

Abstract
Hydrogen sulfide (H2S), methanethiol (MeSH) and dimethylsulfide (DMS) are abundant sulfur gases with roles in biogeochemical cycling, chemotaxis and/or climate regulation. Catabolism of the marine osmolyte dimethylsulfoniopropionate (DMSP) is a major source of DMS and MeSH, but both also result from S-methylation of H2S via MddA, an H2S and MeSH S-methyltransferase whose gene is abundant in soil but scarce in marine environments. Here we identify the S-adenosine methionine (SAM)-dependent MeSH and H2S S-methyltransferase ‘MddH’, which is widespread in diverse marine bacteria and some freshwater and soil bacteria. mddH is predicted in up to ~5% and ~15% of seawater and coastal sediment bacteria, respectively, which is considerably higher than mddA. Furthermore, marine mddH transcript levels are similar to those for the most abundant DMSP lyase gene dddP. This study implies that the importance of H2S and MeSH S-methylation pathways in marine environments is significantly underestimated.
Similar content being viewed by others


Main
Dimethylsulfide (DMS) is the largest natural sulfur source transferred from Earth’s oceans to the atmosphere (~13–37 Tg annually)1,2, and its oxidation products can act as cloud condensation nuclei that potentially impact climate3. DMS also plays important roles in microbial metabolism, global sulfur cycling4 and chemotaxis5. Dimethylsulfoniopropionate (DMSP) is thought to be the major bio-source of DMS via microbial DMSP lyase enzymes6 (Fig. 1a). However, there are other aerobic and anaerobic DMSP-independent DMS production pathways, such as hydrogen sulfide (H2S) and methanethiol (MeSH) S-methylation7,8,9 (Fig. 1a). Microorganisms can produce MeSH from methionine (Met) cleavage10, H2S S-methylation11, DMS degradation12 and DMSP demethylation13 (Fig. 1a), which accounts for >70% of marine DMSP catabolism14. Indeed, the ability to produce MeSH from Met (via MegL) and the DMSP demethylation intermediate methylmercaptopropionate (MMPA, via DmdBCD) is widespread in bacteria15. H2S is also abundant in diverse environments, present at mM levels in, for example, sediment and hydrothermal environments16,17.

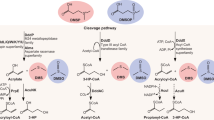
a, A simplified schematic representation of MeSH and DMS-related metabolic pathways and enzymes, only showing the molecules that contain the sulfur component that ends up in DMS. Green and grey arrows/fonts predict pathways and enzymes that exist or do not exist in Halomonas strains, respectively. The Halomonas strains were predicted to cleave DMSP via DMSP lyase enzymes (DddD) and oxidize DMS to DMSO (via DdhA and not Tmm). They were not predicted to reduce DMSO to DMS and lacked homologues to known DMSO reductase enzymes (Dms, Dor). The Halomonas strains lacked the potential to demethylate DMSP (via DmdA) but contained DmdBC and AcuH that convert 3-methylmercaptopropionate (MMPA) to MeSH. The MddA enzyme was absent in Halomonas strains used in this study, but some of Halomonas strains contained MddH, an isoform H2S and MeSH S-methyltransferase enzyme. Halomonas strains were grown in MBM and the following assays conducted: b, MeSH and DMS production with 0.5 mM l-Met added; c, DMS production with 0.5 mM MeSH added; d, DMS production with 0.5 mM DMSP added; e, MeSH and DMS production with 0.5 mM MMPA added; f, MeSH and DMS production with 0.5 mM H2S added. The values for DMS and MeSH production are shown as mean ± s.d. of 3 biological replicates. No MeSH or DMS was detected in the blank MBM media control.
MddA is a microbial SAM-dependent S-methyltransferase that S-methylates MeSH8 and H2S9 to yield DMS (Fig. 1a), which can function to detoxify H2S and MeSH9. Although abundant in soil (relative abundance (RA) 5–76%) and surface saltmarsh sediment (RA 9.6%) bacteria, mddA is less common in seawater bacteria (RA ≤ 0.5%)8,18,19. However, Carrión et al.19 identified marine bacteria with MeSH-dependent DMS production (Mdd) activity that lacked mddA, implying the existence of unidentified Mdd enzymes. Recently, thiol methyltransferases, termed TMT1A and TMT1B, capable of methylating H2S were identified in humans, other mammals and fish20,21, but not yet in bacteria. Here we identify a bacterial thiol methyltransferase that allowed reevaluation of the importance of Mdd pathways as sources of DMS in marine settings.
Results
Methanethiol-dependent DMS production by Halomonas species
Halomonas alimentaria EF61 (isolated from Mariana Trench seawater22) produced MeSH and DMS when grown with l-Met (Fig. 1b). EF61 did not produce DMSP; thus its DMS production was not due to DMSP cleavage despite this bacterium exhibiting DMSP cleavage with exogenous DMSP (Fig. 1c). EF61 produced 11.3 ± 3.1 nmol DMS mg−1 total protein h−1 from MeSH (Fig. 1d). EF61 also produced MeSH and DMS from MMPA (Fig. 1e), the primary catabolite of the DMSP demethylase predicted to be in ~20% of marine bacteria15, and from H2S (Fig. 1f). DMS production from l-Met, MMPA, MeSH and H2S was also observed in 3 of 5 other tested Halomonas strains (SCS19, H33-56 and RT37) from diverse marine environments (Fig. 1 and Supplementary Table 1). In contrast, H. alimentaria H10-9-1 and H. saccharevitans H10-59 could not generate DMS from any of these four sulfur compounds but could produce MeSH from l-Met, MMPA and H2S (Fig. 1). These data implied that H10-9-1 and H10-59 lacked or did not express key MeSH S-methyltransferase enzyme(s) that were active in the four other Halomonas strains.
MddH, an H2S and MeSH S-methyltransferase in Halomonas
The genomes of all six Halomonas isolates contained megL encoding Met γ-lyase, consistent with their ability to liberate MeSH from l-Met (Fig. 1a,b). They contained dmdBC and acuH, encoding enzymes that catabolize MMPA and release MeSH15, consistent with their observed MMPA-dependent MeSH production phenotype (Fig. 1a,e). Similar to Halomonas HTNK1 (ref. 23), the strains contained the DMSP lyase gene dddD and generated DMS from exogenously added DMSP and acetyl CoA, probably produced intracellularly24 (Fig. 1a,c). None of the isolates contained mddA encoding the only known microbial H2S and MeSH S-methyltransferase, or proteins with >63% coverage and >34% amino acid identity to the human thiol S-methyltransferases TMT1A and TMT1B, indicating that the four Halomonas isolates with Mdd activity probably contained an unidentified Mdd enzyme.
There were 84 genes unique to the four Halomonas strains with Mdd activity compared with the two lacking this phenotype and only one encoded a candidate methyltransferase. This gene, termed mddH, was associated with multicopper oxidase genes and those predicted to be involved in metal transport and resistance, with no obvious link to sulfur metabolism (Supplementary Fig. 1). MddH shared no protein sequence identity with MddA. Instead, it encoded a ubiquinone methyltransferase UbiE family protein (COG2226) with only 37% amino acid identity to Escherichia coli UbiE, a SAM-dependent methyltransferase involved in menaquinone synthesis25. Notably, all six Halomonas strains contained a different UbiE homologue, with 74–75% protein identity to E. coli UbiE. EF61 MeSH S-methylation activity was cytosolic (2.46 ± 0.3 nmol DMS mg−1 total protein h−1) and not membranous, consistent with the EF61 MddH protein lacking a signal peptide. This differs from MddA which has 4–6 membrane-spanning helices and whose activity is enriched in Pseudomonas deceptionensis membrane fractions8. Indeed, MeSH S-methylation activity was detected in the cytosolic (2.46 ± 0.3 nmol DMS mg−1 total protein h−1) but not in membrane fractions of H. alimentaria EF61.
The MddH protein shared 31/50% and 34/53% amino acid sequence identity/similarity to the human thiol S-methyltransferases TMT1A and TMT1B, respectively, and their AlphaFold26 predicted structures were similar, particularly in their central and C-terminal regions, containing the conserved central GxGxG binding motif21 for SAM binding (Supplementary Fig. 2a). The predicted MddH structure was comparatively more compact than for TMT1A and TMT1B, lacked an extended N-terminal ‘hooked’ helical region and a conserved aspartate residue at position 98 previously implicated in SAM binding21, which was a glutamate at position 63 in MddH (Supplementary Fig. 2b). These observations support the hypothesis that MddH was a thiol S-methyltransferase similar to TMT1A and TMT1B.
E. coli cell extracts containing EF61 MddH protein showed in vitro SAM-dependent Mdd activity (46.4 ± 4.0 nmol DMS mg−1 total protein h−1) and H2S-dependent MeSH (14.4 ± 1.5 nmol mg−1 total protein h−1) and DMS (10.3 ± 0.2 nmol mg−1 total protein h−1) production (Table 1). Furthermore, an EF61 ΔmddH mutant (Supplementary Fig. 3a) overproduced MeSH when grown with l-Met or H2S compared with the wild-type strain and completely lacked Mdd activity (Supplementary Fig. 3b,c). These mutant phenotypes were restored to wild-type levels by cloned mddH, consistent with MddH being the Halomonas spp. SAM-dependent MeSH S-methyltransferase enzyme. These data and those in Fig. 1 indicated that there were also MddH-independent pathways converting H2S to MeSH in the tested Halomonas strains studied here, potentially through l-cysteine and l-Met as intermediates27,28.
Importantly, when incubated in sterilized coastal seawater with 4 nM H2S or MeSH, EF61 but not the ΔmddH mutant showed DMS production (Supplementary Table 2) compared with the seawater control. The H2S or MeSH levels used were physiologically relevant for seawater and sediment samples29,30, implying that MddH yields DMS in marine environments.
MddH is widespread in diverse bacteria
Proteins with >45% amino acid identity to MddH were identified in diverse bacterial taxa, mainly Gammaproteobacteria and Alphaproteobacteria¸ but also some Betaproteobacteria, Deltaproteobacteria, Acidobacteria and Bacteroidetes (Fig. 2). When cloned, candidate mddH genes from marine and soil bacteria, but not Vibrio ubiE (negative control) conferred H2S and Mdd S-methylation activity to E. coli (Table 1). This included MlMddH from Marinobacter litoralis Sw-45, characterized below, which shared 60.3% amino acid identity to EF61 MddH. The diverse natural host bacteria containing mddH genes also had H2S and MeSH S-methylation activity (Supplementary Fig. 4). We predict that the candidate MddH enzymes in Fig. 2, which are distinct from the UbiE outgroup, constitute the ‘MddH’ family of H2S and MeSH S-methyltransferase enzymes. Several SAM-dependent methyltransferases were structurally similar to MddH (predicted by AlphaFold), but they had <21% amino acid identity to MddH and broad substrate specificity where characterized (Supplementary Table 3), and their activity on H2S/MeSH required further examination.

The tree is drawn to scale, with branch lengths measured in number of substitutions per site. The scale bar indicates 0.5 amino acid substitutions per site. Different coloured circles at the end of each branch indicate bacterial taxonomy (see Taxonomy key). Different label colours indicate the source of the bacterial strain (see Source key). Proteins with experimentally ratified Mdd activity are marked with a yellow star near the labels. MddH from Halomonas alimentaria EF61 is highlighted by a red star. A putative UbiE protein from Vibrio sp. with no Mdd activity was used as the outgroup (shown in a black box).
Interestingly, M. litoralis Sw-45 mddH was adjacent to the cydABCD operon encoding a cytochrome bd oxidase complex (CydAB) and cysteine transporter (CydDC) involved in the regulation of intracellular cysteine and redox levels and H2S production31,32. However, mddH was not associated with any genes obviously linked to H2S or MeSH generation in other bacteria (Supplementary Fig. 1).
mddH and mddA were found in distinct but similarly diverse host bacteria. The mddA gene was mostly found in actinobacterial, alphaproteobacterial Rhizobiales and, except for Pseudomonas, was far less common in gammaproteobacteria than mddH8. The key difference between bacteria containing mddA and those with mddH was not in their host taxonomy, but more prominently in the environments they inhabit. Most bacteria with mddA were isolated from terrestrial soil or freshwater and not marine environments8. In contrast, mddH was predominantly in diverse bacteria from marine seawater or sediment such as Halomonas, Marinobacter, Novosphingobium and Erythrobacter (Fig. 2). mddH was also found but far less frequently in bacteria from soil, lake, spring and other sources such as wastewater plants, compost, fruits or animals (Fig. 2).
Characterization of the MddH enzyme
The purified MlMddH enzyme (Fig. 3a) showed SAM-dependent S-methylation of H2S and MeSH, producing both MeSH and slightly lower amounts of DMS from H2S (Fig. 3b). MlMddH had an optimal pH of ~9.0 (Supplementary Fig. 5a) and temperature of 45 °C (Supplementary Fig. 5b) for MeSH and H2S, and showed high activities at ~pH 8 and at 10–20 °C, physiologically relevant seawater pH and temperature values, respectively (Supplementary Fig. 5). MlMddH had Km (the Michaelis constant) and kcat (enzyme turnover number) values of 0.23 mM and 0.08 s−1, respectively, for MeSH, and 0.07 mM and 0.06 s−1, respectively, for the SAM co-substrate. The Km of MlMddH for H2S (0.22 mM) was similar to that for MeSH (0.23 mM), while the kcat value of 0.16 s−1 measured for H2S was ~2-fold higher than that for MeSH (Fig. 3e,f). Overall, MlMddH was ~2-fold more efficient when using H2S (kcat/Km ≈ 727 M−1 s−1) over MeSH (kcat/Km ≈ 347 M−1 s−1) as substrate.

a, SDS–PAGE of purified MlMddH. b, In vitro DMS and/or MeSH production by purified MlMddH with MeSH or H2S as substrates. The units (nmol mg−1 h−1) represent the nanomolar amount of DMS or MeSH produced by MlMddH per milligram per hour. c, The ability of MlMddH to S-methylate a range of substrates (as detailed) as monitored by the formation of S-adenosyl homocysteine (SAH) from S-adenosyl methionine (SAM). d, The effect of EDTA addition on MlMddH activity. e, Michaelis–Menten curves of purified MlMddH for H2S S-methylation and SAM. f, Michaelis–Menten curves of purified MlMddH for MeSH S-methylation and SAM. Initial rates were determined with 0.27 µM MlMddH (molecular weight: 24.24 kDa) and 0–2 mM SAM (1 mM MeSH/H2S), or 0–2 mM MeSH/H2S (1 mM SAM) at 45 °C and pH 9 for 30 min. Kinetic parameters for MddH were determined by nonlinear fitting using the Michaelis–Menten equation in the form v/[E] = kcat×[S]/(Km + [S]) based on the initial rates of DMS production (or DMS and MeSH production) in triplicate experiments. Data are shown as mean ± s.d.
MlMddH turnover rates for H2S (0.16 s−1) and MeSH (0.08 s−1) were consistent with enzymes involved in secondary metabolism33 and the SAM-dependent S-methyltransferase MddA with MeSH (~0.09 s−1) and H2S (~0.01 s−1)9. Compared with MddA, the specificity constants for MlMddH with H2S and MeSH were substantially higher by around an order of magnitude, indicating higher catalytic efficiency. The modestly lower kcat/Km values observed for MlMddH relative to the lower limit expected for most enzymes33 may be due to the reactive nature of these gaseous substrates and/or substrate diffusion limitation in assays. MlMddH showed no S-methylation activity towards most other tested sulfur compounds including glutathione (GSH), cysteine (l-Cys), coenzyme A (CoA), 2-mercaptoethanesulfonate (Coenzyme M) or the DMSP synthesis intermediates l-Met and 4-methylthio-2-hydroxybutyrate (MTHB) (Fig. 3c). However, S-adenosyl homocysteine was formed from SAM when MlMddH was incubated with ethanethiol and 1-propanethiol at levels ~23 and ~40% less, respectively, compared with MeSH (Supplementary Fig. 5e). This is consistent with MddH being able to S-methylate other short-chain low molecular weight alkyl thiols.
The purified MlMddH protein contained only up to 0.15 Zn and 0.038 Ca metals per protein and addition of mM levels of the metal-chelator EDTA only slightly reduced its activity (Fig. 3d). Furthermore, MddH activity was not enhanced by the addition of various metals and was even reduced by mM levels of Mn2+, Zn2+ and Co2+ (Supplementary Fig. 5c,d). Thus, despite Halomonas mddH being linked to candidate metal transporters and metalloenzymes (Supplementary Fig. 1), MddH does not likely require a metal co-factor for activity.
The role of MddH in bacteria
The wild-type EF61, ΔmddH mutant and genetically complemented strains were assessed for their ability to grow with mM l-Met, H2S, MeSH, cysteine, H2O2, cobalt or zinc levels. These compounds and metals can be cytotoxic if allowed to accumulate, cause oxidative stress31,32,34 and/or were associated with the action of gene products situated near mddH in microbial genomes (Supplementary Fig. 1). Except for MeSH, none of these compounds or metals affected the growth or yield of the ΔmddH compared with the wild-type strain (Supplementary Fig. 6). In contrast, despite having a similar initial growth rate to the wild-type and complemented strains, the ΔmddH mutant had reduced final biomass when grown with 2 mM MeSH compared with the wild-type and complemented strains (Supplementary Fig. 6). Furthermore, mddH transcription was significantly 2.5-fold upregulated by growth with MeSH but not with l-Met or H2S (Supplementary Fig. 6e). These data are consistent with MddH having a role to detoxify MeSH when it reaches high environmental levels, through generation of non-toxic DMS, as was recently shown for MddA9. Although MeSH is potentially abundant in Earth’s oceans due to the prominence of DMSP demethylation, it is rarely likely to reach mM levels35. Thus, if MddH does have a role in MeSH detoxification, it is probably minor under physiologically relevant marine conditions. Alternatively, we hypothesize that there are other detoxification strategies for the MeSH and/or the other tested stress-inducing molecules in EF61 that compensate for the loss of MddH in the EF61/ΔmddH mutant. This hypothesis was supported by the MddH-independent S-methylation of H2S observed in all Halomonas strains tested here (Fig. 1).
MddH is abundant in marine environments
mddH was found in 242 out of 243 Tara Oceans samples in the OM-RCG marine metagenome database36, comprising 68 sampling locations in epipelagic and mesopelagic waters across the globe. In the 178 prokaryote-enriched samples (>0.22 μm size fractionated), the percentage of mddH normalized by cell numbers ranged between 0.09 and 5.2% (with an average of 2.19 ± 0.93%) (Fig. 4 and Supplementary Table 4). Marine samples with abundant mddH-containing bacteria (>4%) were from the South/North Atlantic Ocean, South/North Pacific Ocean, Indian Ocean and Mediterranean Sea. The relative abundance of mddH in surface water (SRF, median: 2.26%) and the deep chlorophyll maximum layers (DCM, median: 2.21%) were similar but significantly higher than in the mesopelagic zone (MES, median: 1.60%) (Kruskal–Wallis test, Chi square = 16.0, d.f. = 2, P < 0.05) (Supplementary Fig. 7). Surprisingly, many copies of mddH were also identified in virus-enriched samples (<0.22 μm) (5.79 × 10−8 to 4.06 × 10−5 per mapped read). Indeed, 32 distinct MddH homologues were identified from marine viruses in Tara Oceans Viromes37 data. Many of these were highly homologous to bacterial mddH genes (Supplementary Table 5 and Supplementary Fig. 8), supporting the hypothesis of mddH horizontal gene transfer between viruses and bacteria. In contrast, mddA was detected in far less Tara Oceans (190 of the 243) and marine prokaryote-enriched samples (169 of 178) than mddH. mddA was significantly less abundant than mddH in these samples (~50-fold lower, Mann–Whitney test, P < 0.05) with on average only 0.04 ± 0.07% of bacteria predicted to contain mddA (Supplementary Table 4). In addition, unlike mddH, the percentage of bacteria with mddA was highest in the MES samples (Kruskal–Wallis test, Chi square = 34.8, d.f. = 2, P < 0.05). The 216 mddH sequences retrieved from Tara Oceans metagenomes were all from Proteobacteria (73.1% Gammaproteobacteria, 12.0% Alphaproteobacteria, 0.5% Betaproteobacteria and 14.4% others) (Fig. 4b). In contrast, the 25 mddA sequences were distributed in more diverse bacterial taxa, including Bacteroidetes, Cyanobacteria, Planctomycetes and Alpha-, Gamma- and Epsilon-proteobacteria (Fig. 4b).

a, The relative abundance of mddH and mddA in Tara Oceans metagenome samples from the OM-RGCv1 database (normalized by cell numbers). b, Taxonomic assignment of MddH and MddA sequences in Tara Oceans metagenome samples from the OM-RGCv1 database. c, The relative abundance of mddH and mddA transcripts in Tara Oceans metatranscriptome samples from the OM-RGCv2 database (normalized by per cent of mapped reads). d, The relative abundance of mddH and mddA in sediment metagenome samples from the Yellow Sea and the Bohai Sea (normalized by cell numbers). e, Taxonomy assignment of MddH and MddA sequences from sediment metagenome samples from the Yellow Sea and the Bohai Sea.
mddH and mddA transcripts were respectively found in 186 and 63 of the 187 metatranscriptomes in the OM-RGCv2 database38 (Fig. 4c). Consistent with their gene abundance, the abundance of mddH transcripts (2.80 × 10−7 to 5.33 × 10−5 per mapped read) was far higher than for mddA (4.84 × 10−9 to 8.03 × 10−7 per mapped read). These data are consistent with MddH being an important enzyme in Earth’s oceans and marine H2S and MeSH S-methylation, being more crucial than previously predicted8.
In contrast, the most abundant DMSP lyase gene dddP was predicted to be in 12.4 ± 6.7% (0.4%–29.3%) of bacteria in Tara Oceans metagenomes, which was ~5-fold more than those with MddH (Supplementary Table 4). dddP transcript levels (2.78 × 10−7 to 9.98 × 10−5 per mapped read) were also slightly higher than those of mddH (1.86 × 10−7 to 5.32 × 10−5 per mapped read). These data imply that the Mdd pathway is probably a less important source of DMS than DMSP-dependent DMS production in marine systems.
As much as ~15% of bacteria (0.88–14.74%, normalized by cell number) in surface sediments from the Bohai and Yellow Seas near China39 were predicted to contain mddH (Fig. 4d). Indeed, mddH was far more abundant than mddA (predicted in 0.39–3.34% of bacteria) in most samples. These omics data again suggest that bacteria with mddH and mddA are generally more abundant in sediment than in aquatic marine samples, and that mddH is the dominant gene in these marine settings (Fig. 4d). Sediment mddH genes were mainly gammaproteobacterial but were also in Acidobacteria and some sulfate-reducing Deltaproteobacteria, whereas mddA was found in more diverse phyla including Ignavibacteriae, Nitrospirae, Planctomycetes and Bacteroidetes (Fig. 4e). These sediment environments probably contain higher physiological levels of l-Met, MeSH and, more prominently, H2S (that can be present at mM levels) than seawater environments22,39. However, the genetic potential to cleave DMSP was still higher in these sediments, with 10.0–29.5% of bacteria predicted to contain dddP. Once again, this implies DMSP cleavage as the likely dominant DMS-producing pathway. Nevertheless, considering the large number of bacteria in marine sediment40 and the often-high substrate availability22,39, H2S- and MeSH-dependent DMS production pathways are probably important sources of DMS in marine sediment environments.
Given that Carrión et al.8 predicted from metagenomic analysis that 5–76% of soil bacteria contained mddA, we also examined the abundance of mddH in these soil metagenomes (Supplementary Table 6)8. No reliable mddH sequence was identified in these soil metagenomes possibly due to their sequence depth. Thus, we also investigated the abundance of mddA and mddH in larger metagenome datasets from rhizosphere soil samples of different plants (Glycine soja, Sesbania cannabina and Sorghum bicolor)41. Only 0.1–1.67% of bacteria in these soil samples were predicted to contain mddH, whereas mddA was far more abundant (8.74–13.11%) (Supplementary Fig. 9). These data are consistent with MddA being the major H2S and MeSH S-methylation enzyme in terrestrial soils, while MddH probably dominates in marine settings.
Discussion
MddH is a SAM-dependent H2S and MeSH S-methyltransferase, which is phylogenetically distinct from bacterial MddA and human TMT1A and TMT1B (Supplementary Fig. 10). Importantly, MddH was found in up to ~5% and ~15% of bacteria in seawater and coastal sediments, respectively, which correspondingly equates to 2.6 × 104 bacteria ml−1 and 2.85 × 108 bacteria g−1 (refs. 22,42), respectively, containing mddH and the capacity to S-methylate H2S and MeSH to yield DMS. These findings challenged the view that Mdd processes are only likely dominant in soil bacteria8,9, emphasizing a potentially important and unexpected role for this pathway in global marine sulfur cycling.
Notably, l-Met is potentially toxic to cells if allowed to accumulate43 and is a substrate for both DMSP biosynthesis and Mdd via the MegL enzyme which liberates MeSH and is common to most bacteria. Thus, both these pathways potentially alleviate the cellular toxicity of excess cellular l-Met since DMSP and DMS are non-toxic molecules. Far less marine bacteria contain the dominant DMSP biosynthesis gene dsyB (0.5% of bacteria44) than megL and mddH, implying that MddH may have a more prominent role in the management of free l-Met in marine bacteria. H2S and MeSH are also cytotoxic, which animals and plants detoxify by enzymatic S-methylation, in many cases, to DMS45,46. While the data presented here support the role of MddH in the detoxification of MeSH, its role in detoxification of excess H2S is not supported, since the Halomonas mddH− mutant showed no growth or yield impairment compared with the wildtype in the presence of this toxic molecule. There were probably other H2S S-methylation pathways that might have compensated for the loss of mddH. Nevertheless, bacterial DMSP biosynthesis genes and mddH were generally more abundant in marine sediments than in waters, as are l-Met, MeSH and H2S, indicating that diverse sediments might be environments with high levels of not only DMSP but also DMS produced through the MeSH and H2S S-methylation and DMSP cleavage pathways.
Considering the enormous scale of DMSP production47 and the high abundance of diverse microbial ddd genes6 in marine settings, DMSP cleavage is still probably the dominant DMS-producing pathway in marine aquatic environments. However, it should not be forgotten that DMSP demethylation, which produces MeSH, is thought to account for 70% of marine DMSP catabolism48 and that H2S can reach mM levels in marine sediments16,17, indicating the presence of considerable amounts of the MeSH and H2S substrates for MddA and MddH in marine settings. Compared with DMS, there are few measurements of MeSH and H2S from aerobic marine environments, but they are generally thought to be far less abundant than DMS49, which may be due in part to an active marine MeSH and H2S S-methylation pathway, particularly via MddH. Further studies evaluating MeSH and H2S S-methylation and its flux in diverse marine settings are required to establish its impact on global DMS production and sulfur cycling, but this study implies that these methylation reactions are far more important in marine environments than previously predicted.
Methods
Bacterial strains and culturing
Detailed information of the strains used in this study are listed in Supplementary Table 1. Escherichia coli strains were grown on LB media overnight at 37 °C. Halomonas strains and other marine bacterial isolates were routinely grown on Marine Agar (MA) medium (per litre seawater: 1 g yeast extract, 5 g peptone, 0.01 g ferric phosphate, 20 g agar, pH 7.6) for 24 h at 28 °C. These isolates were also cultivated in marine basal medium (MBM) minimal medium (salinity 35 PSU)50 supplemented with a mixed carbon source (10 mM from a 1 M stock of 200 mM succinate, glucose, pyruvate, sucrose and glycerol).
Quantification of DMS and MeSH production
To measure DMS and MeSH production by Halomonas strains, colonies from fresh agar plates were inoculated into 200 μl MBM medium supplemented with or without l-Met and MMPA (0.5 mM final concentration) in 2 ml sealed glass vials and incubated at 28 °C and 170 r.p.m. for 24 h. The headspace MeSH and DMS was monitored by gas chromatography (GC) with a flame photometric detector (Agilent 7890B GC fitted with a 7693A autosampler) and an HP-INNOWax 30 m × 0.320 mm capillary column (Agilent Technologies J&W). DMS and MeSH calibration curves were produced as described in ref. 8. The detection limits for DMS and MeSH were 0.2 and 5 nmol, respectively. Cellular protein content was estimated using Bradford assays (BioRad). Rates of MeSH and DMS production were expressed as nmol mg−1 total protein h−1. Experiments were carried out in three biological replicates. To measure DMS production from MeSH, Halomonas strains were cultured in MBM supplemented with 0.5 mM MeSH as described above. To measure MeSH and DMS production from H2S, Halomonas strains were cultured in Marine Broth (MB) medium (per litre seawater: 1 g yeast extract, 5 g peptone, pH 7.6) supplemented with 0.5 mM H2S, since H2S is reactive with Fe(III)EDTA in the MBM.
To measure DMSP production, Halomonas strains were cultured in MBM with l-Met (0.5 mM) in 1.5 ml Eppendorf tubes at 28 °C and 170 r.p.m. for 24 h, then 200 μl of NaOH (10 M) was added to 200 μl of cultures, immediately sealed and mixed in 2 ml glass vials to chemically cleave the DMSP and yield DMS. Vials were incubated in the dark overnight and the DMS derived from DMSP was measured by GC, as above. To measure DMSP catabolism, strains were cultured in MBM with DMSP (0.5 mM) in 2 ml sealed glass vials at 28 °C and 170 r.p.m. for 24 h. The resulting DMS was measured and quantified as described above.
Genome sequencing and comparative genomic analysis
Genomic DNA from six Halomonas strains was extracted following the phenol-chloroform-isoamylic alcohol extraction protocol. Genomic DNA sequencing and quality control was performed by the Beijing Genomics Institute using Illumina Hiseq 4000 with a 270 bp paired-end library and PacBio with a 20 kb library. The PacBio reads and Illumina reads were assembled using Unicycle (v.0.4.8). Annotation of these genomes was conducted using Prokka51 (for comparative genomic analysis) and the RASTtk online service52 with default settings. The general features of the six Halomonas genomes are listed in Supplementary Table 7. Protein sequences whose activity had been confirmed of MddA, MegL and other enzymes involved in DMS cycling described in ref. 39 were used as query sequences to perform BLASTP against the six Halomonas genomes. The programme GET_HOMOLOGUES (v.3.0.3)53, with three clustering algorithms (bidirectional best hit, COGtriangles and OrthoMCL), was used for clustering orthologous genes and identifying core- and pan-genomes under default parameter values. Unique genes belonging to DMS-producing Halomonas strains compared with non-DMS-producing isolates were extracted.
Construction of in-frame deletion mutant of mddH
An in-frame deletion mutation of mddH (ΔmddH) was constructed in H. alimentaria EF61 by double-crossover allelic exchange. The plasmids and primers used are listed in Supplementary Tables 8 and 9. Briefly, the up- and downstream region of mddH was amplified by polymerase chain reaction (PCR) using primer pair mddH-UO/UI (product size: 660 bp) and mddH-DO/DI (product size: 766 bp), respectively. The two PCR products above were further used as templates in overlapping PCR using mddH-UO/DO to yield a final product of 1,426 bp comprising the up- and downstream region of mddH. The resulting product was cloned into the pK18mobsacB suicide vector and conjugated into H. alimentaria EF61 by triparental mating using the E. coli helper strain 803/pRK2013. Transconjugants with a single-crossover insertion in EF61 chromosome were obtained by screening on MA plates containing rifampicin and kanamycin. Allelic exchange to delete a 483 bp core region within mddH (621 bp) was achieved by a second crossover event, which was selected on MA containing 20% (w/v) sucrose. The resultant mutant, EF61/ΔmddH, was selected by antibiotic sensitivity (kanamycin sensitive and rifampicin resistant) and confirmed by PCR assay followed by nucleotide sequence analysis (Supplementary Fig. 3a).
To complement the mddH mutation, the mddH gene containing its promoter region was amplified using primers mddHcom-F and mddHcom-R (Supplementary Table 9). The fragment was cloned into a low-copy plasmid pBBR1MCS-5, verified by sequencing and transformed into the EF61/ΔmddH mutant by electroporation. The complemented strain (EF61/ΔmddH/pBBR1MCS-5-mddH) was selected as gentamycin-resistant transformants and the presence of the plasmid was confirmed by PCR analysis and sequencing. The EF61/ΔmddH mutant and complemented strain EF61/ΔmddH/pBBR1MCS-5-mddH were grown on MA for 20 generations to ensure that they were stably maintained.
Seawater incubation experiments
H. alimentaria EF61 and the ΔmddH mutant were grown at 28 °C and 170 r.p.m. for 24 h in MB. Bacterial cells were collected, washed three times and resuspended in sterilized seawater (collected from the Qingdao coast, October 2023). The resuspended cultures were 10-fold diluted into 2.5 ml sterilized seawater (with 4 nM MeSH or H2S) in triplicate, followed by incubation at 16 °C for a further 24 h. The resulting DMS was quantified using a modified purge and trap method and GC54.
Construction of the MddH phylogenetic tree
The H. alimentaria EF61 MddH protein sequence was used as a query sequence to perform a BLASTP search against the representative genome database in NCBI. Candidate MddH sequences with >45% identity and an E-value ≤ 1 × 10−50 were chosen to construct an MddH phylogenetic tree. All protein sequences were aligned using the Muscle method in the MEGA 7.0.26 package55. The maximum-likelihood tree of MddH homologues was constructed using IQ-TREE (v.1.6.1)56 under the LG + F + R6 model with 1,000 bootstrap replications. The UbiE protein sequence from Vibrio sp. ZXX013 (ON685883), a strain with no Mdd activity, was used as the outgroup. The resulting tree was visualized in iTol57.
Prediction of MddH cell location in H. alimentaria EF61
The signal peptide and cell location of MddH were predicted using SignalP (6.0)58, Cello59 and PortB60. The membrane and cytoplasmic proteins of H. alimentaria EF61 were extracted using the Bacterial Membrane Protein/Cytoplasmic Protein Extraction kit (Solarbio) following manufacturer instructions, resulting in 24.3 mg ml−1 membrane proteins and 23.3 mg ml−1 cytoplasmic proteins. Approximately 600 μg of membrane, cytoplasmic and total proteins were added to Tris-HCl (pH 9.0) with 1 mM MeSH and SAM to a final volume of 150 μl and incubated at 37 °C for 3 h. DMS production was quantified as described above.
Heterologous expression of MddH and homologous enzymes
The mddH gene from H. alimentaria EF61 (621 bp, WP_013333065.1) was amplified by PCR using the primer set MddHPE-F/MddHPE-R (Supplementary Table 9) and cloned into the E. coli expression vector, pET-24a. The cloned gene was confirmed by sequencing at Sangon Biotech (China). The pET-24a construct containing mddH and empty vector controls were transformed into E. coli BL21 (DE3) cells and grown at 37 °C and 150 r.p.m. in LB broth supplemented with kanamycin (100 μg ml−1). At the mid-exponential growth phase, isopropylthio-β-galactoside (IPTG) was added at a final concentration of 0.1 mM, and the cells were then incubated at 28 °C and 150 r.p.m. for a further 3 h. The E. coli cells were then pelleted by centrifugation, resuspended in PBS buffer and sonicated as described in ref. 8. The supernatants were collected and cell pellets were resuspended. Triplicate 200 μl supernatants were incubated with 1 mM SAM and 1 mM MeSH (or 1 mM H2S) for 2 h before quantifying the DMS (and MeSH) produced in the headspace and protein concentrations, as described above.
To define the functionality of the MddH protein family, candidate mddH genes from Marinobacter litoralis Sw-45 (WP_114333749.1), Algiphilus aromaticivorans DG1253 (WP_043766208.1), Pseudomonas pelagia CL-AP6 (WP_022964284.1), Hyphomonas adhaerens MHS-3 (WP_035568920.1), Pyruvatibacter mobilis CGMCC_1.15125 (WP_160588566.1), Novosphingobium colocasiae KCTC 32255 (WP_189621457.1), Erythrobacter ramosus DSM 8510 (WP_160761066.1), Ramlibacter aquaticus LMG 30558 (WP_193782355.1) and Caulobacter henricii CB4 (WP_062144739.1) were obtained from NCBI (Supplementary Table 10), synthesized by Sangon Biotech and cloned into pET-24a. The ubiE gene from Vibrio sp. ZXX013 with no Mdd activity was also synthesized and cloned into pET-24a. Heterologous expression of these genes/proteins in E. coli as well as H2S and MeSH S-methyltransferase assays were performed as described above.
Characterization of MddH enzyme properties
Heterologously expressed MlMddH protein was purified using NTA-Ni (Qiagen) according to manufacturer recommendations. The purified MlMddH was assessed using 12% sulfate-polyacrylamide gel electrophoresis (SDS–PAGE) and stored at −20 °C with 25% glycerol. The enzymatic activity of MlMddH was determined in a standard reaction system containing 20 mM Tris-HCl buffer (pH 9.0), 1 mM SAM, 1 mM MeSH (or H2S) and 0.27 μM of enzyme in a final volume of 150 μl. After incubation at 37 °C for 30 min, the reaction was terminated by adding 100 μl HCl (10%) and the production of DMS (and MeSH) was measured by GC as described above.
To determine the optimal catalysis temperature, the activity of MlMddH was measured at 4–65 °C at different intervals. The optimal pH values of MlMddH were measured between pH 4 and 10.6 with buffer systems described in ref. 61. To analyse the effects of metal ions on MlMddH activity, Mn2+, Na+, K+, Zn2+, Ca2+, Mg2+ and Co2+ were added at final concentrations of 0.1 and 1 mM. The effects of metal-chelator EDTA, and protein-denaturant SDS and urea were examined at final concentrations of 0.1 and 1 mM. In addition, the metal concentrations (Zn, Pb, Mn, Mg, K, Fe, Cu, Co, Ca, Al and Na) of the purified MlMddH were analysed using Agilent 7500c inductively coupled plasma mass spectrometry (ICP–MS). To test substrate specificity, l-Met, MTHB, glutathione (GSH), cysteine and CoA were added to a final concentration of 1 mM, and the resulting S-adenosyl-l-homocysteine (SAH) from SAM was detected using high performance liquid chromatography (HPLC) analyses9.
Km and kcat values were determined by nonlinear analysis of kinetic data using 0.27 µM MddH and 0–2 mM SAM (1 mM MeSH/H2S), or 0–2 mM MeSH/H2S (1 mM SAM) in 20 mM Tris-HCl (pH 9.0). The reaction mixture was incubated at 45 °C for 30 min and terminated by the addition of 100 μl 10% HCl before detection of DMS (and MeSH) by GC. MlMddH methyltransferase activity was reported as the amount of DMS produced from MeSH, and as the summed amount of MeSH and twice the DMS production (since two methyl groups are transferred by MlMddH to produce DMS) with H2S as substrate. No background DMS production was detected with either SAM and MeSH or SAM and H2S in solution at 2 mM under these experimental conditions. Kinetic parameters were calculated by fitting a nonlinear regression directly to the Michaelis–Menten equation using GraphPad Prism 8.
Growth of Halomonas strains under different conditions
To compare the growth of H. alimentaria EF61, ΔmddH and the complemented strain, overnight cultures of these strains were adjusted to the same absorbance at 590 nm and inoculated (1:100) into MBM, and MBM supplemented with 2 mM of MeSH and H2S. The cultures were incubated at 28 °C and the absorbance at 595 nm was measured hourly in 96-well plates. Each experiment was conducted in triplicate.
To investigate the direct influence of Co2+, Zn2+, H2O2 and cysteine on growth, overnight cultures of H. alimentaria EF61, ΔmddH and the complemented strain were adjusted to the same absorbance at 595 nm and inoculated (1:100) into 5 ml MBM with Co2+, Zn2+, H2O2 or cysteine, added to a final concentration of 1 mM. The cultures were incubated at 28 °C and 170 r.p.m., and the growth of each strain was recorded after 24 h by measuring absorbance at 595 nm. Each experiment was conducted in triplicate.
RT–qPCR analysis
H. alimentaria EF61 was cultured in MB at 28 °C until mid-exponential phase. Cultures were collected by centrifugation at 4,000 g for 5 min, rinsed and resuspended in fresh MB. MeSH and H2S were added to a final concentration of 1 mM and the cells were incubated at 28 °C for 2 h. The same volume of distilled water was added to the control. Total RNA extraction was performed using the E.Z.N.A Bacterial RNAkit (Omega). Reverse transcription was performed using the TransScript One-Step gDNA Removal and cDNA Synthesis SuperMix Transcript (TransGen). RT–qPCR was performed on an ABI 7500 real-time PCR (Applied Biosystems) using SYBR Premix Ex Taq (TaKaRa) and primers are listed in Supplementary Table 9. The housekeeping recA gene of H. alimentaria EF61 was used as a reference control for sample normalization.
Metagenomic and metatranscriptome analysis
Validated protein sequences of MddH listed in Supplementary Table 10, as well as validated protein sequences of MddA from Mycobacterium tuberculosis H37Rv (NP_217755.1), Bradyrhizobium diazoefficiens USDA 110 Blr1218 (NP_767858.1) and Blr5741 (NP_772381.1), Pseudomonas sp. GM41 (WP_008148420.1), P. deceptionensis M1T (AJE75769) and Sulfurovum sp. NBC37-1 (YP_001358232.1)8, were used as query sequences to perform hidden Markov Model (HMM)-based searches (HMMER 3.1b2) for homologues in seawater metagenome and metatranscriptome data using Ocean Gene Atlas62, as well as in marine sediment metagenomic data from the Bohai Sea and Yellow Sea39 and soil metagenomes41. The cut-off E-value for HMM searches was set as <1 × 10−80. The Tara Oceans Microbiome Reference Gene Catalog (OM-RGCv1) database containing data from 243 Tara Oceans samples was chosen to analyse the abundance of mddH and mddA in seawater metagenomes36. Cell numbers were estimated using the observed median abundance of ten prokaryotic single marker genes36, and the abundance of mddH and mddA in metagenome data was normalized by cell number in each sample. The Tara Oceans Viromes data were obtained from iVirus (https://www.ivirus.us/data), and MddH homologues were identified using HMM searches with E-value < 1 × 10−80. The metagenomes from the Bohai and Yellow Sea sediment samples (top 5 cm surface sediment) were collected, sequenced and analysed in ref. 39. The metagenomes from rhizosphere soil samples of Glycine soja, Sesbania cannabina and Sorghum bicolor were collected, sequenced and analysed in ref. 41. For the Bohai and the Yellow Sea sediment metagenome47 and soil metagenome data41, the relative abundances of mddH and mddA were calculated by normalizing to recA abundance using a cut-off of E < 1 × 10−50, as described in ref. 39. mddH and mddA transcript abundances were analysed against the Tara Oceans Microbiome Reference Gene Catalog with Arctic data (OM-RGCv2)38 and normalized by per cent of mapped read. The abundances of dddP and dmdA in Tara Oceans metagenomes/metatranscriptomes and sediment metagenomes were also evaluated as above, with specific cut-off E-value and corresponding validated proteins described in ref. 39.
Statistics and reproducibility
All measurements of MeSH and DMS levels (in bacterial strains or enzyme assays) and RT–qPCR were based on the mean of three biological replicates per strain/condition tested and the error bars indicate standard deviations. To identify statistically significant differences between standard and experimental conditions in Supplementary Fig. 6e, a two-sided independent Student’s t-test was applied to the data. To compare the gene abundance between different groups in Supplementary Fig. 7, statistical analysis was carried out in Origin Pro 2021 and the normality of data in each group was tested. Non-normally distributed data were compared using Mann–Whitney test (between two groups) or Kruskal–Wallis test (between three groups). For Fig. 3a and Supplementary Fig. 3a, at least three independent experiments were performed and the results shown are from one representative experiment. No statistical method was used to predetermine sample size and no data were excluded from the analyses.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The genomes of six Halomonas strains were deposited in WGS Batch at NCBI under accession number JAMSHM000000000, JAMSHN000000000, JAMSHO000000000, JAMSHP000000000, CP098827 and CP098828 (PRJNA844217). The ubiE gene from Vibrio sp. ZXX013 was deposited at NCBI with accession number ON685883. Verified functional MddH protein sequences are listed in Supplementary Table 10. The Tara Oceans Microbiome Reference Gene Catalog (OM-RGCv1) database was obtained from Ocean Gene Atlas (https://tara-oceans.mio.osupytheas.fr/). The Tara Oceans Viromes data were obtained from iVirus (https://www.ivirus.us/data). Source data are provided with this paper.
References
Ksionzek, K. B. et al. Dissolved organic sulfur in the ocean: biogeochemistry of a petagram inventory. Science 354, 456–459 (2016).
Kiene, R. P. & Bates, T. S. Biological removal of dimethyl sulphide from sea water. Nature 345, 702–705 (1990).
Vallina, S. M. & Simó, R. Strong relationship between DMS and the solar radiation dose over the global surface ocean. Science 315, 506–508 (2007).
Sievert, S., Kiene, R. & Schulz-Vogt, H. The sulfur cycle. Oceanography 20, 117–123 (2007).
Li, C. Y. et al. Dimethylsulfoniopropionate and its catabolites are important chemical signals mediating marine microbial interactions. Trends Microbiol. 31, 992–994 (2023).
Zhang, X.-H. et al. Biogenic production of DMSP and its degradation to DMS—their roles in the global sulfur cycle. Sci. China Life Sci. 62, 1296–1319 (2019).
Stets, E. G., Hines, M. E. & Kiene, R. P. Thiol methylation potential in anoxic, low-pH wetland sediments and its relationship with dimethylsulfide production and organic carbon cycling. FEMS Microbiol. Ecol. 47, 1–11 (2004).
Carrión, O. et al. A novel pathway producing dimethylsulphide in bacteria is widespread in soil environments. Nat. Commun. 6, 6579 (2015).
Li, C. Y. et al. Aerobic methylation of hydrogen sulfide to dimethylsulfide in diverse microorganisms and environments. ISME J. 17, 1184–1193 (2023).
Tanaka, H., Esaki, N. & Soda, K. Properties of l-methionine γ-lyase from Pseudomonas ovalis. Biochemistry 16, 100–106 (1977).
Lomans, B. P. et al. Obligate sulfide-dependent degradation of methoxylated aromatic compounds and formation of methanethiol and dimethyl sulfide by a freshwater sediment isolate, Parasporobacterium paucivorans gen. nov., sp. nov. Appl. Environ. Microbiol. 67, 4017–4023 (2001).
Schäfer, H., Myronova, N. & Boden, R. Microbial degradation of dimethylsulphide and related C1-sulphur compounds: organisms and pathways controlling fluxes of sulphur in the biosphere. J. Exp. Bot. 61, 315–334 (2010).
Howard, E. C. et al. Bacterial taxa that limit sulfur flux from the ocean. Science 314, 649–652 (2006).
Kiene, R. P., Linn, L. J. & Bruton, J. A. New and important roles for DMSP in marine microbial communities. J. Sea Res. 43, 209–224 (2000).
Reisch, C. R. et al. Novel pathway for assimilation of dimethylsulphoniopropionate widespread in marine bacteria. Nature 473, 208–211 (2011).
Miyazaki, J. et al. Deepest and hottest hydrothermal activity in the Okinawa trough: the Yokosuka site at Yaeyama Knoll. R. Soc. Open Sci. 4, 171570 (2017).
Wang, F. & Chapman, P. M. Biological implications of sulfide in sediment—a review focusing on sediment toxicity. Environ. Toxicol. Chem. 18, 2526–2532 (1999).
Carrión, O. et al. Methanethiol-dependent dimethylsulfide production in soil environments. ISME J. 11, 2379–2390 (2017).
Carrión, O. et al. Methanethiol and dimethylsulfide cycling in Stiffkey saltmarsh. Front. Microbiol. 10, 1040 (2019).
Maldonato, B. J., Russell, D. A. & Totah, R. A. Human METTL7B is an alkyl thiol methyltransferase that metabolizes hydrogen sulfide and captopril. Sci. Rep. 11, 4857 (2021).
Dalmasy, J. M. G. et al. The thiol methyltransferase activity of TMT1A (METTL7A) is conserved across species. Preprint at bioRxiv https://doi.org/10.1101/2023.11.17.567538 (2023).
Williams, B. T. et al. Bacteria are important dimethylsulfoniopropionate producers in coastal sediments. Nat. Microbiol. 4, 1815–1825 (2019).
Todd, J. D. et al. Molecular dissection of bacterial acrylate catabolism – unexpected links with dimethylsulfoniopropionate catabolism and dimethyl sulfide production. Environ. Microbiol. 12, 327–343 (2010).
Alcolombri, U., Laurino, P., Lara-Astiaso, P., Vardi, A. & Tawfik, D. S. DddD is a CoA-transferase/lyase producing dimethyl sulfide in the marine environment. Biochemistry 53, 5473–5475 (2014).
Lee, P. T., Hsu, A. Y., Ha, H. T. & Clarke, C. F. A C-methyltransferase involved in both ubiquinone and menaquinone biosynthesis: isolation and identification of the Escherichia coli ubiE gene. J. Bacteriol. 179, 1748–1754 (1997).
Jumper, J. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021).
Maier, T. H. P. Semisynthetic production of unnatural l-α-amino acids by metabolic engineering of the cysteine-biosynthetic pathway. Nat. Biotechnol. 21, 422–427 (2003).
Kessler, D. Enzymatic activation of sulfur for incorporation into biomolecules in prokaryotes. FEMS Microbiol. Rev. 30, 825–840 (2006).
Midilli, A., Ay, M., Kale, A. & Veziroglu, T. N. A parametric investigation of hydrogen energy potential based on H2S in Black Sea deep waters. Int. J. Hydrogen Energy 32, 117–124 (2007).
Gros, V. et al. Concentrations of dissolved dimethyl sulfide (DMS), methanethiol and other trace gases in context of microbial communities from the temperate Atlantic to the Arctic Ocean. Biogeosciences 20, 851–867 (2023).
Mironov, A. et al. CydDC functions as a cytoplasmic cystine reductase to sensitize Escherichia coli to oxidative stress and aminoglycosides. Proc. Natl Acad. Sci. USA 117, 23565–53570 (2020).
Mironov, A. et al. Mechanism of H2S-mediated protection against oxidative stress in Escherichia coli. Proc. Natl Acad. Sci. USA 114, 6022–6027 (2017).
Bar-Even, A. et al. The moderately efficient enzyme: evolutionary and physicochemical trends shaping enzyme parameters. Biochemistry 50, 4402–4410 (2011).
Linder, D. P. & Rodgers, K. R. Methanethiol binding strengths and deprotonation energies in Zn(II)-imidazole complexes from M05-2X and MP2 theories: coordination number and geometry influences relevant to zinc enzymes. J. Phys. Chem. B 119, 12182–12192 (2015).
Wilkening, J. V. et al. The production and fate of volatile organosulfur compounds in sulfidic and ferruginous sediment. J. Geophys. Res. Biogeosci. 124, 3390–3402 (2019).
Sunagawa, S. et al. Structure and function of the global ocean microbiome. Science 348, 1261359 (2015).
Brum, J. R. et al. Patterns and ecological drivers of ocean viral communities. Science 348, 1261498 (2015).
Salazar, G. et al. Gene expression changes and community turnover differentially shape the global ocean metatranscriptome. Cell 179, 1068–1083.e21 (2019).
Song, D. et al. Metagenomic insights into the cycling of dimethylsulfoniopropionate and related molecules in the eastern China marginal seas. Front. Microbiol. 11, 157 (2020).
Kallmeyer, J., Pockalny, R., Adhikari, R. R., Smith, D. C. & D’Hondt, S. Global distribution of microbial abundance and biomass in subseafloor sediment. Proc. Natl Acad. Sci. USA 109, 16213–16216 (2012).
Zheng, Y. et al. Patterns in the microbial community of salt-tolerant plants and the functional genes associated with salt stress alleviation. Microbiol. Spectr. 9, e0076721 (2021).
Whitman, W. B., Coleman, D. C. & Wiebe, W. J. Prokaryotes: the unseen majority. Proc. Natl Acad. Sci. USA 95, 6578–6583 (1998).
Tuite, N. L., Fraser, K. R. & O’Byrne, C. P. Homocysteine toxicity in Escherichia coli is caused by a perturbation of branched-chain amino acid biosynthesis. J. Bacteriol. 187, 4362–4371 (2005).
Curson, A. R. J. et al. Dimethylsulfoniopropionate biosynthesis in marine bacteria and identification of the key gene in this process. Nat. Microbiol. 2, 17009 (2017).
Sun, Y. et al. Adaption to hydrogen sulfide-rich environments: strategies for active detoxification in deep-sea symbiotic mussels, Gigantidas platifrons. Sci. Total Environ. 804, 150054 (2022).
Itoh, N. et al. Involvement of S-adenosylmethionine-dependent halide/thiol methyltransferase (HTMT) in methyl halide emissions from agricultural plants: isolation and characterization of an HTMT-coding gene from Raphanus sativus (daikon radish). BMC Plant Biol. 9, 116 (2009).
Curson, A. R. J., Todd, J. D., Sullivan, M. J. & Johnston, A. W. B. Catabolism of dimethylsulphoniopropionate: microorganisms, enzymes and genes. Nat. Rev. Microbiol. 9, 849–859 (2011).
Kiene, R. P. & Linn, L. J. The fate of dissolved dimethylsulfoniopropionate (DMSP) in seawater: tracer studies using 35S-DMSP. Geochim. Cosmochim. Acta 17, 2797–2810 (2000).
Nightingale, P. D. & Liss, P. S. in Treatise on Geochemistry Vol. 6 (ed. Elderfield, H.) 49–81 (Elsevier, 2003).
Liu, J. et al. Bacterial dimethylsulfoniopropionate biosynthesis in the East China Sea. Microorganisms 9, 657 (2021).
Seemann, T. Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069 (2014).
Brettin, T. et al. RASTtk: a modular and extensible implementation of the RAST algorithm for building custom annotation pipelines and annotating batches of genomes. Sci. Rep. 5, 8365 (2015).
Contreras-Moreira, B. & Vinuesa, P. GET_HOMOLOGUES, a versatile software package for scalable and robust microbial pangenome analysis. Appl. Environ. Microbiol. 79, 7696–7701 (2013).
Zhang, S. H., Yang, G. P., Zhang, H. H. & Yang, J. Spatial variation of biogenic sulfur in the south Yellow Sea and the East China Sea during summer and its contribution to atmospheric sulfate aerosol. Sci. Total Environ. 488–489, 157–167 (2014).
Kumar, S., Stecher, G. & Tamura, K. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874 (2016).
Nguyen, L. T., Schmidt, H. A., von Haeseler, A. & Minh, B. Q. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–274 (2015).
Letunic, I. & Bork, P. Interactive Tree Of Life (iTOL) v4: recent updates and new developments. Nucleic Acids Res. 47, W256–W259 (2019).
Teufel, F. et al. SignalP 6.0 predicts all five types of signal peptides using protein language models. Nat. Biotechnol. 40, 1023–1025 (2022).
Yu, C. S. et al. CELLO2GO: a web server for protein subcellular localization prediction with functional gene ontology annotation. PLoS ONE 9, e99368 (2014).
Yu, N. Y. et al. PSORTb 3.0: improved protein subcellular localization prediction with refined localization subcategories and predictive capabilities for all prokaryotes. Bioinformatics 26, 1608–1615 (2010).
He, X. et al. Characterization of multiple alginate lyases in a highly efficient alginate-degrading Vibrio strain and its degradation strategy. Appl. Environ. Microbiol. 88, e0138922 (2022).
Villar, E. et al. The Ocean Gene Atlas: exploring the biogeography of plankton genes online. Nucleic Acids Res. 46, W289–W295 (2018).
Acknowledgements
This work was supported by the National Natural Science Foundation of China (92251303 and 32370118, Principal Investigator (PI): X.-H.Z.; 42376101, PI: Y. Zhang), the Scientific and Technological Innovation Project of Qingdao Marine Science and Technology Center (LSKJ202203206, LSKJ202203201 and 2022QNLM030004-3, PI: X.-H.Z.), the Fundamental Research Funds for the Central Universities (202172002, PI: X.-H.Z.), Biotechnology and Biological Sciences Research Council, UK (BB/X005968, PI: J.D.T.), Natural Environmental Research Council, UK (NE/P012671, NE/S001352, NE/V000756/1, NE/X000990 and NE/X014428, PI: J.D.T.), Leverhulme Trust (RPG-2020-413, PI: J.D.T.), and the Biotechnology and Biological Sciences Research Council, UK (BB/M00256X/1 and BB/S008942/1, PI: A.J.G.).
Author information
Authors and Affiliations
Contributions
X.-H.Z. and Y. Zhang conceived the work; X.-H.Z., Y. Zhang and J.D.T. designed experiments; Y. Zhang performed bioinformatic analysis and with J.D.T. and X.-H.Z. wrote the paper; Y. Zhang, C.S. and Z.G. carried out most of the experiments; L.L. helped construct the in-frame deletion mutant; K.S. and X.Z. assisted with bioinformatic analysis; A.J.G. analysed the enzyme kinetic and structural data. Y. Zheng analysed soil metagenomes. All authors edited and approved the paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Microbiology thanks Uria Alcolombri, Christopher Reisch, David Zechel and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Information
Supplementary Figs. 1–10 and Tables 1–10.
Supplementary Data 1
Source data for Supplementary Figs. 3–7 and 9 and Table 2.
Source data
Source Data Fig. 1
Statistical source data.
Source Data Fig. 3
Statistical source data.
Source Data Fig. 3a
Unprocessed protein gels.
Source Data Fig. 4
Statistical source data.
Source Data Table 1
Statistical source data.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhang, Y., Sun, C., Guo, Z. et al. An S-methyltransferase that produces the climate-active gas dimethylsulfide is widespread across diverse marine bacteria. Nat Microbiol 9, 2614–2625 (2024). https://doi.org/10.1038/s41564-024-01788-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41564-024-01788-6
- Springer Nature Limited