

Abstract
Gram-negative bacteria are extraordinarily difficult to kill because their cytoplasmic membrane is surrounded by an outer membrane that blocks the entry of most antibiotics. The impenetrable nature of the outer membrane is due to the presence of a large, amphipathic glycolipid called lipopolysaccharide (LPS) in its outer leaflet1. Assembly of the outer membrane requires transport of LPS across a protein bridge that spans from the cytoplasmic membrane to the cell surface. Maintaining outer membrane integrity is essential for bacterial cell viability, and its disruption can increase susceptibility to other antibiotics2,3,4,5,6. Thus, inhibitors of the seven lipopolysaccharide transport (Lpt) proteins that form this transenvelope transporter have long been sought7,8,9. A new class of antibiotics that targets the LPS transport machine in Acinetobacter was recently identified. Here, using structural, biochemical and genetic approaches, we show that these antibiotics trap a substrate-bound conformation of the LPS transporter that stalls this machine. The inhibitors accomplish this by recognizing a composite binding site made up of both the Lpt transporter and its LPS substrate. Collectively, our findings identify an unusual mechanism of lipid transport inhibition, reveal a druggable conformation of the Lpt transporter and provide the foundation for extending this class of antibiotics to other Gram-negative pathogens.
Similar content being viewed by others


Main
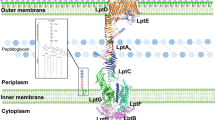
The outer membrane of Gram-negative bacteria is an asymmetric bilayer containing phospholipids in its inner leaflet and lipopolysaccharide (LPS) in its outer leaflet1,10,11,12,13. The biosynthesis of LPS is completed inside the cell at the inner membrane. LPS must be extracted from the inner membrane, moved across the periplasmic compartment and delivered through the outer membrane to the cell surface1,12,14,15,16 (Fig. 1a). To accomplish outer membrane biogenesis, the inner membrane components of the lipopolysaccharide transporter, LptB2FGC, form a subcomplex that couples ATP hydrolysis to extraction of LPS from the bilayer17,18,19,20,21,22, passing it to the protein bridge formed by the connected β-jellyroll domains of LptF, LptC, the soluble periplasmic protein LptA and the periplasmic portion of the integral membrane protein LptD (refs. 6,17,23,24,25,26,27,28,29). LptD, together with its associated lipoprotein LptE, form the outer membrane translocon that serves as a conduit for LPS to pass directly from the bridge into the outer leaflet of the outer membrane5,23,25,30,31.

a, Schematic of the seven protein LPS transport machine. b, Structures of macrocyclic peptides that prevent growth of Acinetobacter strains. The compounds were selected from those prepared during the drug discovery–development process32: compound 1 (RO7196472) was a potent hit found early in the discovery process; compound 2 (Zosurabalpin) is a clinical candidate; and compound 3 (RO7075573) was an important preclinical lead. Compound 2a, the epimer of compound 2 at the starred position, is an inactive compound that was used as a negative control. c, Cryo-EM structure of the inner membrane A. baylyi LptB2FG complex bound to LPS and 1. The drug has 500 Å2 contact with LptFG and 230 Å2 contact with LPS. Postprocessing of the map was carried out using DeepEMhancer. The unsharpened map is shown as an outline to show the positioning of the detergent micelle. Inset shows a close-up view of LPS and 1. LptB, LptF, LptG, LPS and 1 are coloured tan, green, blue, yellow and purple, respectively. d, Cryo-EM structure of Acinetobacter LptB2FG with Acinetobacter LPS and 1 bound in the lumen of the transporter in white superimposed with the structure of Acinetobacter LptB2FG bound to E. coli LPS and 1 (LptF, LptG, LPS and 1 are coloured green, blue, yellow and purple respectively). The overall r.m.s.d. is 0.44 Å over 7,999 atoms.
A family of macrocyclic peptides (Fig. 1b, 1–3) proposed to target the lipopolysaccharide transport machinery was recently identified (1, RO7196472; 2, Zosurabalpin; 3, RO7075573)32. These macrocyclic peptides all have potent and selective activity against Acinetobacter strains, including carbapenem-resistant A. baumannii. One macrocyclic peptide, Zosurabalpin (compound 2), is now undergoing clinical trials. Resistance mutations to compound 2 map to lptFG and biochemical experiments have shown that 2 blocks LPS extraction from liposomes containing Acinetobacter LptB2FGC (ref. 32). To determine the molecular mechanism by which these antibiotics inhibit transport of LPS, we sought to solve a structure of LptB2FG bound to a macrocyclic peptide. A. baumannii proteins expressed poorly and tended to aggregate, so we instead solved structures of A. baylyi LptB2FG with compounds 1–3 to high resolution using cryo-electron microscopy (cryo-EM). A. baylyi LptB2FG is about 85% identical to A. baumannii (Extended Data Fig. 1), is similarly susceptible to macrocyclic peptides 1–3 and mutations that provide resistance in A. baumannii also confer resistance to A. baylyi (see below; ref. 32).
Drug binds LPS within the transporter
We first solved a structure of LptB2FG in the presence of LPS and compound 1 to 3.0 Å resolution. Unexpectedly, compound 1 was found to form extensive contacts with both LptB2FG and a bound LPS molecule, which is a unique mode of inhibition (Fig. 1c and Extended Data Fig. 2). Because LptB2FG was heterologously expressed in Escherichia coli, the structure we obtained contained copurified E. coli LPS. To rule out the possibility that structural differences between E. coli and Acinetobacter LPS affect how LPS binds or how compound 1 interacts with the LptB2FG–LPS complex, we also purified LptB2FG from A. baylyi to trap the native Acinetobacter LPS (Extended Data Fig. 3). We obtained a structure of A. baylyi LptB2FG with Acinetobacter LPS and 1 and found only minor differences compared to the complex with E. coli LPS (Fig. 1d). The sugars attached to the Acinetobacter LPS are better resolved and an ordered detergent molecule observed in the structure solved using E. coli LPS is displaced to accommodate the extra lipid chain that is present on Acinetobacter LPS (Extended Data Fig. 3g,h). Notably, the drug contact interface is nearly identical regardless of the LPS chemotype (see below) and the protein conformation is also unaltered.
The compound binding pocket is lined by side chains of several amino acids in the transmembrane (TM) helices of LptF (Glu58, Glu249, Trp271, Val314, Ile317, Arg320 and Thr321) and LptG (Leu36) (Fig. 2). Previous morbidostat experiments carried out with compound 2 identified mutations that altered the corresponding residues in A. baumannii LptFG (ref. 32). To verify that alterations in these residues reduced susceptibility to the macrocyclic peptide antibiotics, we constructed A. baylyi strains encoding each LptFG variant identified from the morbidistat experiments and measured minimum inhibitory concentrations (MICs) for compounds 1–3 against A. baylyi. All the mutations decreased A. baylyi susceptibility to compounds 1, 2 and 3 (Fig. 2b and Supplementary Table 1), some by two to three orders of magnitude. We also purified two A. baylyi LptB2FGC complexes with individual LptF substitutions (E249K or I317N) that conferred high-level resistance to the macrocyclic peptides (Fig. 2b) and tested inhibition of LPS release in the presence of 1 (Fig. 2c,d). Because E. coli LPS is readily available and there are well-validated commercial antibodies to it, the biochemical experiments were conducted using E. coli LPS. Compound 1 blocked LPS release from the wild-type complex to LptA but did not block LPS release from either of these mutant complexes. These results confirm the importance of the contacts observed between the macrocyclic peptides and LptFG for inhibiting growth of Acinetobacter strains.

a, View of the ternary complex highlighting key contacts from LptF (bolded) to both LPS and 1. LptF, LptG, LPS and 1 are coloured green, blue, yellow and purple, respectively. b, Table showing the MICs of 1 against A. baylyi containing various LptF variants. MIC values were consistent across three cultures started from individual colonies. c,d, 1 inhibits LPS transport to LptA by wild-type LptB2FGC (c) but not by LptB2FE249KGC or LptB2FI317NGC (d). Lipopolysaccharide transport from LptB2FGC to LptA modified with a photocrosslinkable amino acid (I36pBPA) was monitored in the presence of the indicated dose of 1 by exposing the samples to UV light after 60 min of transport, quenching by addition of SDS-loading buffer, PAGE to separate LPS-LptA adducts from LPS and western blotting against LPS. Data shown are representative of experiments conducted in biological triplicate. e, Cryo-EM structure of LptB2FG with LPS bound in the lumen of the transporter in white superimposed with the LptB2FG-LPS-1 structure, which is coloured as in a. The two structures have an r.m.s.d. of 0.31 Å over 8,010 atoms.
Drug traps LPS during transport
The LptB2FG–LPS–1 structure suggested that 1 traps an intermediate state of the LPS-bound Lpt transporter. To assess whether the conformation of LPS in the drug-bound structure reflects an on-pathway intermediate, we solved structures of A. baylyi LptB2FG bound to either E. coli or Acinetobacter LPS but now in the absence of 1 (Fig. 2e and Extended Data Fig. 4). In both cases, the overall conformation and contacts of LPS to the transporter are nearly identical to those determined in the presence of compound 1 (Fig. 2e). These findings are consistent with a mechanism in which compound 1 binds a pre-existing, LPS-loaded state of the transporter complex, identifying this state as a druggable conformation for antibiotic development.
We observed contacts from LptF (Arg30 and Arg55) to the same phosphate group on LPS that is coordinated by a primary amine from 1 (bold in Fig. 2a). To assess their functional importance, we separately mutated them to Ala and Gly, respectively (LptF R30A and LptF R55G). Neither of these variants produced viable cells in A. baylyi, suggesting that these residues are critical for protein folding or function. We found that we could express and purify complexes with these LptF substitutions, and both of the resulting LptB2FG variants had ATPase activity comparable to wild type but neither transferred LPS to LptA (Extended Data Fig. 4). Therefore, we concluded that both LptF Arg30 and Arg55 are critical for the function of the complex in Acinetobacter, probably because they help to position LPS during transport.
The ternary complex structure also showed an extensive interface between LPS and 1. We therefore sought to determine whether changing LPS structure in Acinetobacter would affect the inhibitory potency of 1. LPS biosynthesis involves more than 100 genes1,12,33 but mutations that confer decreased susceptibility to the compound were loss of function mutations in lpxM (Extended Data Fig. 5). LpxM performs the final acylation steps during LPS biosynthesis and of the total contact area of about 230 Å2 between LPS and 1, 94 Å2 (41%) involves contact area between the drug and the acyl chain installed by LpxM (refs. 34,35,36) (Fig. 3a). In a biochemical reconstitution, transport of LPS isolated from an E. coli LpxM deletion strain was possible at 20-fold higher concentrations of compound 1 than that required to inhibit the transport of the matching wild-type LPS structure (Fig. 3b). E. coli and Acinetobacter ΔlpxM LPS chemotypes have identical acylation patterns. Consistent with the biochemical experiments using E. coli ΔlpxM LPS, we found the A. baylyi lpxM deletion strain to be 30-fold less susceptible to 1 than was wild type. Previous studies in E. coli and A. baumannii have established that the loss of the fatty acyl chains installed by LpxM does not prevent LPS transport to the outer membrane but does reduce outer membrane barrier function37,38. Consistent with this, our A. baylyi ΔlpxM strain was as much as 1,000-fold more susceptible to a broad range of other antibiotics (Supplementary Table 2). Because ΔlpxM strains are known to have reduced virulence, loss of lpxM may not cause reduced susceptibility to macrocyclic peptides in vivo38,39.

a, The acyl chain that is added by LpxM, highlighted in salmon, nestles between LptF helices 2, 4 and 5. Residues contacting this acyl chain are labelled. Residues that contact this acyl chain and elicited resistance in spontaneous mutation studies32 are bolded. LptF is shown in green, LPS in yellow and LptG in blue. b, LPS isolated from a ΔLpxM strain renders LptB2FGC resistant to 1 (ref. 36). Lipopolysaccharide transport from LptB2FGC to LptA was measured as described in Fig. 2. Data shown are representative of experiments conducted in biological triplicate. c, Cryo-EM structure of Acinetobacter LptB2FGC superimposed with the structure of Acinetobacter LptB2FG in complex with LPS and 1. The LptB2FGC structure is shown in pink, whereas the LptB2FG-1-LPS structure is coloured as in Fig. 2a. The observed positioning of the TM helix of LptC sterically clashes with the compound 1 binding site observed in the LptB2FG structures. The positioning of LptF helices 2–5 are also shifted relative to what was observed in the LptB2FG structures.
LptC helix movement allows drug binding
LptC, a member of the inner membrane Lpt complex, contains a periplasmic β-jellyroll domain that plays an essential role in transfer of LPS from LptF to LptA (refs. 19,40). LptC also contains a TM helix that exists in two states. In one, the TM helix is sandwiched between the TM helices of LptG (helix 1) and LptF (helices 5 and 6), which form the gate through which LPS enters the transporter lumen19,41,42. In a second state, the LptC helix has moved away from the transporter42,43. Movement of the LptC helix between these states is thought to be important in coordinating LPS transport with the catalytic cycle of ATP binding and hydrolysis19,41,42,43. The LptB2FG–LPS–1 structure revealed that 1 protrudes into the gate formed between helix 5 of LptF and helix 1 of LptG, suggesting that its binding competes with the LptC TM helix for binding to the complex. We obtained a structure of A. baylyi LptB2FGC in the presence of LPS, although density for the LPS molecule was poorly resolved. In this structure, the LptC helix was sandwiched between LptG helix 1 and LptF helix 5, similar to the previously determined structures from other species19,41,42,44 (Fig. 3c and Extended Data Fig. 6). Despite several attempts, we were unable to obtain an LptB2FGC complex in which 1 was also bound. These data are compatible with a model in which binding of 1 and binding of the TM helix of LptC at the LptFG gate are mutually exclusive. Superimposing the LptB2FG–LPS–1 structure with the LptB2FGC–LPS structure identified shifts in the conformation of LptFG, particularly in helices 4, 5 and 6 of LptF (Fig. 3). This conformation of LptF creates a pocket between helices 4 and 5 that better accommodates acyl chain 6 of LPS. Although the specific sequence of events is uncertain, we propose that C-helix movement away from LptFG during the transport cycle permits LPS to bind in the intermediate transport state observed in the structure of the LptB2FG–LPS complex and that this is the state required for inhibitor recognition.
A model that requires C-helix movement for binding to occur would explain an otherwise perplexing observation. Compounds 1–3 have comparable cellular potency against wild-type strains and lptFG mutations cause similar reductions in susceptibility to all three compounds (Supplementary Table 1). However, when we tested 1–3 in a biochemical assay that monitors LPS release from the LptB2FGC complex to LptA, we found that compound 3 was 100-fold less effective than 1 and 2 (Fig. 4). To identify the structural basis for these differences in biochemical behaviour, we determined the cryo-EM structures of compounds 2 and 3 bound to LptB2FG–LPS. Both 2 and 3 bound in nearly identical positions as compound 1 (Fig. 4a–c and Extended Data Fig. 7). Compounds 1 and 2 contain relatively large substituents—an amino pyridine and a similarly sized benzoate, respectively—that overlap with the predicted position of the LptC TM helix in the gate-occluded state (dashed circles, Fig. 4a,b). These substituents would be predicted to compete more effectively with the LptC TM helix than compound 3, which contains a smaller chlorine atom at that position (Fig. 4c). If varying degrees of LptC TM helix competition are responsible for the different biochemical activities, 3 should block LPS release as well as 1 and 2 when the LptC TM helix is absent (LptB2FGΔTM-LptC). It has previously been demonstrated that ΔTM-LptC is able to support LPS transport even though it lacks the TM helix, allowing us to test this in vitro19,40. Consistent with our prediction, compounds 1–3 were similarly effective at blocking release from the ΔTM-LptC-containing complex (Fig. 4d). Because 3 is as effective as 1 and 2 in vivo, we conclude that the macrocyclic peptides target a conformational state in vivo in which the C-helix has moved.

a–c, Cryo-EM structures of 1 (a), 2 (b) and 3 (c) bound to LptB2FG. The observed positioning of the LptC TM helix from Fig. 3d is highlighted in pink. d, Transport of LPS to LptA by LptB2FGC is not inhibited by 3 in vitro but LPS transport by LptB2FG-ΔTM-C is inhibited by 1, 2 and 3 in vitro. Data shown are representative of experiments conducted in biological triplicate. e, 1 treatment increases the ATPase activity of LptB2FG, LptB2FGC and LptB2FG-ΔTM-C in an LPS-dependent manner. This effect is reduced in the presence of the TM helix of LptC. ATP hydrolysis was monitored by measuring concentrations of inorganic phosphate. Experiments were conducted in biological triplicate and data are presented as mean values ± standard deviation. f, 2 binds Lpt in the presence of LPS and absence of LptC. The binding of radiolabelled 2 to His-tagged LptB2FG and LptB2FGC in the presence and absence of LPS was measured in a SPA. Data are presented as counts per minute (c.p.m.) in arbitrary units (a.u.) and are from three biological replicates. g, The cellular activity of 1-derivatives correlates to their observed binding to LptB2FG through SPA. The ability of 1-derivatives to displace radiolabelled 2 from His-tagged LptB2FG was measured in the presence of LPS. Active compounds 1–3 showed potent binding to LptB2FG, whereas the inactive control compound 2a did not. 2a is the epimer of 2 at the highlighted (*) carbon; Fig. 1. Uncertainties represent the standard deviation of three biological replicates.
Drug uncouples ATPase activity from LPS transport
LPS loading in the LptB2FG transporter has been shown to stimulate ATPase activity45. Because our data have shown that these macrocyclic peptides trap LPS in the transporter, we sought to determine if these compounds would increase ATPase activity above the LPS-bound baseline. The addition of compounds 1–3 to the LptB2FG and LptB2FGΔTM-LptC complexes led to a large increase in ATPase activity (Fig. 4e and Extended Data Fig. 8). This activity was dependent on the presence of LPS, supporting the importance of LPS for drug binding to the transporter. The results of the ATPase activity and LPS release assays suggested that the best proxy for in vivo activity of the macrocyclic peptides is the affinity of a compound for LptB2FG, in the absence of LptC. Indeed, a scintillation-proximity assay (SPA) showed that radiolabelled 2 bound to LptB2FG but not LptB2FGC and that binding was dependent on the presence of LPS (Fig. 4f). Consistent with their similar potency in cellular assays, 1, 2 and 3 had similar abilities to competitively displace H3-2 from LptB2FG (Ki = 35, 35 and 56 nM, respectively). The ATPase and binding assays with complexes lacking LptC or its TM helix therefore recapitulated the in vivo findings that 1–3 have comparable activity, again suggesting that the macrocyclic peptides described here target a state in which the LptC TM helix has dissociated from the complex.
We have shown that a new family of macrocyclic peptide antibiotics kills Acinetobacter by trapping LPS as this substrate is in transit within the lipopolysaccharide transporter. Because LPS is not required for viability in Acinetobacter, a comment is warranted here on the mechanism of cell death. Previous work has shown that many genes involved in LPS biogenesis in Acinetobacter are conditionally essential and can only be deleted if initiation of LPS biosynthesis is blocked46,47. Toxic accumulation of LPS biosynthesis intermediates results when LPS transport initiates but cannot proceed to completion. We found that A. baylyi strains lacking LPS (ΔlpxC) can grow in vitro in the presence of very high concentrations of 1 (Supplementary Table 3). Therefore, the drug does not act by depleting LPS from the outer membrane because these cells can live without any LPS in the outer membrane, but through its toxic accumulation within the cell. Although loss of LPS in Acinetobacter provides a mechanism to escape drug susceptibility, it significantly decreases both fitness and virulence46,48,49. It remains to be seen whether elimination of LPS is a viable strategy to reduce susceptibility to macrocyclic peptide treatment in vivo.
The macrocyclic peptide inhibitors are very potent against Acinetobacter strains but are inactive against other Gram-negative organisms and we have wondered what lessons our structures and mechanistic experiments hold for understanding this narrow drug susceptibility. We have shown that the ability of macrocyclic peptides to bind Acinetobacter LptB2FG requires LPS but the structures reveal that these inhibitors contact only the most conserved regions of the LPS lipid A core. Thus, variance of LPS structure alone does not explain the species-selectivity of these drugs (Fig. 1d). The drug pose observed in the ternary structures of A. baylyi LptB2FG in complex with E. coli LPS fully explained the resistance mutations that were isolated in Acinetobacter, indicating that contacts with LptFG are critical for drug recognition. Furthermore, results of biochemical transport assays using E. coli LPS were consistent with cellular assays that were performed in Acinetobacter strains. Therefore, it seems likely that the species-selectivity is due to differences in the Lpt proteins. Homologous bacterial proteins from different genera often have low sequence conservation. Although the sequences of A. baumannii and A. baylyi LptFG are 82% identical and share almost all the residues (16 of 18) that contact either LPS or the macrocyclic peptide, E. coli LptFG proteins are only 25% identical to their Acinetobacter counterparts and most of the residues that contact LPS are different (Extended Data Fig. 1). Structures of LptB2FG from E. coli previously determined in complex with E. coli LPS show that LPS occupies a different position in the central cavity of LptFG (refs. 41,42) (Fig. 5). Moreover, there are differences in the positions of some of the LptF helices that would result in a clash with the macrocyclic peptides, as well as differences in the electrostatic surface surrounding the LptB2FG–LPS binding pockets (Fig. 5c,d). In fact, purified E. coli LptB2FGC is more than 1,000-fold more resistant to inhibition by the clinical candidate, 2, than its Acinetobacter homologue (Fig. 2c and Extended Data Fig. 8d). The narrow spectrum therefore reflects the differences in the proteins, which affect how they bind LPS. We note that there are other binding pockets surrounding LPS in E. coli LptB2FG and it may therefore be possible to design analogous inhibitors for this or other Gram-negative pathogens that trap an intermediate LPS-bound state (Fig. 5e,f). More broadly, the mechanism of these molecular glues provides a roadmap for the development of other compounds that bind a transporter and its substrate simultaneously to block lipid transport in prokaryotic and eukaryotic systems50.

a–f, Three different representations of the structures of either Acinetobacter LptB2FG (a,c,e) or E. coli LptB2FG (b,d,f) in complex with E. coli LPS. In the case of Acinetobacter, the positioning of 1 is as observed experimentally. In the case of E. coli, 1 is placed based on alignment to the Acinetobacter LptB2FG structure. a,b, The binding site for macrocyclic peptide 1 that is present in Acinetobacter (a) is not present in E. coli (b). As highlighted, the drug has steric clashes with both LPS and helix 5 of E. coli LptF. c,d, The electrostatic surface of Acinetobacter (c) and E. coli (d) LptB2FG, with negative surfaces shown in red and positive surfaces in blue. Note that the primary amine of the macrocyclic peptides lodge into a negative pocket in Acinetobacter LptB2FG that does not exist in E. coli LptB2FG. e,f, Both Acinetobacter and E. coli LptB2FG have extra cavities formed between LPS and Lpt protein. In this Article, we have validated that drug binding to a composite surface between Acinetobacter LptB2FG and LPS (purple pocket, e) can block LPS transport. Analogous pockets exist in other species (pink, f), providing opportunities for future drug design.
Methods
No statistical methods were used to predetermine sample size. The experiments were not randomized and investigators were not blinded to allocation during experiments and outcome assessment.
SDS–PAGE and immunoblotting
Homemade Tris-HCl 4–20% polyacrylamide gradient gels or 4–20% Mini-PROTEAN TGX precast protein gels (Bio-Rad) were used with Tris-glycine running buffer. The 2× SDS sample loading buffer refers to a mixture containing 125 mM Tris (pH 6.8), 4% (w/v) SDS, 30% (v/v) glycerol, 0.005% bromophenol blue and 5% (v/v) β-mercaptoethanol. SDS–polyacrylamide gel electrophoresis (SDS–PAGE) gels were run for 45–60 min at 200 V. Protein complexes purified for cryo-EM were analysed by SDS–PAGE followed by staining with Coomassie blue (Alfa Aesar) and imaging using the Gel feature of an Azure Biosystems C400 imager. For western blotting, proteins were transferred onto Immun-Blot PVDF membranes (Bio-Rad). Membranes were then blocked using sterile-filtered Casein blocking buffer (Sigma-Aldrich) for 1 h and subsequently incubated with the appropriate antibodies. The following primary antibodies were used: mouse anti-His HRP conjugate (Biolegend, 652504, 1:10,000 dilution) and anti-LPS core mouse monoclonal (Hycult Biotechnology, HM6011, clone WN1 222-5, lot no. 18419M0715-A, 1:5,000 dilution). The following secondary antibodies were used: donkey-anti-rabbit RP conjugate (GE Amersham, NA934-1ML, lot no. 16801031, 1:10,000 dilution), sheep-anti-mouse HRP conjugate (GE AMersham, LNA931V/AH, lot no. 14251045, 1:10,000 dilution). Bands were visualized using ECL Prime western blotting detection reagent (GE Amersham) and an Azure c400 imaging system. Uncropped immunoblots are available in Supplementary Fig. 1.
Plasmids, strains and oligonucleotides
Genes encoding the LptB, LptC and LptFG were amplified by polymerase chain reaction (PCR) from Acinetobacter baylyi ADP1 (ATCC 33305) genomic DNA. lptB and lptFG PCR products were inserted into pCDFduet by Gibson assembly (New England Biolabs) to generate plasmids analogous to those used for other LptB2FG homologues19. Similar design was used for the modified plasmid pTRAB-FLAG-LptB-LptFG for purification of the same complex from the native host, which was constructed by combining the gDNA amplicons of the same open reading frames incorporating a linker-less FLAG tag at the N terminus of LptB, a modified trp promoter of E. coli and adjacent regions from pTRC99a, a hybrid pBR322WH1266 replicon and a spectinomycin resistance cassette from pCDFduet by Gibson assembly. A linker-less N-terminal His7 tag was added to LptB in pCDFduet using NEBuilder HiFi DNA assembly (New England Biolabs). lptC PCR products were inserted into pET22/42 with a C-terminal thrombin cleavage site and a His7 tag. Oligonucleotide primers were purchased from Eton Biosciences, Genewiz or Integrated DNA Technologies. Plasmids and strains used in this study are reported in Supplementary Tables 4 and 5, respectively. Plasmid sequences are below.
Construction and use of mutant A. baylyi strains
Culture, genetic manipulation and MIC measurements of A. baylyi ADP1 were conducted according to previously reported procedures46,51. Point mutants were constructed in a two-step procedure following ref. 52 with the introduction and excision of the integration cassette at codon 66 of pepA, wherein the excising fragment of otherwise wild-type chromosomal DNA sequence from codon 406 of pepA to codon 193 of lptG bore the desired mutation and the resulting clones were screened by amplicon sequencing from codon 81 of HolC to codon 501 of GpmI, whereas lpxM deletion was achieved following the same procedure except that the integration cassette insertion and excision removed codons 79–279 of lpxM to avoid interference with neighbouring and overlapping genes that a larger deletion may risk and replaced codon 78 with an ochre stop codon to prevent a readthrough resulting in an aberrant fusion, with the excising fragment sequence spanning from codon 211 of sppA to codon 497 of ComA and verified by amplicon sequencing from codon 94 of MhpC to codon 327 of ComA as well as by absence of a PCR product corresponding to a region spanning codons 79 to 279 of lpxM to check for duplications. A deletion of the lon protease was made in the same manner to produce the stain used for expression and purification of LptB2FG to mimic the BL21 strain of E. coli used in the rest of purifications, for which the region encompassing 72 base pairs (bp) upstream of the lon start codon and 1 bp downstream of the lon stop codon was excised after being replaced with the same integration cassette, yielding a markerless deletion, with the excising fragment sequence spanning from codon 491 of ArnT to codon 40 of 45_DOPA_Dioxygenase and verified by amplicon sequencing spanning from codon 322 of ArnT to codon 221 of 45_DOPA_Dioxygenase as well as by absence of a PCR product corresponding to a region spanning from codons 328 to 768 of lon to check for duplications. Following amplicon confirmation, three validated isolates of each constructed mutant were tested for susceptibility to a panel of antibiotics with known antibiotics with known mechanisms of action as a further validation step to ensure congruence of phenotypes across replicates, which was confirmed in all cases and one of the validated replicates was later used for MIC measurements reported here. In the case of R30A and R55G, no colonies that incorporated these mutations could be isolated, whereas the identical approach readily introduced conservative R30K and R55K substitutions, which resulted in increased antibiotic sensitivity in spite of their mild nature, indicating that impairments caused by substituting dissimilar residues at those positions are not survived.
MIC determination
MIC determinations were performed by broth microdilution in line with CLSI guidelines (CLSI M07-A11 2018). Bacterial inocula were prepared by diluting overnight liquid cultures in LB. Antibacterial panels containing antibacterial solutions were inoculated with an appropriate volume of inoculum to give a final inoculum of about 5 × 105 c.f.u. ml−1 and desired test concentrations of antibacterial agents in standard 96-well plates with 0.1 ml of culture per well. The test plates were incubated for 20–24 h and optical density (OD600) was recorded using a plate reader. MIC values corresponded to the lowest compound concentration inhibiting bacterial growth beyond which OD ceased to decrease.
Purification of LptB2FG complexes for cryo-EM
LptB2FG complexes were purified as previously described, with slight modifications20. Overnight cultures of Bl21(λDE3) E. coli containing pCDFduet-His7LptB-LptFG or A. baylyi containing pTRAB-FLAGLptB-LptFG were diluted 1:100 into LB or terrific broth containing 50 mg l−1 of spectinomycin. Cells were grown at 37 °C (or 30 °C for A. baylyi) to an OD600 of about 0.8. Then 200 µM IPTG and 0.2% glucose (or 500 µM IPTG for A. baylyi) were added and cells were allowed to grow for another 2–3 h. Cells were harvested by centrifugation (4,200g, 20 min, 4 °C). Cell pellets were flash frozen using liquid nitrogen and stored at −80 °C. All subsequent steps were carried out at 4 °C unless otherwise noted.
Thawed cell pellets were resuspended in lysis buffer (50 mM Tris (pH 7.4), 300 mM NaCl, 1 mM PMSF, 100 μg ml−1 of lysozyme, 50 μg ml−1 of DNase I, 1 cOmplete Protease Inhibitor Cocktail tablet per 40 ml) homogenized and subjected to passage through an EmulsiFlex-C3 high-pressure cell disruptor three times. The cell lysate was centrifuged (10,000g, 10 min) and the supernatant was further centrifuged (100,000g, 1 h). The resulting pellets were resuspended and solubilized in solubilization buffer (20 mM Tris (pH 7.4), 300 mM NaCl, 15% glycerol, 5 mM MgCl2, 1% (wt/vol) DDM (Anatrace Maumee), 100 μM PMSF, 2 mM ATP) and rocked at 4 °C for 2 h. (A. baylyi cell lysate was immediately subjected to detergent solubilization without the preceding centrifugation steps or ATP addition but 0.35 µM 1 was used to supplement some batches from the solubilization step onward). The mixture was centrifuged (100,000g, 30 min), the supernatant was spiked with imidazole to a final concentration of 15 mM and then rocked with Ni-NTA Superflow resin (Qiagen) for 1 h. (A. baylyi supernatant was also filtered through a 0.45 µM pore size PVDF Durapore (Millipore-Sigma) membrane and incubated with M2-FLAG agarose resin (Millipore-Sigma) without imidazole supplementation instead of Ni-NTA Superflow resin). The resin was then washed with 2 × 10 column volumes affinity buffer (300 mM NaCl, 20 mM Tris (pH 7.4), 15% glycerol, 0.01% (wt/vol) DDM, 0.04% (wt/vol) GDN (Anatrace Maumee)) containing 20 mM imidazole followed by 2 × 15 column volumes of affinity buffer containing 35 mM imidazole. (A. baylyi-derived batches were washed with 3 × 10 column volumes of affinity buffer). Protein was eluted with 2 × 2 column volumes of affinity buffer containing 200 mM imidazole (12.5 column volumes of affinity buffer supplemented with 0.2 mg ml−1 of FLAG peptide (Genscript) for A. baylyi-derived batches) concentrated using a 100 kDa molecular weight cutoff Amicon Ultra centrifugal filter (Millipore) and purified by size-exclusion chromatography on a Superdex 200 increase column in SEC buffer (300 mM NaCl, 20 mM Tris (pH 7.4), 0.02% GDN, 0.25 mM tris(hydroxypropyl)phosphine). Fractions were pooled and concentrated to 7–8 mg ml−1 using a 100 kDa molecular weight cutoff Amicon Ultra centrifugal filter. Protein was then prepared for microscopy as described below.
Purification of LptB2FGC complexes for cryo-EM
Purification was conducted largely as described for LptB2FG with the following modifications. Expression was conducted using C43(λDE3) E. coli containing pCDFduet-LptB-LptFG and pET22/42-LptC-thrombin-His7. Cultures were grown in the presence of 50 mg l−1 of spectinomycin and 50 mg l−1 of carbenicillin. The rest of the expression and purification was conducted identically to the LptB2FG purification until the size-exclusion chromatography step. Fractions collected after size-exclusion chromatography were incubated overnight with restriction-grade thrombin (Sigma) to cleave the His tag. The solution was spiked with 8 mM imidazole and the uncleaved protein was removed by passage through Ni-NTA resin and benzamidine Sepharose. Fractions were pooled and concentrated to 7–8 mg ml−1 using a 100 kDa molecular weight cutoff Amicon Ultra centrifugal filter. Protein was then prepared for microscopy as described below.
Electron microscopy data collection
Protein was purified as described above and then incubated on ice with 0.2 mg ml−1 of lipopolysaccharides from E. coli EH100 (Ra mutant; Sigma-Aldrich) and 0.25 mM drug (if applicable) for 45 min with gentle agitation. For proteins purified out of Acinetobacter, E. coli lipopolysaccharides were not added. Sample was then applied to glow-discharged C-flat 20 nm holey carbon 1.2 μm hole diameter, 1.3 μm hole spacing, 400-mesh copper grids (Protochips). Grids were blotted for 6.5 s at 4 °C and 100% humidity with the blot force set to 12 and flash frozen by liquid nitrogen-cooled liquid ethane using a Thermo Fisher Scientific Vitrobot Mark IV (Thermo Fisher Scientific). The grid was then loaded onto a Titan Krios G3i electron cryo-microscope (Thermo Fisher) operated at 300 kV accelerating voltage. Image stacks (videos) were recorded on a Gatan Bioquantum K3 Imaging Filter (Gatan), using counting mode and a calibrated magnification of ×105,000 and a pixel size of 0.825 Å, using SerialEM53. The slit of the energy filter was set to 20 eV with a defocus range between 1.1 and 2.2 μm. The subframe time was set to allow the collection of 50 subframes per image stack with an electron dose rate of about 1 e− per Å2 per frame. The total electron dose was about 50 e− per Å2. The multishot scheme in SerialEM was used for data collection, with settings of nine holes per stage move and two shots per hole. The data collections for all structures were performed in the same manner.
Image processing and three-dimensional reconstruction
The video frames were motion-corrected and dose-weighted and the contrast transfer function (CTF) parameters were estimated using CryoSPARC Live54. Particle picking was carried out using the cryoSPARC blob-picker and junk particles were filtered out by successive rounds of two-dimensional classification in cryoSPARC. Initial models were generated using the ab initio reconstruction in cryoSPARC and then particles were filtered by successive rounds of heterogeneous refinement. After an initial non-uniform refinement job, particles were subject to local motion correction, patch CTF estimation, local CTF refinement and global CTF refinement (fit for beam tilt, beam trefoil and spherical aberration). The particles were then subject to non-uniform refinement to yield the final global reconstruction. Maps were further refined using particle subtraction and local refinement with a mask focused on the TM and nucleotide-binding domains of the transporter. For all maps, we also tried classification without alignment in Relion. At best this only yielded nominal improvements in resolution after reimporting into cryoSPARC and conducting non-uniform refinement when compared to the preclassification maps. 3D classification without alignments in cryoSPARC revealed several possible conformations of the drug within the transporter, as highlighted in Extended Data Fig. 3b,f55,56. Maps used for figures were either filtered according to local resolution with B-factor sharpening within cryoSPARC or using postprocessing carried out in DeepEMhancer57. Structural biology applications used in this project were compiled and configured by SBGrid58.
Model building, refinement and validation
Initial models for LptB, LptF and LptG were generated using SwissModel59. The resulting structures were docked into the LptBFG map using Chimera60. Cif restraints for E. coli lipopolysaccharide were generated using the sketcher tool in CCP4 (ref. 61). Cif restraints for Acinetobacter lipopolysaccharide were generated using the Grade2 web server from Global Phasing Limited. Cif restraints for the macrocyclic peptides were generated using eLBOW62. The coordinates were then refined using Phenix63,64. The model was further optimized using ISOLDE65, accessed through ChimeraX66. Manual model building was carried out in Coot67. The final model was visually inspected for general fit to the map and further inspected using MolProbity and the residue-wise local quality estimation DAQ68,69. All residues in our models had >0 DAQ scores, except those contained in the helix of LptC. The helix of LptC is modelled as poly-alanine because our maps were not of sufficient quality to allow unambiguous assignment of the helix register. The model validation statistics are summarized in Extended Data Table 1.
Purification of LptB2FG complexes for biochemical reconstitution
LptB2FG used for biochemical experiments was purified as described for cryo-EM with the following modifications. The affinity buffer was 300 mM NaCl, 20 mM Tris (pH 7.4), 10% glycerol, 0.015% (wt/vol) DDM. The SEC buffer was 300 mM NaCl, 20 mM Tris (pH 7.4), 5% glycerol, 0.05% DDM, 0.5 mM tris(hydroxypropyl)phosphine.
Purification of LptB2FGC complexes for biochemical reconstitution
LptB2FGC used for biochemical experiments was purified as described for cryo-EM with the following modifications. The affinity buffer was 300 mM NaCl, 20 mM Tris (pH 7.4), 10% glycerol, 0.015% (wt/vol) DDM. The SEC buffer was 300 mM NaCl, 20 mM Tris (pH 7.4), 5% glycerol, 0.05% DDM, 0.5 mM tris(hydroxypropyl)phosphine. E. coli LptB2FGC complexes were purified as described previously20.
Purification of LptAI36pBPA
LptAI36pBPA was purified as described previously20. Briefly, Bl21 (λDE3) E. coli cells containing pSup-BpaRS-6TRN and pET22b-LptA(I36Am) were grown to an OD600 of approximately 0.6 at 37 °C in LB media containing 50 μg ml−1 of carbenicillin, 30 μg ml−1 of chloramphenicol and 0.8 mM pBPA (BaChem). Cells were then induced with 50 μM IPTG; allowed to grow for 2 h; harvested; resuspended in a mixture containing 50 mM Tris-HCl (pH 7.4), 250 mM sucrose and 3 mM EDTA; incubated on ice for 30 min; and pelleted (6,000g, 10 min). The supernatant was supplemented with 1 mM PMSF and 10 mM imidazole and pelleted (100,000g, 30 min). The supernatant was incubated with Ni-NTA resin, which was then washed twice (20 column volumes of 20 mM Tris-HCl (pH 8.0), 150 mM NaCl, 10% (vol/vol) glycerol and 20 mM imidazole). LptA was eluted twice (2.5 column volumes of wash buffer supplemented with a further 180 mM imidazole), concentrated using a 10-kDa-cutoff Amicon centrifugal concentrator (Millipore), flash frozen and stored at −80 °C until use.
Preparation of LptB2FG or LptB2FGC liposomes
Proteoliposomes were prepared as described previously20. Aqueous E. coli polar lipid extract (Avanti Polar Lipids) (30 mg ml−1) and aqueous LPS from E. coli EH100 (Ra mutant; Sigma) (2 mg ml−1) were sonicated briefly for homogenization. For experiments testing the effect of LPS structure, we used LPS isolated from either GKM374 (BL21DE3 eptA::catR arnA::kanR eptC::gentR) or TXM418 (BL21DE3 eptA::catR arnA::FRT eptB::gentR lpxM::kanR) as described previously36. A mixture of 20 mM Tris-HCl (pH 8.0), 150 mM NaCl, 7.5 mg ml−1 of E. coli polar lipids, 0.5 mg ml−1 of LPS and 0.25% DDM was prepared and kept on ice for 10 min. Purified LptB2FGC or LptB2FG was added to a final concentration of 0.86 μM and the mixture was left on ice for 20 min. The mixture was diluted 100-fold with cold 20 mM Tris-HCl (pH 8.0) and 150 mM NaCl and kept on ice for 20 min. The proteoliposomes were pelleted (300,000g, 2 h, 4 °C), resuspended in 20 mM Tris-HCl (pH 8.0) and 150 mM NaCl, diluted 100× and centrifuged (300,000g, 2 h, 4 °C). The pellets were resuspended in a mixture of 20 mM Tris-HCl (pH 8.0), 150 mM NaCl and 10% glycerol (250 μl per 100 μl of the original predilution solution), homogenized by sonication, flash frozen and stored at −80 °C until use.
Purification of LptC(ΔTM)
LptC(ΔTM) was purified largely as previously described19. Briefly, Bl21 (λDE3) E. coli cells containing pET22/42-LptC(ΔTM)-His7 were grown to an OD600 of approximately 0.6 at 37 °C in LB media containing 50 μg ml−1 of carbenicillin. Cells were then induced with 50 μM IPTG; allowed to grow for 2 h; harvested and resuspended in lysis buffer (50 mM Tris pH 7.4, 300 mM NaCl, 0.1 mM EDTA). Lysozyme, DNaseI and PMSF were added to final concentrations of 100 µg ml−1, 50 µg ml−1 and 1 mM, respectively. Cells were homogenized and subjected to passage through an EmulsiFlex-C3 high-pressure cell disruptor three times. The cell lysate was centrifuged (10,000g, 10 min) and the supernatant was further centrifuged (100,000g, 1 h). The supernatant was spiked with imidazole to a final concentration of 15 mM and then rocked with Ni-NTA Superflow resin (Qiagen) for 1 h. The resin was then washed with 2 × 10 column volumes of affinity buffer (300 mM NaCl, 20 mM Tris (pH 7.4), 15% glycerol) containing 20 mM imidazole followed by 2 × 15 column volumes of affinity buffer containing 35 mM imidazole. Protein was eluted with 2 × 2 column volumes of affinity buffer containing 200 mM imidazole, concentrated using a 10 kDa molecular weight cutoff Amicon Ultra centrifugal filter (Millipore) and purified by size-exclusion chromatography on a Superdex 200 increase column in SEC buffer (300 mM NaCl, 20 mM Tris (pH 7.4), 5% glycerol). Fractions were pooled and stored at −80 °C.
LPS release assay
The amounts of release of LPS from proteoliposomes to LptA were measured as previously described20. Assays used 60% proteoliposomes (by volume) in a solution containing 50 mM Tris-HCl (pH 8.0), 500 mM NaCl, 10% glycerol and 2 µM LptAI36pBPA. Reaction mixtures were incubated with drug for 10 min at room temperature, as applicable. Reactions were then initiated by the addition of ATP and MgCl2 (final concentrations of 5 mM and 2 mM, respectively) and proceeded at 30 °C. Aliquots (25 µl) were removed from the reaction mixtures and irradiated with ultraviolet (UV) light (365 nm) on ice for 10 min using a B-100AP lamp (Fisher Scientific). Following UV irradiation, 25 µl of 2× SDS–PAGE sample loading buffer was added, samples were boiled for 10 min and proteins were separated using Tris-HCl 4–20% polyacrylamide gradient gels with Tris-glycine running buffer. Immunoblotting was conducted as described above.
ATPase assay
ATPase assays were done using a modified molybdate method, as previously reported, with slight modifications20. Assays used 30% proteoliposomes (by volume) in a mixture containing 50 mM Tris-HCl (pH 8.0), 500 mM NaCl, 10% glycerol and 2 mM MgCl2. Proteoliposome-containing reaction mixture was incubated with drug at room temperature for 10 min, as applicable. Reactions were initiated by the addition of ATP to a final concentration of 5 mM and run at 30 °C. Aliquots (5 µl) were taken at 0, 15, 30 and 45 min. Reactions were quenched with an equal volume of 12% SDS. The amounts of Pi were determined using a colorimetric method and potassium phosphate was used as a standard46. Reagents were obtained from Sigma-Aldrich. After the addition of SDS, a mixture containing 10 µl of 30 mg ml−1 of ascorbic acid, 0.5 N HCl, 5 mg ml−1 of ammonium molybdate and 6% SDS was added. The samples were incubated at room temperature for 7 min and 15 µl of an aqueous solution containing 20 mg ml−1 of sodium citrate tribasic dihydrate, 2 mg ml−1 of sodium arsenite and 2% (vol/vol) acetic acid was added. The absorbance at 850 nm was measured using a Spectramax Plus 384 (Molecular Devices) after 20 min. Error bars indicate the standard deviations of the average rates measured over three biological replicates.
Scintillation-proximity assay
The binding of radioligand [3H]-RO7223280 to ablLptB2FG and ablLptB2FGC was measured by bead-based SPA. All steps were performed in SEC buffer (20 mM Tris pH 7.5, 300 mM NaCl, 5% glycerol, 0.5 mM TCEP, 0.05% DDM ± 10 µM E.coli J5 LPS(Rc) TLRGRADE (Enzo Life Sciences)) and at 4 °C unless otherwise indicated. Purified protein was first incubated with Copper PVT HIS-tag beads (Perkin Elmer) for 1.5 h under gentle rotation. Twelve radioligand concentrations were added to the PVT–protein mix and incubated for another 30 min. The mixture was diluted into SPA buffer without or with 500 nM cold RO7223280 to measure total and non-specific binding respectively into an Optiplate-384 microplate (Perkin Elmer). Each well contained 25 ul of total volume, 18 nM protein, 5% dimethylsulfoxide and 6% v/v PVT beads. The SPA plates were sealed (TopSeal, Perkin Elmer) and stored at 4 °C overnight. Before the measurement, plates were mixed on a shaker for 20 min, 750 rpm at room temperature and the seal was thoroughly wiped with antistatic spray to reduce electrostatic events. Scintillation data were recorded with a Topcount NXT C384, in the form of three independent replicates each consisting of three technical triplicates. Specific binding was calculated by subtracting non-specific binding raw counts from total binding raw counts. The dissociation constant Kd and standard deviation of the three independent replicates are reported and were calculated by the GraphPad Prism ‘One site – specific binding’ tool.
Radioligand displacement experiments were similarly conducted, applying a constant concentration of 25 nM [3H]-RO7223280 and 16 concentrations of cold ligands in the presence of 10 µM LPS(Rc). Displacement values were normalized by including nine wells containing no radioligand (defined as 100% competition) and nine wells containing 8 uM radioligand only (defined as 0% competition). The inhibitory constant Ki was calculated with the ‘One Site – Fit Ki‘ tool using the concentration (25 nM) and Kd (86 nM) of [3H]-RO7223280 as constraints. The Hill coefficient was used as a quality control metric (theoretically, nH = 1 for a 1:1 competitive inhibitor) and determined with the ‘[Inhibitor] vs. response -- Variable slope (four parameters)’ tool.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The atomic coordinates of abLptB2FG-ecLPS-1, abLptB2FG-ecLPS, abLptB2FG-ecLPS-2, abLptB2FG-ecLPS-3, abLptB2FGC, abLptB2FG-abLPS and abLptB2FG-abLPS-1 are deposited at the Protein Data Bank with accession codes 8FRL, 8FRM, 8FRN, 8FRO, 8FRP, 8UFG and 8UFH, respectively. Cryo-EM density maps of abLptB2FG-LPS-1, abLptB2FG-LPS, abLptB2FG-ecLPS-2, abLptB2FG-ecLPS-3, abLptB2FGC, abLptB2FG-abLPS and abLptB2FG-abLPS-1 are deposited at the Electron Microscopy Data Bank at accession codes EMD-29400, EMD-29401, EMD-29402, EMD-29403, EMD-29404, EMD-42206 and EMD-42207, respectively.
Change history
11 July 2024
A Correction to this paper has been published: https://doi.org/10.1038/s41586-024-07645-0
10 January 2024
A Correction to this paper has been published: https://doi.org/10.1038/s41586-024-07035-6
References
Whitfield, C. & Trent, M. S. Biosynthesis and export of bacterial lipopolysaccharides. Annu. Rev. Biochem. 83, 99–128 (2014).
García-Quintanilla, M. et al. Inhibition of LpxC increases antibiotic susceptibility in Acinetobacter baumannii. Antimicrob. Agents Chemother. 60, 5076–5079 (2016).
Mandler, M. D. et al. Novobiocin enhances polymyxin activity by stimulating lipopolysaccharide transport. J. Am. Chem. Soc. 140, 6749–6753 (2018).
Vaara, M. Antibiotic-supersusceptible mutants of Escherichia coli and Salmonella typhimurium. Antimicrob. Agents Chemother. 37, 2255–2260 (1993).
Sampson, B. A., Misra, R. & Benson, S. A. Identification and characterization of a new gene of Escherichia coli K-12 involved in outer membrane permeability. Genetics 122, 491–501 (1989).
Wu, T. et al. Identification of a protein complex that assembles lipopolysaccharide in the outer membrane of Escherichia coli. Proc. Natl Acad. Sci. USA 103, 11754–11759 (2006).
Srinivas, N. et al. Peptidomimetic antibiotics target outer-membrane biogenesis in Pseudomonas aeruginosa. Science 327, 1010–1013 (2010).
Vetterli, S. U. et al. Thanatin targets the intermembrane protein complex required for lipopolysaccharide transport in Escherichia coli. Sci. Adv. 4, eaau2634 (2018).
Moura, E. C. C. M. et al. Thanatin impairs lipopolysaccharide transport complex assembly by targeting LptC–LptA interaction and decreasing LptA stability. Front. Microbiol. 11, 909 (2020).
Muhlradt, P. F. & Golecki, J. R. Asymmetrical distribution and artifactual reorientation of lipopolysaccharide in the outer membrane bilayer of Salmonella typhimurium. Eur. J. Biochem. 51, 343–352 (1975).
Kamio, Y. & Nikaido, H. Outer membrane of Salmonella typhimurium: accessibility of phospholipid head groups to phospholipase c and cyanogen bromide activated dextran in the external medium. Biochemistry 15, 2561–2570 (1976).
Raetz, C. R. H. & Whitfield, C. Lipopolysaccharide endotoxins. Annu. Rev. Biochem. 71, 635–700 (2002).
Nikaido, H. Molecular basis of bacterial outer membrane permeability revisited. Microbiol. Mol. Biol. Rev. 67, 593–656 (2003).
Zhou, Z., White, K. A., Polissi, A., Georgopoulos, C. & Raetz, C. R. H. Function of Escherichia coli MsbA, an essential ABC family transporter, in lipid A and phospholipid biosynthesis. J. Biol. Chem. 273, 12466–12475 (1998).
Ho, H. et al. Structural basis for dual-mode inhibition of the ABC transporter MsbA. Nature 557, 196–201 (2018).
Mi, W. et al. Structural basis of MsbA-mediated lipopolysaccharide transport. Nature 549, 233 (2017).
Okuda, S., Freinkman, E. & Kahne, D. Cytoplasmic ATP hydrolysis powers transport of lipopolysaccharide across the periplasm in E. coli. Science 338, 1214–1217 (2012).
Sherman, D. J. et al. Decoupling catalytic activity from biological function of the ATPase that powers lipopolysaccharide transport. Proc. Natl Acad. Sci. USA 111, 4982–4987 (2014).
Owens, T. W. et al. Structural basis of unidirectional export of lipopolysaccharide to the cell surface. Nature 567, 550–553 (2019).
Simpson, B. W. et al. Combining mutations that inhibit two distinct steps of the ATP hydrolysis cycle restores wild-type function in the lipopolysaccharide transporter and shows that ATP binding triggers transport. mBio 10, e01931-01919 (2019).
Narita, S.-i & Tokuda, H. Biochemical characterization of an ABC transporter LptBFGC complex required for the outer membrane sorting of lipopolysaccharides. FEBS Lett. 583, 2160–2164 (2009).
Wang, Z. et al. Structural and functional studies of conserved nucleotide-binding protein LptB in lipopolysaccharide transport. Biochem. Biophys. Res. Commun. 452, 443–449 (2014).
Sherman, D. J. et al. Lipopolysaccharide is transported to the cell surface by a membrane-to-membrane protein bridge. Science 359, 798–801 (2018).
Okuda, S., Sherman, D. J., Silhavy, T. J., Ruiz, N. & Kahne, D. Lipopolysaccharide transport and assembly at the outer membrane: the PEZ model. Nat. Rev. Microbiol. 14, 337–345 (2016).
Braun, M. & Silhavy, T. J. Imp/OstA is required for cell envelope biogenesis in Escherichia coli. Mol. Microbiol. 45, 1289–1302 (2002).
Sperandeo, P., Pozzi, C., Dehò, G. & Polissi, A. Non-essential KDO biosynthesis and new essential cell envelope biogenesis genes in the Escherichia coli yrbG-yhbG locus. Res. Microbiol. 157, 547–558 (2006).
Sperandeo, P. et al. Characterization of lptA and lptB, two essential genes implicated in lipopolysaccharide transport to the outer membrane of Escherichia coli. J. Bacteriol. 189, 244–253 (2007).
Sperandeo, P. et al. Functional analysis of the protein machinery required for transport of lipopolysaccharide to the outer membrane of Escherichia coli. J. Bacteriol. 190, 4460–4469 (2008).
Ruiz, N., Gronenberg, L. S., Kahne, D. & Silhavy, T. J. Identification of two inner-membrane proteins required for the transport of lipopolysaccharide to the outer membrane of Escherichia coli. Proc. Natl Acad. Sci. USA 105, 5537–5542 (2008).
Bos, M. P., Tefsen, B., Geurtsen, J. & Tommassen, J. Identification of an outer membrane protein required for the transport of lipopolysaccharide to the bacterial cell surface. Proc. Natl Acad. Sci. USA 101, 9417–9422 (2004).
Xie, R., Taylor, R. J. & Kahne, D. Outer membrane translocon communicates with inner membrane ATPase to stop lipopolysaccharide transport. J. Am. Chem. Soc. 140, 12691–12694 (2018).
Zampaloni, C. et al. A novel antibiotic class targeting the lipopolysaccharide transporter. Nature https://doi.org/10.1038/s41586-023-06873-0 (2024).
Raetz, C. R. H., Reynolds, C. M., Trent, M. S. & Bishop, R. E. Lipid A modification systems in Gram-negative bacteria. Annu. Rev. Biochem. 76, 295–329 (2007).
Dovala, D. et al. Structure-guided enzymology of the lipid A acyltransferase LpxM reveals a dual activity mechanism. Proc. Natl Acad. Sci. USA 113, E6064–E6071 (2016).
Clementz, T., Bednarski, J. J. & Raetz, C. R. Function of the htrB high temperature requirement gene of Escherichia coli in the acylation of lipid A: HtrB catalyzed incorporation of laurate. J. Biol. Chem. 271, 12095–12102 (1996).
Komazin, G. et al. Substrate structure–activity relationship reveals a limited lipopolysaccharide chemotype range for intestinal alkaline phosphatase. J. Biol. Chem. 294, 19405–19423 (2019).
Vorachek-Warren, M. K., Ramirez, S., Cotter, R. J. & Raetz, C. R. H. A triple mutant of Escherichia coli lacking secondary acyl chains on lipid A. J. Biol. Chem. 277, 14194–14205 (2002).
Boll, J. M. et al. Reinforcing lipid A acylation on the cell surface of Acinetobacter baumannii promotes cationic antimicrobial peptide resistance and desiccation survival. mBio 6, e00478-15 (2015).
Somerville, J. E., Cassiano, L. & Darveau, R. P. Escherichia coli msbB gene as a virulence factor and a therapeutic target. Infect. Immun. 67, 6583–6590 (1999).
Villa, R. et al. The Escherichia coli Lpt transenvelope protein complex for lipopolysaccharide export is assembled via conserved structurally homologous domains. J. Bacteriol. 195, 1100–1108 (2013).
Li, Y., Orlando, B. J. & Liao, M. Structural basis of lipopolysaccharide extraction by the LptB2FGC complex. Nature 567, 486–490 (2019).
Tang, X. et al. Cryo-EM structures of lipopolysaccharide transporter LptB2FGC in lipopolysaccharide or AMP-PNP-bound states reveal its transport mechanism. Nat. Commun. 10, 4175 (2019).
Wilson, A. & Ruiz, N. The transmembrane α-helix of LptC participates in LPS extraction by the LptB2FGC transporter. Mol. Microbiol. 118, 61–76 (2022).
Luo, Q., Shi, H. & Xu, X. Cryo-EM structures of LptB2FG and LptB2FGC from Klebsiella pneumoniae in complex with lipopolysaccharide. Biochem. Biophys. Res. Commun. 571, 20–25 (2021).
Sherman, D. J. Reconstitution of Bacterial Lipopolysaccharide Transport from Purified Components (Harvard Univ., 2015).
Zhang, G. et al. Cell-based screen for discovering lipopolysaccharide biogenesis inhibitors. Proc. Natl Acad. Sci. USA 115, 6834–6839 (2018).
Richie, D. L. et al. Toxic accumulation of LPS pathway intermediates underlies the requirement of LpxH for growth of Acinetobacter baumannii ATCC 19606. PLoS ONE 11, e0160918 (2016).
Kamoshida, G. et al. Lipopolysaccharide-deficient Acinetobacter baumannii due to colistin resistance is killed by neutrophil-produced lysozyme. Front. Microbiol. 11, 573 (2020).
Beceiro, A. et al. Biological cost of different mechanisms of colistin resistance and their impact on virulence in Acinetobacter baumannii. Antimicrob. Agents Chemother. 58, 518–526 (2014).
Schreiber, S. L. The rise of molecular glues. Cell 184, 3–9 (2021).
Metzgar, D. et al. Acinetobacter sp. ADP1: an ideal model organism for genetic analysis and genome engineering. Nucleic Acids Res. 32, 5780–5790 (2004).
de Berardinis, V. et al. A complete collection of single-gene deletion mutants of Acinetobacter baylyi ADP1. Mol. Syst. Biol. 4, 174 (2008).
Mastronarde, D. N. Automated electron microscope tomography using robust prediction of specimen movements. J. Struct. Biol. 152, 36–51 (2005).
Punjani, A., Rubinstein, J. L., Fleet, D. J. & Brubaker, M. A. cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 14, 290–296 (2017).
Zivanov, J. et al. New tools for automated high-resolution cryo-EM structure determination in RELION-3. eLife 7, e42166 (2018).
Zhong, E. D., Bepler, T., Berger, B. & Davis, J. H. CryoDRGN: reconstruction of heterogeneous cryo-EM structures using neural networks. Nat. Methods 18, 176–185 (2021).
Sanchez-Garcia, R. et al. DeepEMhancer: a deep learning solution for cryo-EM volume post-processing. Commun. Biol. 4, 874 (2021).
Morin, A. et al. Collaboration gets the most out of software. eLife 2, e01456 (2013).
Waterhouse, A. et al. SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res. 46, W296–W303 (2018).
Pettersen, E. F. et al. UCSF Chimera-a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 (2004).
Potterton, E., Briggs, P., Turkenburg, M. & Dodson, E. A graphical user interface to the CCP4 program suite. Acta Crystallogr. D 59, 1131–1137 (2003).
Moriarty, N. W., Grosse-Kunstleve, R. W. & Adams, P. D. Electronic ligand builder and optimization workbench (eLBOW): a tool for ligand coordinate and restraint generation. Acta Crystallogr. D 65, 1074–1080 (2009).
Adams, P. D. et al. PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr. D 58, 1948–1954 (2002).
Afonine, P. V. et al. Real-space refinement in PHENIX for cryo-EM and crystallography. Acta Crystallogr. D 74, 531–544 (2018).
Croll, T. I. ISOLDE: a physically realistic environment for model building into low-resolution electron-density maps. Acta Crystallogr. D 74, 519–530 (2018).
Goddard, T. D. et al. UCSF ChimeraX: meeting modern challenges in visualization and analysis. Protein Sci. 27, 14–25 (2018).
Emsley, P. & Cowtan, K. Coot: model-building tools for molecular graphics. Acta Crystallogr. D 60, 2126–2132 (2004).
Chen, V. B. et al. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D 66, 12–21 (2010).
Terashi, G., Wang, X., Maddhuri Venkata Subramaniya, S. R., Tesmer, J. J. G. & Kihara, D. Residue-wise local quality estimation for protein models from cryo-EM maps. Nat. Methods 19, 1116–1125 (2022).
Acknowledgements
We thank S. Sterling, R. Walsh, M. Mayer and S. Rawson for assistance with electron microscopy. We also thank S. Walker, R. Taylor and D. Tomasek for providing feedback on the manuscript. T. Meredith (Penn State) provided the kind gift of purified LPS variants. We thank W. Zhang for chemical synthesis and C. Bartelmus for small-molecule mass spectrometry. K. Pahil thanks H. Pahil. Cryo-EM data were collected at the Harvard Cryo-EM Center for Structural Biology at Harvard Medical School. Structural biology applications used in this project were compiled and configured by SBGrid. Funding for this work was provided by National Institutes of Health grants U19 AI158028 (to A.C.K. and D.K.), R01 AI149778 (to D.K.), R01 AI081059 (to D.K.) and R01 AI153358 (to D.K. and A.C.K.). M.G. is supported by the HHMI Hanna H. Gray Fellows Program.
Author information
Authors and Affiliations
Contributions
D.K. and K.S.P. conceptualized the project. K.S.P., V.B. and D.K. designed experiments. K.S.P. constructed the strains for protein expression from E. coli reported in this work and developed the protocol for expressing and purifying A. baylyi Lpt proteins. K.S.P. performed cryo-EM experiments. K.S.P. and M.G. solved and refined the structures. T.C. and F.D. provided input on model geometries. K.S.P. and D.K. conceptualized biochemical experiments. K.S.P. performed biochemical experiments. V.B. constructed and characterized all reported A. baylyi strains. P.M. supplied compounds for biochemical assays. D.M. synthesized 3H-Zosurabalpin. T.C. and R.B. designed SPA experiments and analysed data with input from D.K. and K.S.P. T.C. and R.B. performed SPA experiments. D.K. and A.C.K. supervised the project. K.S.P., M.G. and D.K. wrote the manuscript with input from V.B., T.C., P.M., C.B., F.D., M.L., K.B. and A.C.K.
Corresponding authors
Ethics declarations
Competing interests
T.C., P.M., C.B., F.D., R.B., M.L. and K.B. are current or former employees of F. Hoffmann-La Roche. A.C.K. is a cofounder and consultant for Tectonic Therapeutic and Seismic Therapeutic and for the Institute for Protein Innovation, a non-profit research institute.
Peer review
Peer review information
Nature thanks Russell Bishop, Paul Hergenrother and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Fig. 1 Sequences of Lpt proteins from Acinetobacter baylyi ADP1, Acinetobacter baumannii 19606 and E. coli K12.
Percent coverage and identities when compared to the Acinetobacter baylyi ADP1 sequence are shown. Alignments were made using ClustalO and then coloured in MView (https://www.ebi.ac.uk/Tools/msa/clustalo/).
Extended Data Fig. 2 Expression, purification and cryo-EM data processing for LptB2FG in complex with LPS and 1.
a. Representative size-exclusion chromatogram of Acinetobacter baylyi LptB2FG. b. SDS–PAGE gel showing peak fractions from the size-exclusion chromatography of the complex in a and used in subsequent structural experiments described in this figure. Four independent purifications of this construct gave similar size-exclusion and SDS–PAGE results. c. Representative (n = 12,402) micrographs of LptB2FG in complex with 1 and LPS embedded in vitreous ice. d. Scheme of three-dimensional classification and refinement of cryo-EM particle images. The initial models used for 3D classification were generated by ab initio reconstruction in cryoSPARC. e. Representative selected two-dimensional class averages of cryo-EM particle images. (f-g) Gold-standard Fourier shell correlation (FSC) curves calculated with different masks in cryoSPARC from the overall (f) and locally refined (g) structures of 1-bound LptB2FG. The resolution was determined at FSC = 0.143 (horizontal blue line). The final corrected mask gave an overall resolution of 3.0 Å. h. LPS and 1 fit in the final locally refined, sharpened map. Post processing of the map was done using DeepEMhancer.
Extended Data Fig. 3 Cryo-EM data processing and analysis for Acinetobacter LptB2FG with Acinetobacter LPS in the presence and absence of 1.
a. Structures of E. coli and Acinetobacter LPS. The parts that were not modelled in the structures presented in this work are coloured in red. b. Scheme of three-dimensional classification and refinement of cryo-EM particle images for Acinetobacter LptB2FG in the presence of Acinetobacter LPS and 1. The initial models used for 3D classification were generated by ab initio reconstruction in cryoSPARC. c. Representative selected two-dimensional class averages of Acinetobacter LptB2FG in the presence of Acinetobacter LPS and 1. (d-e) Gold-standard Fourier shell correlation (FSC) curves calculated with different masks in cryoSPARC from the overall (d) and locally refined (e) structures of Acinetobacter LptB2FG in the presence of Acinetobacter LPS and 1. The resolution was determined at FSC = 0.143 (horizontal blue line). The final corrected mask gave an overall resolution of 3.15 Å. f. 3D classification without alignments of the dataset detailed in (b–e) revealed conformational flexibility in the lysine of 1. There is a major population (purple box in b, transparent surface in f, state 1) in which said lysine is in contact with the 2’-phosphate of LPS and a minor population (pink box in b, solid surface in f, state 2) where the lysine is oriented towards the carboxylate of the branched KDO from LPS. This KDO is conserved in E. coli and Acinetobacter LPS, as shown in panel a. (g-h). There is an ordered DDM molecule in the Acinetobacter LptB2FG + 1 structure obtained with E. coli LPS, but not in the structure obtained using Acinetobacter LPS. LptF, LptG, LPS and 1 are coloured green, blue, yellow and purple. The structure obtained using E. coli LPS is shown in g with the DDM coloured violet. The Acinetobacter LPS acyl chain that overlaps with the location of the DDM highlighted in salmon (h).
Extended Data Fig. 4 Cryo-EM data processing and analysis for drug-free Acinetobacter baylyi LptB2FG.
(a-d). Processing for the structure of Acinetobacter baylyi LptB2FG in the presence of E. coli LPS. a Scheme of three-dimensional classification and refinement of cryo-EM particle images. The initial models used for 3D classification were generated by ab initio reconstruction in cryoSPARC. b. Representative selected two-dimensional class averages of cryo-EM particle images. (c-d) Gold-standard Fourier shell correlation (FSC) curves calculated with different masks in cryoSPARC from the overall (c) and locally refined (d) structures of drug-free LptB2FG. The resolution was determined at FSC = 0.143 (horizontal blue line). The final corrected mask gave an overall resolution of 3.0 Å. (e-h) Processing for the structure of Acinetobacter baylyi LptB2FG in the presence of Acinetobacter baylyi LPS. e Scheme of three-dimensional classification and refinement of cryo-EM particle images. The initial models used for 3D classification were generated by ab initio reconstruction in cryoSPARC. f. Representative selected two-dimensional class averages of cryo-EM particle images. (g-h) Gold-standard Fourier shell correlation (FSC) curves calculated with different masks in cryoSPARC from the overall (g) and locally refined (h) structures of drug-free LptB2FG. The resolution was determined at FSC = 0.143 (horizontal blue line). The final corrected mask gave an overall resolution of 3.1 Å. i. Cryo-EM structure of Acinetobacter LptB2FG with Acinetobacter LPS bound in the lumen of the transporter in white superimposed with the structure of Acinetobacter LptB2FG bound to 1 and Acinetobacter LPS (LptF, LptG and LPS are coloured green, blue and yellow, respectively). The overall r.m.s.d. is 0.37 over 7714 atoms. j. Cryo-EM structure of Acinetobacter LptB2FG with Acinetobacter LPS bound in the lumen of the transporter in white superimposed with the structure of Acinetobacter LptB2FG bound to E. coli LPS (LptF, LptG and LPS are coloured green, blue and yellow, respectively). The overall r.m.s.d. is 0.51 over 8138 atoms. (k-l). LptB2F(R55G)G and LptB2F(R30A)G have comparable ATPase hydrolysis rates to wild-type LptB2FG (k) but are unable to transport LPS to LptA (l). ATPase rates were measured using liposomes containing LptB2FG complexes and LPS and the presented data represent averages and standard deviations of results determined using three different proteoliposomes preparations for each LptF variant. Inorganic phosphate release was measured using a molybdate assay. LPS transport from LptB2FG to LptAI36pBPA in the presence of LptC-ΔTM was measured by detecting UV-dependent LptA-LPS crosslinks by LPS immunoblotting. Data shown is representative of experiments conducted in biological triplicate.
Extended Data Fig. 5 Loss of function mutations in LpxM render cells resistant to 1.
5 distinct mutations in LpxM were observed to provide resistance to 1. The location of the mutated residues relative to the active site (active-site residues are bolded) is shown.
Extended Data Fig. 6 Cryo-EM data processing and analysis for LptB2FGC.
a. Representative size-exclusion chromatogram of Acinetobacter baylyi LptB2FGC. b. SDS–PAGE gel showing peak fractions from the size-exclusion chromatography of the complex in a and used for the structural experiments. Three independent purifications of this construct gave similar size-exclusion and SDS–PAGE results. c. Representative (n = 18,862) micrograph of LptB2FGC embedded in vitreous ice. d. Scheme of three-dimensional classification and refinement of cryo-EM particle images. The initial models used for 3D classification were generated by ab initio reconstruction in cryoSPARC. e. Representative selected two-dimensional class averages of cryo-EM particle images. (f-g) Gold-standard Fourier shell correlation (FSC) curves calculated with different masks in cryoSPARC from the overall (f) and locally refined (g) structures of LptB2FGC. The resolution was determined at FSC = 0.143 (horizontal blue line). The final corrected mask gave an overall resolution of 3.6 Å. h. Electron density of the LptB2FGC structure with LptF coloured in green, LptG in blue and LptC in pink. The lipopolysaccharide positioning from our LptB2FG structure is overlayed with the LptB2FGC electron density. Post processing of the map was carried out using DeepEMHancer.
Extended Data Fig. 7 Cryo-EM data processing and analysis for 2- and 3-bound LptB2FG.
a. Scheme of three-dimensional classification and refinement of cryo-EM particle images for 2-bound LptB2FG. The initial models used for 3D classification were generated by ab initio reconstruction in cryoSPARC. b. Representative selected two-dimensional class averages of 2-bound LptB2FG. (c-d) Gold-standard Fourier shell correlation (FSC) curves calculated with different masks in cryoSPARC from the overall (c) and locally refined (d) structures of 2-bound LptB2FG. The resolution was determined at FSC = 0.143 (horizontal blue line). The final corrected mask gave an overall resolution of 3.25 Å. e. Scheme of three-dimensional classification and refinement of cryo-EM particle images for 3-bound LptB2FG. The initial models used for 3D classification were generated by ab initio reconstruction in cryoSPARC. f. Representative selected two-dimensional class averages of 3-bound LptB2FG. (g-h) Gold-standard Fourier shell correlation (FSC) curves calculated with different masks in cryoSPARC from the overall (f) and locally refined (g) structures of 3-bound LptB2FG. The resolution was determined at FSC = 0.143 (horizontal blue line). The final corrected mask gave an overall resolution of 3.1 Å.
Extended Data Fig. 8 Biochemical determinants of compound binding.
a) 2 treatment increases the ATPase activity of LptB2FG, LptB2FGC and LptB2FG-ΔTM-C but only in the presence of LPS. b) 3 treatment increases the ATPase activity of LptB2FG, LptB2FG-ΔTM-C but only in the presence of LPS. 3 does not affect the ATPase of LptB2FGC. c) LPS isolated from a ΔLpxM strain renders LptB2FGC resistant to 1. For panels a–c, ATPase rates were measured using liposomes containing the indicated Lpt complex indicated LPS variant and the presented data represent averages and standard deviations of results determined using three different proteoliposomes preparations for each condition variant. Inorganic phosphate release was measured using a molybdate assay. d) 2 does not inhibits LPS transport to LptA by wild-type E. coli LptB2FGC. Lipopolysaccharide transport from LptB2FGC to LptA modified with a photocrosslinkable amino acid (I36pBPA) was monitored in the presence of the indicated dose of 2 by exposing the samples to UV light after 60 min of transport, quenching by addition of SDS-loading buffer, PAGE to separate LPS-LptA adducts from LPS and western blotting against LPS. Data shown is representative of experiments conducted in biological triplicate.
Supplementary information
Supplementary Information
Supplementary Fig. 1, Tables 1–5 and Methods.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Pahil, K.S., Gilman, M.S.A., Baidin, V. et al. A new antibiotic traps lipopolysaccharide in its intermembrane transporter. Nature 625, 572–577 (2024). https://doi.org/10.1038/s41586-023-06799-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41586-023-06799-7
- Springer Nature Limited
This article is cited by
-
Considering the host in host–pathogen interactions
Nature Microbiology (2024)
-
Macrocyclic peptides: up-and-coming weapons to combat antimicrobial resistance
Signal Transduction and Targeted Therapy (2024)
-
A new type of antibiotic targets a drug-resistant bacterium
Nature (2024)
-
Macrocyclic peptides thwart Gram-negative bacteria
Nature Reviews Drug Discovery (2024)
-
Stability study in selected conditions and biofilm-reducing activity of phages active against drug-resistant Acinetobacter baumannii
Scientific Reports (2024)