br. J. Pharmacol. 177, 4563–4580 (2020).
Medjouel Khlifi, H., Guia, S., Vivier, E. & Narni-Mancinelli, E. Role of the ITAM-bearing receptors expressed by natural killer cells in cancer. Front. Immunol. 13, 898745 (2022).
Pesce, S. et al. B7-H6-mediated downregulation of NKp30 in NK cells contributes to ovarian carcinoma immune escape. Oncoimmunology 4, e1001224 (2015).
Berrien-Elliott, M. M., Jacobs, M. T. & Fehniger, T. A. Allogeneic natural killer cell therapy. Blood 141, 856–868 (2023).
Piccinelli, S., Romee, R. & Shapiro, R. M. The natural killer cell immunotherapy platform: an overview of the landscape of clinical trials in liquid and solid tumors. Semin. Hematol. 60, 42–51 (2023).
Nahi, H. et al. Autologous NK cells as consolidation therapy following stem cell transplantation in multiple myeloma. Cell Rep. Med. 3, 100508 (2022).
Li, L. et al. Loss of metabolic fitness drives tumor resistance after CAR-NK cell therapy and can be overcome by cytokine engineering. Sci. Adv. 9, eadd6997 (2023).
Foltz, J. A., Moseman, J. E., Thakkar, A., Chakravarti, N. & Lee, D. A. TGFβ imprinting during activation promotes natural killer cell cytokine hypersecretion. Cancers 10, 423 (2018).
Terrén, I. et al. Cytokine-induced memory-like NK cells: from the basics to clinical applications. Front. Immunol. 13, 884648 (2022).
Fehniger, T. A. & Cooper, M. A. Harnessing NK cell memory for cancer immunotherapy. Trends Immunol. 37, 877–888 (2016).
Kerbauy, L. N. et al. Combining AFM13, a bispecific CD30/CD16 antibody, with cytokine-activated blood and cord blood-derived NK cells facilitates CAR-like responses against CD30+ malignancies. Clin. Cancer Res. 27, 3744–3756 (2021). This study describes the advantages of combining NK cell infusions and NK cell engagers.
Shapiro, R. M. et al. Expansion, persistence, and efficacy of donor memory-like NK cells infused for posttransplant relapse. J. Clin. Invest. 132, e154334 (2022).
Oyer, J. L. et al. Natural killer cells stimulated with PM21 particles expand and biodistribute in vivo: Clinical implications for cancer treatment. Cytotherapy 18, 653–663 (2016).
Denman, C. J. et al. Membrane-bound IL-21 promotes sustained ex vivo proliferation of human natural killer cells. PLoS ONE 7, e30264 (2012).
Oyer, J. L. et al. Cryopreserved PM21-particle-expanded natural killer cells maintain cytotoxicity and effector functions in vitro and in vivo. Front. Immunol. 13, 861681 (2022).
Coyle, K. M., Hawke, L. G. & Ormiston, M. L. Addressing natural killer cell dysfunction and plasticity in cell-based cancer therapeutics. Cancers 15, 1743 (2023).
Balzasch, B. M. & Cerwenka, A. Microenvironmental signals shaping NK-cell reactivity in cancer. Eur. J. Immunol. 53, e2250103 (2023).
Tong, L. et al. NK cells and solid tumors: therapeutic potential and persisting obstacles. Mol. Cancer 21, 206 (2022).
Burga, R. A. et al. Engineering the TGFβ receptor to enhance the therapeutic potential of natural killer cells as an immunotherapy for neuroblastoma. Clin. Cancer Res. 25, 4400–4412 (2019).
Solocinski, K. et al. Overcoming hypoxia-induced functional suppression of NK cells. J. Immunother. Cancer 8, e000246 (2020).
Zhu, H. et al. Metabolic reprograming via deletion of CISH in human iPSC-derived NK cells promotes in vivo persistence and enhances anti-tumor activity. Cell Stem Cell 27, 224–237 (2020).
Daher, M. et al. Targeting a cytokine checkpoint enhances the fitness of armored cord blood CAR-NK cells. Blood 137, 624–636 (2021).
Loftus, R. M. et al. Amino acid-dependent cMyc expression is essential for NK cell metabolic and functional responses in mice. Nat. Commun. 9, 2341 (2018).
Guedan, S., Calderon, H., Posey, A. D. Jr & Maus, M. V. Engineering and design of chimeric antigen receptors. Mol. Ther. Methods Clin. Dev. 12, 145–156 (2019).
Biederstädt, A. & Rezvani, K. Engineering the next generation of CAR-NK immunotherapies. Int. J. Hematol. 114, 554–571 (2021).
Liu, E. et al. Use of CAR-transduced natural killer cells in CD19-positive lymphoid tumors. N. Engl. J. Med. 382, 545–553 (2020). This study describes the clinical efficacy of CAR NK cells against B cell malignancies.
Leem, G. et al. Safety and efficacy of allogeneic natural killer cells in combination with pembrolizumab in patients with chemotherapy-refractory biliary tract cancer: a multicenter open-label phase 1/2a trial. Cancers 14, 4229 (2022).
Marotel, M., Hasim, M. S., Hagerman, A. & Ardolino, M. The two-faces of NK cells in oncolytic virotherapy. Cytokine Growth Factor Rev. 56, 59–68 (2020).
Ruscetti, M. et al. NK cell-mediated cytotoxicity contributes to tumor control by a cytostatic drug combination. Science 362, 1416–1422 (2018).
Pan, R., Ryan, J., Pan, D., Wucherpfennig, K. W. & Letai, A. Augmenting NK cell-based immunotherapy by targeting mitochondrial apoptosis. Cell 185, 1521–1538 (2022).
Walle, T. et al. Radiotherapy orchestrates natural killer cell dependent antitumor immune responses through CXCL8. Sci. Adv. 8, eabh4050 (2022).
Delconte, R. B. et al. CIS is a potent checkpoint in NK cell-mediated tumor immunity. Nat. Immunol. 17, 816–824 (2016).
Cichocki, F. et al. Quadruple gene-engineered natural killer cells enable multi-antigen targeting for durable antitumor activity against multiple myeloma. Nat. Commun. 13, 7341 (2022).
Cichocki, F. et al. Nicotinamide enhances natural killer cell function and yields remissions in patients with non-Hodgkin lymphoma. Sci. Transl. Med. 15, eade3341 (2023).
Lordo, M. R., Stiff, A. R., Oakes, C. C. & Mundy-Bosse, B. L. Effects of epigenetic therapy on natural killer cell function and development in hematologic malignancy. J. Leukocyte Biol. 113, 518–524 (2023).
Young, R. M., Engel, N. W., Uslu, U., Wellhausen, N. & June, C. H. Next-generation CAR T-cell therapies. Cancer Discov. 12, 1625–1633 (2022).
Reighard, S. D. et al. Therapeutic targeting of follicular T cells with chimeric antigen receptor-expressing natural killer cells. Cell Rep. Med. 1, 100003 (2020).
Acknowledgements
We thank the colleagues at Innate-Pharma and CIML for help and advice, and M.-A. Rarivoson and K. Lam for help in the preparation of the manuscript. The E.V. laboratory at CIML and Assistance-Publique des Hôpitaux de Marseille was supported by funding from the European Research Council (ERC) under the EU Horizon 2020 research and innovation program (TILC, grant agreement no. 694502), the Agence Nationale de la Recherche including the PIONEER Project (ANR-17-RHUS-0007), MSDAvenir, Innate Pharma, and institutional grants awarded to the CIML (INSERM, CNRS and Aix-Marseille University) and Marseille Immunopole.
Author information
Authors and Affiliations
Contributions
All of the authors participated in writing the manuscript.
Corresponding author
Ethics declarations
Competing interests
E.V. and S.C. are employees of Innate Pharma. R.Y.I. and V.R.F. are employees of SANOFI. The other authors declare no competing interests.
Peer review
Peer review information
Nature thanks Hergen Spits and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Vivier, E., Rebuffet, L., Narni-Mancinelli, E. et al. Natural killer cell therapies. Nature 626, 727–736 (2024). https://doi.org/10.1038/s41586-023-06945-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41586-023-06945-1
- Springer Nature Limited