

Abstract
Hyperfluorescence shows great promise for the next generation of commercially feasible blue organic light-emitting diodes, for which eliminating the Dexter transfer to terminal emitter triplet states is key to efficiency and stability. Current devices rely on high-gap matrices to prevent Dexter transfer, which unfortunately leads to overly complex devices from a fabrication standpoint. Here we introduce a molecular design where ultranarrowband blue emitters are covalently encapsulated by insulating alkylene straps. Organic light-emitting diodes with simple emissive layers consisting of pristine thermally activated delayed fluorescence hosts doped with encapsulated terminal emitters exhibit negligible external quantum efficiency drops compared with non-doped devices, enabling a maximum external quantum efficiency of 21.5%. To explain the high efficiency in the absence of high-gap matrices, we turn to transient absorption spectroscopy. It is directly observed that Dexter transfer from a pristine thermally activated delayed fluorescence sensitizer host can be substantially reduced by an encapsulated terminal emitter, opening the door to highly efficient ‘matrix-free’ blue hyperfluorescence.
Similar content being viewed by others
Main
The ‘blue OLED problem’—the quest for a stable and efficient blue organic light-emitting diode (OLED), has plagued academic and industrial researchers alike for over 20 years1,2. Thermally activated delayed fluorescence (TADF)-sensitized fluorescent OLEDs (coined as Hyperfluorescent in other work3,4) are considered one of the most promising contemporary solutions, combining the high efficiency of a TADF donor with the stability and colour purity of a conventional fluorescent acceptor2,5,6.
In current hyperfluorescent OLEDs, a high triplet energy matrix is implemented alongside the TADF sensitizer and emitter molecules, affording a three-component emissive layer (EML) (or even four in the case of exciplex/mixed matrices7)—a device structure that we term matrix-containing hyperfluorescence (MCHF)2. Progress in blue MCHF has been swift, with efficient devices now approaching the Commission Internationale de l'Éclairage (CIExy) colour coordinates for display requirements, owing to the concurrent development of efficient TADF sensitizers and narrowband terminal emitters2,7,8,9,10,11,12,13,14,15. However, compared with the typical two-component host–guest EML already adopted by industry, the additional matrices required in MCHF devices unavoidably increases the complexity of device fabrication. Power efficiency is also reduced due to higher turn-on and driving voltages. A simple and efficient matrix-free hyperfluorescent (MFHF)16 device (first introduced in another work17) with a two-component EML solely consisting of a TADF sensitizer (hereafter referred to as the host) alongside a narrowband terminal emitter, would afford a new paradigm in blue OLED technology from a device fabrication perspective.
The underlying obstacle to MFHF is maintaining high device performance and simplifying the device structure. Dilution into a high triplet energy matrix has so far been inescapable to suppress Dexter transfer to long-lived terminal emitter triplets. Although Dexter transfer to terminal emitter triplets has been challenging to directly observe, it has been hypothesized to be highly disadvantageous in hyperfluorescent devices from both efficiency and stability perspectives18,19,20. First, due to their non-radiative nature, the population of terminal emitter triplet states is an efficiency loss pathway. Second, it has been suggested that the operational stability of hyperfluorescent OLEDs is adversely affected by the population of terminal emitter triplets due to a substantial increase in the exciton residence time21. Multiresonant (MR) TADF materials are a popular class of terminal emitter that could, in principle, alleviate the efficiency loss associated with Dexter transfer through their ability to triplet harvest. However, to the best of our knowledge, such a process has not been unequivocally observed1,22. The problem of an elongated exciton residence time through the Dexter population of terminal emitter triplets also persists, as MR TADF reverse intersystem crossing rates are typically orders of magnitude slower than for conventional donor–acceptor TADF materials11,23.
In this work, molecular design provides a new approach to suppress loss through the population of terminal emitter triplet states—the encapsulated ultranarrowband blue emitter. When doped into a pristine TADF host with no additional matrix, transient absorption (TA) studies directly indicate that Dexter transfer can be substantially curtailed by decorating a terminal emitter with insulating alkylene straps. Owing to Dexter transfer suppression, negligible efficiency loss is observed in MFHF OLEDs compared with non-doped TADF devices, with external quantum efficiencies (EQEs) as high as 21.5% and narrow true-blue electroluminescence (EL) (14–15 nm full-width at half-maximum (FWHM)). Hence, simply structured MFHF devices with narrowband blue emission and high efficiency have been realized for the first time.
Molecular design
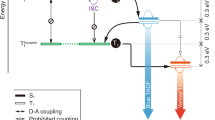
The design and syntheses of new terminal emitters NB-1 and NB-2 are illustrated in Fig. 1. In hyperfluorescence, the working mechanism is an energy cascade from a triplet-harvesting TADF donor (our host) to a fluorescent terminal emitter. To achieve efficient blue MFHF OLEDs, we sought to design emitters with structural and photophysical properties that promote singlet transfer via Förster resonant energy transfer (FRET)18 while suppressing Dexter transfer (Fig. 1a)24,25.

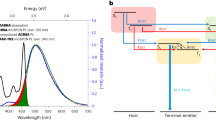
a, Schematic and Jablonski diagram for the suppression of Dexter triplet transfer from a TADF host via emitter encapsulation. b, Structures of the mDICz and encapsulated NB-1 luminophores; relevant absorption and PL spectra for NB-1 and DMAC-DPS. c, Synthesis schemes and structures of NB-1 and NB-2 with X-ray single-crystal structures (H atoms are omitted for clarity).
FRET proceeds via dipole–dipole coupling, with its efficiency linked to the spectral overlap between the host photoluminescence (PL) and the emitter absorption. As the S0 ↔ S1 transitions of the TADF host and fluorescent emitter are spin allowed, the desirable singlet energy transfer is accessible via FRET. FRET follows an R−6 distance dependence, making it efficient over rather long distances—on the molecular scale—of up to around 10 nm (ref. 18). Conversely, as the S0 → Tn transitions of fluorescent emitters are quantum mechanically forbidden, energy transfer to their triplet states is restricted to a Dexter transfer mechanism. Dexter transfer proceeds via direct host–emitter orbital overlap and is therefore restricted to the Ångstrom length scale, with its efficiency dropping off exponentially with distance24,25. Increasing the host–emitter distance in a system with good spectral overlap is, therefore, a plausible strategy to suppress Dexter transfer while preserving efficient FRET, which would prevent triplets accumulating on the terminal emitter to simultaneously improve the device efficiency and stability18,19,20.
When designing NB-1 and NB-2, we first sought to maximize singlet transfer via FRET. A highly rigid and planar indolocarbazole heterocycle (mDICz)26,27,28 was selected as our blue luminophore (Fig. 1b), which displays intense 0–0 absorption (ɛ, ~60,000 M−1 cm−1), a narrow Stokes shift (2–3 nm), an extremely small PL FWHM (~10 nm), unity fluorescence quantum yield (ΦPL) and high photostability (Supplementary Fig. 66). Computations suggest that the narrow emission can be understood in view of its strongly distorted central benzene ring (Supplementary Note 3). The distortion suppresses the Franck–Condon activity of the aromatic breathing vibration, inducing a weak vibronic progression.
The solution absorption and PL spectra of NB-1 are shown in Fig. 1b alongside the neat film PL of DMAC-DPS—an established blue TADF material with high reported non-doped efficiency (ΦPL = ~100%, EQE = ~20%), which functions as one of our host materials29. The photophysical properties of NB-1 are clearly beneficial for maximizing the spectral overlap with the TADF host to promote efficient FRET while preserving pure-blue emission. Our design also allows a subtle tuning of the emitter bandgap for matching with different host materials. Modifying the position of the peripheral tert-butyl groups of NB-1 affords the regioisomer NB-2 (Fig. 1c), for which the solution absorption and emission spectra are redshifted by 6–7 nm (Supplementary Figs. 59 and 60). Supplementary Note 5 presents a full photophysical characterization of the emitters.
Second, we focused on suppressing the Dexter transfer. Previous evidence indicates that alkylene encapsulation can suppress the intermolecular interactions leading to concentration quenching and aggregation30,31,32,33,34,35. We designed NB-1 and NB-2 hypothesizing that the encapsulating straps would similarly shield the luminophore core from the short-range coupling required for Dexter transfer (Fig. 1a)36,37,38. The alkylene straps were installed by either Grubbs metathesis (NB-1) or nucleophilic substitution chemistry (NB-2) (Fig. 1c). Both routes crucially proceed via intermediates that can be obtained in high purity using conventional laboratory techniques. This was achieved by employing bulky alkene (NB-1) or benzyl (NB-2) groups to circumvent the poor solubility of the indolocarbazole core. The luminophore cores of NB-1 and NB-2 are completely enveloped by a continuous alkylene macrocycle (Supplementary Note 2), in contrast to designs that decorate emitters with peripheral steric bulk39,40. Despite high molecular weights (MW = 1,122), NB-1 and NB-2 remain sufficiently volatile for processing via thermal evaporation (240–260 °C at 10−7 mbar; Supplementary Fig. 58). This suggests a suppression of intermolecular interactions, in line with calculations indicating that the non-conjugated straps of NB-1 contribute to over half its molecular volume (Supplementary Table 2). Additionally, although the shielding of emitters with side groups could lead to redshifted and broadened emission1, we note that our design has a negligible effect on the mDICz core photophysics (Supplementary Table 4).
Organic light-emitting devices
OLEDs were fabricated to explore the effect of incorporating encapsulated emitters into MFHF devices (Fig. 2). The EL spectrum of an OLED where the EML consists of a DMAC-DPS host doped with 1 wt% NB-1 is shown in Fig. 2b, alongside that for a non-doped reference device (System A; Fig. 2a). Pertinent device metrics are listed in Table 1. The non-doped DMAC-DPS OLED demonstrates peak EL at 476 nm with a broad FWHM of 88 nm. Owing to singlet energy transfer via FRET, the 1 wt% NB-1 MFHF device displays a far superior EL spectrum, with a desirable 449 nm peak wavelength and an extremely narrow FWHM of 14 nm. Harmful high-energy emission of <450 nm is also reduced. We note that the incorporation of 1 wt% NB-1 is accompanied by only a slight decrease in EQE compared with the non-doped device (16.4% versus 17.0%).

a, Material combinations for matrix-free Systems A, B and C. b, EL spectra of System A, with photographs of OLED emission shown in the insets. c, EL spectra of System B, with En-Per single-crystal X-ray structure shown in the inset. XRD, X-ray diffraction. d, EL spectra of System C, with photographs of OLED emission shown in the insets.
Perylene, the archetypal blue terminal emitter in hyperfluorescent systems, was also encapsulated to afford En-Per (System B; Fig. 2a). In an identical fashion to the results for System A, the 1 wt% En-Per in the DMAC-DPS device affords a maximum EQE of 16.4%, suggesting that encapsulation may be generally applicable to obtain high-MFHF EQEs from different luminophore cores. This efficiency is comparable with the best previously reported values for organic hyperfluorescent systems incorporating perylene terminal emitters, even when matrices are included3,41,42,43.
In System C (Fig. 2a), the slightly redshifted emitter NB-2 exhibits good spectral overlap with the TADF material DPAc-DCzBN (Supplementary Fig. 76), a high-performing emitter in non-doped devices44. In analogy to System A, the incorporation of 1 wt% NB-2 into DPAc-DCzBN in System C affords a large decrease in FWHM compared with the host-only device (86 → 15 nm) (Fig. 2d). The 1 wt% NB-2 device also crosses the 20.0% EQE threshold, affording a high maximum EQE of 21.5% comparable with the non-doped device (maximum EQE = 20.3%). This is a 1.5 times improvement over the best industry-published triplet–triplet annihilation (TTA) blue OLEDs45. Systems A and C are the first examples of blue narrowband MFHF OLEDs, which—to the best of our knowledge—display the highest EQEs and lowest CIExy coordinates yet reported for matrix-free systems36,42. For reference, an MFHF device based on the DMAC-DPS host and an archetypal bis(diphenylamino)pyrene fluorescent emitter affords a maximum EQE of merely 7% in sky blue (Supplementary Fig. 112)46,47. Additionally, even compared with contemporary MCHF and TTA devices, our 14–15 nm FWHM values are record equalling12,27. Supplementary Note 7 presents further detailed device characterization results.
An important observation is that for all the three MFHF systems, there is a negligible difference in the OLED EQEs between doped and non-doped devices. The same trend is additionally seen for a further encapsulated structure based on 9,10-diphenylanthracene (Supplementary Table 21). The lack of any substantial decrease in performance implies that the emitters introduce minimal loss pathways to MFHF OLEDs; for example, quenching via Dexter transfer to the terminal emitter may not be substantial, which would validate the encapsulated design. This prompted us to study the energy transfer dynamics of MFHF blends in further detail.
PL study
To determine the effect of an encapsulated acceptor structure on the energy transfer processes in a TADF host, non-encapsulated analogue NB-3 (Fig. 3a) was synthesized for comparison with NB-1 in photophysical studies on System A. The PL spectra for the concentration series of NB-1 and NB-3 evaporated into DMAC-DPS are shown in Fig. 3, obtained on excitation of the host at 330 nm. Aggregation can plague narrowband emitters, leading to broadened emission and quenched luminescence even in dilute evaporated films14,27,40,48,49,50,51. For NB-3, broad unstructured PL is recorded on increasing the doping concentration (Fig. 3c). In contrast, for NB-1, PL in DMAC-DPS progresses towards its narrow-structured solution spectrum (Fig. 3b), indicating that alkylene encapsulation effectively suppresses the aggregation of mDICz.

a, Molecular structures of NB-1 and NB-3. b,c, PL spectra of the concentration series of NB-1 (b) and NB-3 (c) in DMAC-DPS. d, PL decays for NB-1 and DMAC-DPS.
Next, ΦPL and time-resolved PL experiments were conducted. Data for the films of NB-1 and NB-3 doped into DMAC-DPS are presented in Table 2. In solution, the emitters display only prompt fluorescence with a lifetime (τ) of around 5.0 ns (Fig. 3d and Supplementary Fig. 63), in agreement with previous reports for mDICz (ref. 27), and expectedly shorter than the prompt fluorescence lifetime of pristine DMAC-DPS (13.8 ns). In contrast, when doped into DMAC-DPS, clear biphasic decay is observed, as singlet excitons from the TADF host are transferred to the terminal emitters on both prompt and delayed timescales.
Although the prompt component of ΦPL (ΦPrompt) for both terminal emitters is similar, the overall ΦPL is substantially smaller for NB-3 compared with NB-1 due to a large reduction in the delayed PL (ΦDelayed), as also evidenced by a shorter delayed PL lifetime (τDelayed). As triplets are only present in a PL experiment for TADF hosts on the delayed timescale after intersystem crossing, the selective decrease in delayed PL for NB-3 compared with NB-1 supports the hypothesis of a triplet-mediated pathway such as Dexter transfer, which is suppressed by the encapsulated structure of NB-1. Metrics describing FRET and Dexter transfer were next calculated from the time-resolved PL data (Supplementary Note 5). Crucially, in DMAC-DPS, the rate of Dexter transfer (kDET) for NB-1 is reduced by over a factor of three (3.2) compared with NB-3 at the same wt% doping, supporting the fact that encapsulation can suppress Dexter transfer. Parameters such as FRET rate (kFRET), Förster radius (R0) and average donor–acceptor distance (RAverage) also suggest that FRET is more efficient for NB-3 than NB-1, in line with the encapsulated structure of NB-1 increasing the average intermolecular distances.
TA study
Although supportive of the hypothesis that the encapsulated structure of NB-1 can suppress Dexter transfer to a terminal emitter from a TADF host, calculations based on time-resolved PL data invariably require assumptions stemming from the dark nature of organic triplet states. TA spectroscopy is not restricted to bright states, and therefore, we turned to this technique in the hope of directly observing Dexter transfer to further understand the energy transfer dynamics in DMAC-DPS-hosted films. Experiments were pumped at 355 or 400 nm. The TA spectra of the diluted emitters and a neat film of DMAC-DPS were first assigned (Fig. 4a and Supplementary Figs. 92–97). In a toluene solution, NB-1 and NB-3 display very similar spectra: photoinduced absorption (PIA) centred at around 480 nm, which decays with a lifetime of around 10 ns, is assigned to S1 → Sn, whereas a slower-rising PIA centred between 540 and 560 nm with a lifetime of tens of microseconds and clear oxygen quenching is assigned to T1 → Tn (Supplementary Figs. 92–97). A neat film of DMAC-DPS displays a broad PIA between 600 and 700 nm when probed on timescales between 0.2 ps and 100 μs (Fig. 4a and Supplementary Fig. 101). The kinetics roughly fit biphasic prompt-delayed behaviour, as previously reported for TADF materials52 (Fig. 4b and Supplementary Fig. 104). Fortunately, the PIA of DMAC-DPS has poor overlap with the T1 PIA of the emitters, displaying essentially no long-lived signal of ≤550 nm, which we envisaged should make it possible to directly observe Dexter transfer in doped films.

a, Long-time TA spectra for DMAC-DPS. b, Normalized decays in the PIA region of terminal emitter triplets for doped films and neat DMAC-DPS. c, Long-time TA spectra for DMAC-DPS:1 wt% NB-1. d, Long-time TA spectra for DMAC-DPS:1 wt% NB-3.
Next, 1 wt% NB-1 and NB-3 films in DMAC-DPS were investigated. Owing to the low concentration of the dopants, essentially an exclusive excitation of the DMAC-DPS host is expected. Short-time experiments (<2 ns) indicate a faster initial decay of the DMAC-DPS PIA for the NB-3-doped film compared with NB-1, in agreement with the larger kFRET calculated for NB-3 above (Supplementary Fig. 102). Microsecond-timescale experiments were next performed to probe delayed processes (Fig. 4c,d). Normalized decays of the 540–560 nm region for the doped films are shown in Fig. 4b, with neat DMAC-DPS for comparison. For the NB-3-doped film, the prompt-delayed biphasic kinetics of DMAC-DPS are substantially quenched when probed at either 540–560 nm or 600–700 nm (Fig. 4b and Supplementary Fig. 108), suggesting that delayed fluorescence, and perhaps T → S reverse intersystem crossing, are suppressed (in agreement with the relatively low ΦDelayed value of the NB-3-doped film). Meanwhile, the 540–560 nm PIA levels out within ~200 ns and persists onto the microsecond timescale at an intensity appreciably larger than that for neat DMAC-DPS, consistent with terminal emitter triplets. Although it is plausible to simply ascribe delayed fluorescence quenching to FRET20, this mechanism cannot explain the concomitant population of the terminal emitter triplet state, implying a different quenching pathway. Such a result is consistent with what would be expected for Dexter transfer, which—to the best of our knowledge—is being directly observed for the first time in a hyperfluorescent system.
After validating the potential of TA for directly observing Dexter transfer with NB-3-doped films, DMAC-DPS films doped with the encapsulated NB-1 emitter were investigated. For NB-1-doped films, the prompt-delayed biphasic kinetics of DMAC-DPS are more strongly retained than for NB-3 (Fig. 4b and Supplementary Fig. 106), in agreement with the higher ΦDelayed mentioned above. The more appreciable delayed fluorescence of NB-1-doped films is accompanied by a weaker long-lived 540–560 nm PIA compared with NB-3, suggesting a lower population of terminal emitter triplets. Hence, TA provides direct evidence that the encapsulated molecular structure of NB-1 can appreciably suppress loss via Dexter transfer to terminal emitter triplets in a pristine TADF host, supporting the minimal OLED EQE drop encountered for the MFHF OLED.
Outlook
Current blue hyperfluorescence technology requires OLEDs with complex EMLs of at least three components—a TADF sensitizer, narrowband emitter guest and wide-gap matrix (matrices). High triplet energy matrices have been considered essential to suppress Dexter transfer to the terminal emitter towards high device efficiency and stability.
Here we developed ultranarrowband blue emitters encapsulated by bulky alkylene straps. The ideal spectral properties of the emitters facilitates efficient FRET of singlets from pristine blue TADF hosts. Through comparison with a non-encapsulated analogue using TA spectroscopy, the population of terminal emitter triplets via a Dexter mechanism was directly observed in a hyperfluorescent system for the first time, enabling us to reveal that encapsulating straps can efficiently suppress the Dexter loss pathway to terminal emitter triplets.
Consequently, the new molecular design circumvents the need for a matrix, facilitating efficient blue MFHF OLEDs, with EMLs consisting of TADF sensitizer hosts and encapsulated terminal emitters only. Owing to the efficient suppression of Dexter transfer, MFHF OLEDs demonstrated negligible drops in EQE compared with non-doped reference devices, with a maximum EQE of 21.5%. Simultaneously, the EL FWHM of devices was decreased by a factor of six compared with the non-doped devices from >80 nm to ultranarrow values of 14–15 nm, with desirable deep blue peak wavelengths (449 and 458 nm). Non-doped deep blue TADF research is steadily progressing53,54,55, and with improved TADF hosts, we envisage that this encapsulated ultranarrowband emitter design should enable efficient MFHF OLEDs satisfying BT.2020, similar to the CIEy ≤ 0.05 that can be obtained in TTA-sensitized devices with the NB-1 emitter (Supplementary Table 16). More broadly, it has been suggested that the unsatisfactory stability of current blue MCHF OLEDs is directly linked to the population of terminal emitter triplets21, making the new strategy outlined here for suppressing Dexter transfer highly relevant.
Methods
Molecular synthesis
Supplementary Note 1 discusses the synthesis routes.
S1. This synthesis was inspired by procedures in the literature26,56. 5-Bromoindole (4.07 g, 20.7 mmol, 1.00 eq.), 2-bromo-4-tert-butylbenzaldehyde (5 g, 20.7 mmol, 1.00 eq.) and HI (57 wt% in water, 0.56 ml, 4.23 mmol, 0.20 eq.) were heated overnight to reflux under air in MeCN (150 ml). The reaction mixture was cooled in a −20 °C freezer for 6 h and the precipitate was isolated by filtration and washed with −20 °C MeCN (3 × 20 ml). The isolated solid was suspended in MeCN (150 ml) with iodine (1.95 g, 7.69 mmol, 0.37 eq.), and the resulting mixture heated overnight again to reflux under air. The reaction mixture was cooled in a −20 °C freezer for 6 h and the presumed indolocarbazole intermediate was isolated as a yellow-white powder by filtration after washing with −20 °C MeCN (3 × 20 ml) and drying under suction (3.74 g, 4.47 mmol, 43%). The presumed indolocarbazole intermediate was suspended in dry DMF (150 ml) with TBAOH (40 wt% in methanol, 6.50 g, 11.00 mmol, 2.46 eq.) under argon. The resulting mixture was degassed for 10 min. CuI (852 mg, 4.47 mmol, 1.00 eq.) was added and the mixture was immersed in a 120 °C preheated oil bath overnight. The reaction mixture was cooled to room temperature. The yellow precipitate was isolated via filtration, washed with MeCN (3 × 20 ml) and then suspended in benzonitrile (100 ml). The suspension was refluxed for 10 min and then cooled to room temperature. Filtration afforded S1 as a yellow powder sufficiently pure for the next step (870 mg, 1.29 mmol, 12% overall yield). This compound is too insoluble to obtain the nuclear magnetic resonance (NMR) spectra in common organic solvents. An analytical sample was obtained as fluffy yellow needles after recrystallization from benzonitrile (reflux → room temperature, hot filtered through paper under gravity). The ultraviolet spectra of the crude and recrystallized samples are essentially identical. Anal. Calcd. for C38H30Br2N2: C, 67.67; H, 4.48; N, 4.15. Calcd. for C38H30Br2N2·0.2PhCN: C, 68.08; H, 4.50; N, 4.43. Found: C, 68.09; H, 4.49; N, 4.39 (average of two runs); HRMS (ASAP): m/z 673.0832 [M–H+]. Calcd. for C38H31Br792N2+: 673.0854.
S2. 2-Bromoresorcinol (2.90 g, 15.3 mmol, 1.00 eq.), K2CO3 (12.67 g, 91.7 mmol, 6.00 eq.) and KI (762 mg, 4.59 mmol, 0.30 eq.) were combined in dry DMF (50 ml) under argon and the resulting mixture was heated in a 60 °C oil bath. After 1 h, 8-bromo-1-octene (7.32 g, 38.3 mmol, 2.50 eq.) was added and the heating was continued overnight. The reaction mixture was cooled to room temperature and diluted with hexane (100 ml) and water (300 ml). The layers were separated, and the aqueous layer was extracted with further hexane (3 × 50 ml). The extracts were combined, washed with 2 M NaOH (3 × 50 ml), dried over MgSO4, filtered and the solvent was removed under reduced pressure. The residue was purified by flash chromatography on silica gel (eluent:gradient, 0:1–1:9 EtOAc/n-hexane v/v) to afford S2 as a clear oil (5.84 g, 14.3 mmol, 93%). 1H NMR data were in accordance with those reported in the literature57. 1H NMR (500 MHz, CDCl3) δ (ppm) = 7.18 (t, J = 8.3 Hz, 1H), 6.55 (d, J = 8.3 Hz, 2H), 5.84 (ddt, J = 17.1, 10.2, 6.7 Hz, 2H), 5.03 (ddd, J = 17.1, 3.5, 1.7 Hz, 2H), 4.96 (ddt, J = 10.2, 2.2, 1.7 Hz, 2H), 4.04 (t, J = 6.5 Hz, 4H), 2.13–2.05 (m, 4H), 1.86 (dq, J = 13.0, 6.5 Hz, 4H), 1.58–1.50 (m, 5H), 1.47–1.39 (m, 8H).
S3. A solution of S2 (5.81 g, 14.2 mmol, 1.00 eq.) in dry tetrahydrofuran (THF) (80 ml) was cooled to −78 °C in a dry ice–acetone bath under argon. n-BuLi (2.5 M in hexane, 7.37 ml, 18.4 mmol, 1.30 eq.) was added dropwise and the resulting mixture was stirred for 1 h. B(OMe)3 (5.54 ml, 49.7 mmol, 3.50 eq.) was added dropwise, and the resulting mixture was warmed overnight to room temperature. The reaction mixture was quenched with 1 M HCl (30 ml) and stirred for 1 h. The layers were separated and the aqueous layer was extracted with dichloromethane (DCM) (3 × 50 ml). The extracts were combined, dried over MgSO4, filtered and the solvent was removed under reduced pressure. The residue was purified by flash chromatography on silica gel (eluent:gradient, 0:1–1:0 DCM/n-hexane v/v) to afford S3 as a clear oil that solidified on standing (4.52 g, 12.1 mmol, 85%). 1H NMR data were in accordance with those reported in the literature57. 1H NMR (500 MHz, CDCl3) δ (ppm) = 7.38–7.31 (m, 3H), 6.60 (d, J = 8.4 Hz, 2H), 5.80 (ddt, J = 17.1, 10.2, 6.7 Hz, 2H), 5.00 (ddd, J = 17.1, 3.6, 1.6 Hz, 2H), 4.94 (ddt, J = 10.2, 2.2, 1.2 Hz, 2H), 4.06 (t, J = 6.6 Hz, 4H), 2.06 (td, J = 6.8, 1.2 Hz, 4H), 1.85 (dq, J = 13.2, 6.6 Hz, 4H), 1.51–1.36 (m, 12H).
S4. S1 (1.00 g, 1.48 mmol, 1.00 eq.), S3 (3.33 g, 8.90 mmol, 6.00 eq.), Pd(OAc)2 (20 mg, 88.9 μmol, 6 mol%) and SPhos (73 mg, 178 μmol, 12 mol%) were combined in a 500 ml round-bottomed flask that was evacuated and backfilled with argon five times. Degassed toluene (250 ml, with five drops of Aliquat 336) and a degassed solution of K3PO4 (1.89 g, 8.90 mmol, 6.00 eq.) in water (32 ml) were sequentially added, and the resulting mixture was refluxed overnight in a preheated 130 °C oil bath. The reaction mixture was cooled to room temperature and acidified with 1 M HCl (100 ml). The layers were separated, and the aqueous layer was extracted with DCM (3 × 50 ml). The extracts were combined, dried over MgSO4, filtered and the solvent was removed under reduced pressure. The residue was purified by flash chromatography on silica gel (eluent:gradient, 0:1–35:65 DCM/n-hexane v/v) and then recrystallized (100 ml ethanol and minimal CHCl3, reflux → −20 °C) to afford S4 as yellow needles (950 mg, 0.81 mmol, 55%). 1H NMR (400 MHz, CDCl3) δ (ppm) = 8.59 (s, 2H), 8.42 (d, J = 8.3 Hz, 2H), 8.13–8.05 (m, 4H), 7.67 (d, J = 8.3 Hz, 2H), 7.49 (d, J = 8.2 Hz, 2H), 7.34 (t, J = 8.3 Hz, 2H), 6.77 (d, J = 8.3 Hz, 4H), 5.52–5.39 (m, 4H), 4.73–4.62 (m, 8H), 4.00 (t, J = 6.3 Hz, 8H), 1.75–1.67 (m, 8H), 1.66–1.58 (m, 8H), 1.55 (s, 18H), 1.33–1.24 (m, 8H), 1.13–0.98 (m, 16H); 13C NMR (101 MHz, CDCl3) δ (ppm) = 157.6, 150.1, 142.5, 139.2, 138.8, 137.6, 129.6, 128.9, 128.5, 127.5, 127.4, 126.9, 124.0, 120.9, 119.1, 113.8, 112.4, 112.0, 111.1, 109.1, 105.8, 68.9, 35.4, 33.5, 31.8, 29.2, 28.7, 28.6, 25.9; Anal. Calcd. for C82H96N2O4: C, 83.92; H, 8.24; N, 2.39. Found: C, 83.97; H, 8.14; N, 2.34 (average of two runs); HRMS (ASAP): m/z 1,173.7407 [M–H+]. Calcd. for C82H97N2O4+: 1,173.7448.
S5. S4 (800 mg, 0.68 mmol) and Grubbs second-generation catalyst (36 mg, 41 μmol, 6 mol%) were stirred overnight in degassed dry DCM (200 ml) under argon at reflux. The solvent was evaporated and the residue was purified by flash chromatography on silica gel (eluent:gradient, 0:1–35:65 DCM/n-hexane v/v, a single yellow band was collected) to afford S5 as a yellow powder sufficiently pure for the next step after trituration with methanol and drying under suction (753 mg, 0.66 mmol, 96%), yielding a mixture of E and Z isomers. 1H NMR (400 MHz, CDCl3) δ (ppm) = 8.60 (s, 2H), 8.43 (d, J = 8.2 Hz, 2H), 8.18–8.08 (m, 4H), 7.76–7.67 (m, 2H), 7.50 (d, J = 8.2 Hz, 2H), 7.37 (t, J = 8.3 Hz, 2H), 6.80 (d, J = 8.3 Hz, 4H), 4.19–4.02 (m, 8H), 4.00–3.91 (m, 4H), 1.62–1.53 (m, 8H), 1.32–1.08 (m, 16H), 0.96–0.83 (m, 8H), 0.62–0.42 (m, 8H); 13C NMR (176 MHz, CDCl3) δ (ppm) 157.8, 150.2, 142.6, 139.4, 137.6, 129.9, 129.1, 128.9, 128.7, 127.6, 127.5, 127.1, 124.4, 121.4, 119.1, 112.3, 112.0, 111.3, 109.2, 106.0, 69.2, 35.5, 32.2, 32.0, 29.8, 29.6, 29.3, 26.6; Anal. Calcd. for C78H88N2O4: C, 83.83; H, 7.94; N, 2.51. Found: C, 83.69; H, 7.94; N, 2.49 (average of two runs); HRMS (ASAP): m/z 1,117.6803 [M–H+]. Calcd. for C78H89N2O4+: 1,117.6822. Shoulder peaks in the 1H NMR spectrum are assigned to a mixture of E and Z isomers, considering that no shoulder peaks are present after alkene reduction in the next step. Shoulders are also present in the 13C NMR spectrum. Only peaks for the major compound are listed.
NB-1. A mixture of S5 (710 mg, 0.64 mmol) and Pd/C (10 wt% Pd, 150 mg) in THF (125 ml) was bubbled with H2 for 30 min at room temperature and then refluxed overnight under H2 (balloon). The warm reaction mixture was filtered through celite and the solvent was removed under reduced pressure. The residue was recrystallized (100 ml ethanol and minimal CHCl3, reflux → −20 °C) to obtain NB-1 as a yellow powder (610 mg, 0.54 mmol, 86%). 1H NMR (400 MHz, CDCl3) δ (ppm) = 8.61 (s, 2H), 8.47–8.37 (m, 2H), 8.15–8.01 (m, 4H), 7.70 (d, J = 8.2 Hz, 2H), 7.47 (d, J = 8.1 Hz, 2H), 7.34 (t, J = 8.3 Hz, 2H), 6.78 (t, J = 8.3 Hz, 4H), 4.09–3.92 (m, 8H), 1.56 (s, 18H), 1.35–1.18 (m, 8H), 0.94–0.80 (m, 8H), 0.56–0.32 (m, 16H), 0.21 to −0.02 (m, 8H); 13C NMR (126 MHz, CDCl3) δ (ppm) = 157.8, 150.1, 142.5, 139.3, 137.5, 130.0, 128.9, 128.6, 127.6, 127.5, 127.1, 124.5, 120.9, 119.0, 112.3, 112.0, 111.2, 109.3, 105.7, 69.2, 35.5, 32.0, 29.9, 29.8, 29.8, 29.6, 29.3, 26.9; Anal. Calcd. for C78H92N2O4: C, 83.53; H, 8.27; N, 2.50. Found: C, 83.39; H, 8.26; N, 2.50 (average of two runs); HRMS (ASAP): m/z 1,121.7126 [M–H+]. Calcd. for C78H93N2O4+: 1,121.7135.
S6. This synthesis was inspired by procedures in the literature26,56. 5-Bromoindole (4.07 g, 20.7 mmol, 1.00 eq.), 2-bromo-5-tert-butylbenzaldehyde (5 g, 20.7 mmol, 1.00 eq.) and HI (57 wt% in water, 0.56 ml, 4.23 mmol, 0.20 eq.) were heated overnight to reflux under air in MeCN (50 ml). The reaction mixture was cooled in a −20 °C freezer for 6 h and the presumed indolocarbazole intermediate was isolated as a yellow-white powder by filtration after washing with −20 °C MeCN (3 × 20 ml) and drying under suction (3.29 g, 3.93 mmol, 38%). Aromatization with I2 was not carried out during the synthesis of S6 as it caused the yield to drop by a factor of nearly five. The presumed indolocarbazole intermediate was suspended in dry DMF (100 ml) with TBAOH (40 wt% in methanol, 5.72 g, 9.68 mmol, 2.46 eq.) under argon. The resulting mixture was degassed for 10 min. CuI (750 mg, 3.93 mmol, 1.00 eq.) was added and the mixture was immersed overnight in a 120 °C preheated oil bath. The reaction mixture was cooled to room temperature. The yellow precipitate was isolated via filtration, washed with MeCN (3 × 20 ml) and then suspended in benzonitrile (50 ml). The suspension was refluxed for 10 min and then cooled to room temperature. Filtration afforded S6 as a yellow powder sufficiently pure for the next step (730 mg, 1.08 mmol, 10% overall yield). This compound is too insoluble to obtain the NMR spectra in common organic solvents. HRMS (ASAP): m/z 673.0826 [M–H+]. Calcd. for C38H31Br792N2+: 673.0854.
S7. 2-Bromoresorcinol (13.6 g, 71.6 mmol, 1.00 eq.), K2CO3 (21.8 g, 158 mmol, 2.20 eq.) and KI (1.36 mg, 8.16 mmol, 0.11 eq.) were combined in dry acetone (100 ml) under argon. Benzyl bromide (19.0 ml, 160 mmol, 2.23 eq.) was added and the resulting mixture was heated overnight to reflux. The reaction mixture was cooled to room temperature and filtered through celite eluting with additional acetone (500 ml). The solvent was removed under reduced pressure and the residue was recrystallized from hexane (700 ml, reflux → room temperature) to afford S7 as colourless needles (21.3 g, 57.9 mmol, 81%). 1H NMR data were in accordance with those reported in the literature58. 1H NMR (400 MHz, CDCl3) δ (ppm) = 7.49 (d, J = 7.4 Hz, 4H), 7.41–7.36 (m, 4H), 7.34–7.30 (m, 2H), 7.15 (t, J = 8.3 Hz, 1H), 6.61 (d, J = 8.3 Hz, 2H), 5.17 (s, 4H).
S8. A solution of S7 (9.44 g, 25.6 mmol, 1.00 eq.) in dry THF (200 ml) was cooled to −78 °C in a dry ice–acetone bath under argon. n-BuLi (2.5 M in hexane, 13.3 ml, 33.3 mmol, 1.30 eq.) was added dropwise and the resulting mixture was stirred for 1 h. The mixture set to a solid that could not be stirred. The mixture was transferred to an ice bath until full dissolution was observed, and then transferred back into a dry ice–acetone bath. B(OMe)3 (9.92 ml, 89.0 mmol, 3.48 eq.) was immediately added dropwise, and the resulting mixture was warmed overnight to room temperature. The reaction mixture was quenched with 1 M HCl (30 ml) and stirred for 1 h. The layers were separated and the aqueous layer was extracted with DCM (3 × 50 ml). The extracts were combined, dried over MgSO4, filtered and the solvent removed under reduced pressure. The residue was purified by flash chromatography on silica gel (eluent:gradient, 0:1–1:0 EtOAc/DCM v/v) to afford S8 as an off-white solid (6.84 g, 20.5 mmol, 80%). 1H NMR data were in accordance with those reported in the literature58. 1H NMR (400 MHz, CDCl3) δ (ppm) = 7.45–7.33 (m, 11H), 7.20 (s, 2H), 6.71 (d, J = 8.4 Hz, 2H), 5.15 (s, 4H).
S9. S6 (400 mg, 0.59 mmol, 1.00 eq.), S8 (1.00 g, 3.00 mmol, 5.08 eq.), Pd(OAc)2 (12 mg, 54 μmol, 9 mol%) and SPhos (44 mg, 107 μmol, 18 mol%) were combined in a 250 ml round-bottomed flask that was evacuated and backfilled with argon five times. Degassed toluene (100 ml with three drops of Aliquat 336) and a degassed solution of K3PO4 (637 mg, 3.00 mmol, 5.08 eq.) in water (13 ml) were sequentially added, and the resulting mixture was refluxed overnight in a preheated 130 °C oil bath. The reaction mixture was cooled to room temperature and acidified with 1 M HCl (100 ml). The layers were separated, and the aqueous layer was extracted with DCM (3 × 50 ml). The extracts were combined, dried over MgSO4, filtered and the solvent was removed under reduced pressure. The residue was purified by flash chromatography on silica gel (eluent:gradient, 0:1−3:1 DCM/n-hexane v/v) and then recrystallized (100 ml ethanol and minimal CHCl3, reflux → room temperature) to afford S9 as yellow crystals (360 mg, 0.33 mmol, 56%). 1H NMR (400 MHz, CDCl3) δ (ppm) = 8.82 (s, 2H), 8.53 (s, 2H), 8.11 (d, J = 8.2 Hz, 2H), 8.02 (d, J = 8.3 Hz, 2H), 7.83 (d, J = 8.3 Hz, 2H), 7.65 (d, J = 8.4 Hz, 2H), 7.34 (t, J = 6.8 Hz, 10H), 7.10–7.02 (m, 12H), 6.88 (d, J = 8.4 Hz, 4H), 5.17 (s, 4H), 1.40 (s, 18H); 13C NMR (126 MHz, CDCl3) δ (ppm) = 157.3, 144.6, 142.7, 137.8, 137.3, 136.9, 130.3, 129.2, 128.8, 128.7, 128.5, 127.8, 127.5, 127.1, 126.7, 124.2, 121.5, 121.2, 112.8, 112.7, 111.8, 111.4, 107.1, 70.9, 34.9, 31.9; Anal. Calcd. for C78H64N2O4: C, 85.68; H, 5.90; N, 2.56. Found: C, 85.55; H, 5.79; N, 2.95 (average of two runs); HRMS (ASAP): m/z 1,093.4896 [M–H+]. Calcd. for C78H65N2O4+: 1,093.4944.
S10. S9 (250 mg, 229 μmol), Pd/C (10 wt% Pd, 78 mg), THF (30 ml) and ethanol (5 ml) were sealed in a 50 ml microwave vial with a crimp septum cap. The resulting mixture was bubbled with H2 gas via a balloon for ~30 min. The balloon was removed and the vial was stirred overnight in a preheated 80 °C oil bath. The reaction mixture was cooled to room temperature, diluted with THF (50 ml) and filtered through a plug of silica gel (eluent, ~100 ml THF). The solvent was removed and the residue was triturated with hexane and filtered to obtain S10 as a yellow powder that was used as obtained in the next step after drying under a high vacuum (168 mg, 229 μmol, 100%). 1H NMR (400 MHz, THF) δ (ppm) = 8.81 (d, J = 1.1 Hz, 2H), 8.67 (d, J = 1.7 Hz, 2H), 8.23 (d, J = 8.4 Hz, 2H), 8.18 (d, J = 8.5 Hz, 2H), 7.95 (s, 4H), 7.81 (dd, J = 8.3, 1.5 Hz, 2H), 7.76 (dd, J = 8.5, 1.8 Hz, 2H), 6.99 (t, J = 8.1 Hz, 2H), 6.49 (d, J = 8.1 Hz, 4H); 13C NMR (176 MHz, THF) δ (ppm) = 158.4, 147.0, 144.6, 139.8, 139.3, 132.6, 131.5, 131.0, 130.4, 130.2, 129.8, 126.5, 123.2, 118.8, 114.8, 114.8, 114.0, 113.7, 36.9, 33.7; HRMS (ESI): m/z 733.3053 [M–H+]. Calcd. for C50H41N2O4+: 733.3066.
NB-2. A mixture of S10 (151 mg, 206 μmol, 1.00 eq.) and 1,14-dibromotetradecane (147 mg, 412 206 μmol, 2.00 eq.) was heated to 40 °C in dry DMF (50 ml) under argon until full dissolution was observed (~30 min). K2CO3 (142 mg, 1.03 mmol, 5.00 eq.) was added, and the mixture was stirred at 40 °C for 24 h and then at 80 °C for 48 h. After evaporation of the reaction solvent, the residue was purified by flash chromatography on silica gel (eluent:gradient, 0:1−1:1 DCM/n-hexane v/v, the product is the first fluorescent band to elute and drags, use a small column) to afford NB-2 as a yellow powder after trituration with methanol and drying under suction (162 mg, 144 μmol, 70%). 1H NMR (400 MHz, CDCl3) δ (ppm) = 8.66 (d, J = 1.2 Hz, 2H), 8.55 (d, J = 1.7 Hz, 2H), 8.07 (d, J = 8.3 Hz, 2H), 8.00 (d, J = 8.5 Hz, 2H), 7.75 (dd, J = 8.3, 1.5 Hz, 2H), 7.68 (dd, J = 8.5, 1.8 Hz, 2H), 7.32 (t, J = 8.3 Hz, 2H), 6.75 (d, J = 8.4 Hz, 4H), 4.08 (dt, J = 8.9, 4.5 Hz, 4H), 4.00–3.91 (m, 4H), 1.64–1.55 (m, 8H), 1.53 (s, 18H), 1.44–1.23 (m, 12H), 0.89 (dd, J = 13.6, 6.9 Hz, 8H), 0.48–0.37 (m, 12H), 0.28–0.18 (m, 4H), 0.06 (d, J = 13.0 Hz, 4H); 13C NMR (126 MHz, CDCl3) δ (ppm) = 157.6, 144.3, 142.3, 137.5, 136.9, 130.3, 129.5, 128.6, 128.5, 128.0, 127.2, 124.1, 121.5, 120.3, 112.7, 112.5, 111.8, 111.0, 105.3, 69.0, 35.0, 32.2, 30.1, 29.9, 29.8, 29.7, 29.3, 26.9; Anal. Calcd. for C78H92N2O4: C, 83.53; H, 8.27; N, 2.50. Found: C, 83.37; H, 8.30; N, 2.74; HRMS (ASAP): m/z 1,121.7108 [M–H+]. Calcd. for C78H93N2O4+: 1,121.7135.
NB-3. This synthesis was inspired by procedures in the literature26,56. Indole (1.94 g, 16.6 mmol, 1.00 eq.), 2-bromo-4-tert-butylbenzaldehyde (4.00 g, 16.6 mmol, 1.00 eq.) and HI (57 wt% in water, 0.45 ml, 3.40 mmol, 0.20 eq.) were heated overnight to reflux under air in MeCN (50 ml). The reaction mixture was cooled in a −20 °C freezer for 6 h and the precipitate was isolated by filtration and washed with −20 °C MeCN (3 × 20 ml). The isolated solid was suspended in MeCN (50 ml) with iodine (1.56 g, 6.15 mmol, 0.37 eq.) and the resulting mixture was heated overnight again to reflux under air. The reaction mixture was cooled in a −20 °C freezer for 6 h and the presumed indolocarbazole intermediate was isolated as an off-white powder by filtration after washing with −20 °C MeCN (3 × 20 ml) and drying under suction (890 mg, 1.31 mmol, 16%). The presumed indolocarbazole intermediate was suspended in dry DMF (20 ml) with TBAOH (40 wt% in methanol, 1.89 g, 3.20 mmol, 2.44 eq.) under argon. The resulting mixture was degassed for 10 min. CuI (280 mg, 1.47 mmol, 1.12 eq.) was added and the mixture immersed overnight in a 120 °C preheated oil bath. The reaction mixture was cooled to room temperature. The yellow precipitate was isolated via filtration, washed with MeCN (3 × 20 ml) and recrystallized (100 ml ethanol and minimal CHCl3, reflux → room temperature, hot filtered through paper under gravity) to afford NB-3 as fluffy yellow needles (133 mg, 0.26 mmol, 2% overall yield). 1H NMR (400 MHz, TCE) δ (ppm) = 8.28 (d, J = 7.7 Hz, 2H), 8.23 (d, J = 8.3 Hz, 2H), 7.94–7.88 (m, 4H), 7.64 (td, J = 7.7, 1.2 Hz, 2H), 7.57 (dd, J = 8.3, 1.7 Hz, 2H), 7.48 (td, J = 7.6, 1.0 Hz, 2H), 1.62 (s, 18H); 13C NMR (176 MHz, TCE) δ (ppm) = 150.3, 141.5, 138.6, 138.1, 128.6, 126.2, 126.1, 124.1, 123.8, 121.5, 119.4, 112.2, 111.3, 111.2, 109.1, 35.2, 31.7; Anal. Calcd. for C38H32N2: C, 88.34; H, 6.24; N, 5.42. Calcd. for C38H32N2·0.15CHCl3: C, 85.80; H, 6.02; N, 5.40. Found: C, 85.71; H, 6.06; N, 5.24 (average of two runs); HRMS (ASAP): m/z 517.2625 [M–H+]. Calcd. for C38H33N2+: 517.2624.
S11. In an argon glovebox, perylene (5.00 g, 19.4 mmol, 1.00 eq.), bis(pinacolato)diboron (21.87 g, 85.3 mmol, 4.39 eq.), [Ir(COD)OMe]2 (306 mg, 0.46 mmol, 2.4 mol%) and 4,4′-di-tert-butyl-2,2′-bipyridine (260 mg, 0.92 mmol, 4.7 mol%) were sealed in a 120 ml pressure tube with a Young’s tap having a side port (FengTecEx, product no. P260006). Outside the glovebox, anhydrous degassed THF (60 ml) was added and the tube was heated overnight in an 80 °C oil bath. Initially, everything dissolved on heating before a yellow precipitate slowly formed, which eventually prevented stirring and set to a yellow mass overnight. After the mixture was cooled to room temperature, it was carefully slurried into ~300 ml methanol (warning: H2 evolution). The precipitate was filtered and washed with further methanol (~300 ml) to afford S11 as a yellow solid sufficiently pure for the next step after drying under suction (14.5 g, 19.2 mmol, 99%). 1H NMR data were in accordance with those reported in the literature59. 1H NMR (500 MHz, CDCl3) δ (ppm) = 8.62 (s, 4H), 8.24 (s, 4H), 1.43 (s, 48H).
S12. S11 (2.00 g, 2.64 mmol, 1.00 eq.), S7 (9.75 g, 26.4 mmol, 10.00 eq.), Pd(OAc)2 (60 mg, 0.27 mmol, 10 mol%) and SPhos (217 mg, 0.53 mmol, 20 mol%) were combined in a 250 ml round-bottomed flask that was evacuated and backfilled with argon five times. Degassed toluene (100 ml with five drops Aliquat 336) and a degassed solution of K3PO4 (4.50 g, 21.1 mmol, 8.00 eq.) in water (20 ml) were sequentially added, and the resulting mixture was refluxed overnight in a preheated 130 °C oil bath. The reaction mixture was cooled to room temperature and acidified with 1 M HCl (100 ml). The layers were separated, and the aqueous layer was extracted with toluene (1 × 50 ml). The extracts were combined, dried over Na2SO4, filtered and the solvent was removed under reduced pressure. The residue was purified by flash chromatography on silica gel (eluent:gradient, 0:1−70:30 DCM/n-hexane v/v). The fractions predominantly consisting of the product (the main fluorescent blue band observed in the crude reaction mixture) by thin-layer chromatography were combined and recrystallized (200 ml ethanol and minimal CHCl3, reflux → room temperature) to afford S12 as shiny yellow flakes after drying under suction (1.43 g). The filtrate was combined with impure fractions from the first column and further purified similarly by flash chromatography (eluent:gradient, 0:1−70:30 DCM/n-hexane v/v) and recrystallization (200 ml ethanol and minimal CHCl3, reflux → room temperature) to afford a second crop of comparable purity (970 mg). Total yield (2.40 g, 1.71 mmol, 65%). 1H NMR (400 MHz, CDCl3) δ (ppm) = 8.37 (d, J = 1.5 Hz, 4H), 7.82 (d, J = 1.3 Hz, 4H), 7.26–7.21 (m, 20H), 7.05 (dd, J = 8.4, 6.9 Hz, 16H), 6.99–6.94 (m, 8H), 6.73 (d, J = 8.4 Hz, 8H), 5.02 (s, 16H); 13C NMR (101 MHz, CDCl3) δ (ppm) = 157.0, 137.3, 134.8, 132.1, 130.8, 130.0, 128.5, 128.3, 127.3, 127.3, 126.7, 123.7, 121.7, 106.9, 70.5; HRMS (ESI): m/z 1,405.5581 [M–H+]. Calcd. for C100H77O8+: 1,405.5618.
S13. S12 (2.27 g, 1.61 mmol, 1.00 eq.), Pd/C (5 wt% Pd, 2.17 g), THF (100 ml) and ethanol (20 ml) were sealed in an Ace Glass 500 ml pressure flask (product no. 8415-25). The resulting mixture was bubbled with H2 gas via a balloon for ~30 min. The balloon was removed and the flask was stirred overnight in a preheated 80 °C oil bath. The warm reaction mixture was filtered through celite eluting with additional THF. After evaporation of the solvent, analysis by 1H NMR indicated ~75% debenzylation. The residue was hydrogenated overnight again with Pd/C (5 wt% Pd, 700 mg) in THF (50 ml) and ethanol (10 ml), and worked up similarly. Analysis by 1H NMR indicated complete debenzylation. The residue was purified by flash chromatography on silica gel (eluent:gradient, 0:1−1:1 EtOAc/DCM v/v) to obtain S13 as a yellow powder after trituration with hexane and drying under suction (1.12 g, 1.60 mmol, 100%). 1H NMR (400 MHz, DMSO) δ (ppm) = 9.20 (s, 8H), 8.06 (d, J = 1.5 Hz, 4H), 7.65 (d, J = 1.3 Hz, 4H), 6.98 (t, J = 8.1 Hz, 4H), 6.46 (d, J = 8.2 Hz, 8H); 13C NMR (101 MHz, DMSO) δ (ppm) = 156.2, 134.8, 133.7, 130.0, 129.8, 128.7, 126.1, 123.7, 116.4, 107.3; HRMS (ESI): m/z 683.1729 [M–1]−. Calcd. for C44H27O8−: 683.1706.
En-Per. S13 (1.10 g, 1.61 mmol, 1.00 eq.) and 1,8-dibromooctane (1.19 ml, 6.44 mmol, 4.00 eq.) were dissolved in dry DMF (900 ml) under argon at 40 °C. After 30 min, K2CO3 (4.17 g, 32.2 mmol, 20.0 eq.) was added and the resulting mixture was stirred at 80 °C for a further 48 h. The mixture was cooled to room temperature, filtered and the solvent was removed under reduced pressure. The residue was purified by flash chromatography on silica gel (eluent:gradient, 0:1–1:0 DCM/n-hexane v/v). All of the material that eluted was combined, affording a mixture presumably constituting the material that had been predominantly alkylated as a yellow solid (430 mg, 24% mass yield). Analysis by NMR suggested that S14 constituted the majority of this material (≥60 mol% pure; the NMR spectrum is provided in the Supplementary Information). Then, 120 mg of the material was purified by preparative high-performance liquid chromatography (HPLC) (eluent: 1:1 DCM/n-hexane v/v) to afford En-Per as a yellow powder after trituration with methanol and drying under suction (74 mg, 0.07 mmol, 4%). Based on this, if all the DCM-mobile material was purified by preparative HPLC, a yield of ~14% is anticipated. 1H NMR (400 MHz, CDCl3) δ (ppm) = 8.11 (d, J = 1.4 Hz, 4H), 7.52 (d, J = 1.3 Hz, 4H), 7.25 (t, J = 8.3 Hz, 4H), 6.72–6.64 (m, 8H), 3.98–3.85 (m, 16H), 1.56–1.41 (m, 16H), 1.20 (q, J = 7.3 Hz, 8H), 1.13–0.88 (m, 24H); 13C NMR (101 MHz, CDCl3) δ (ppm) = 157.7, 157.7, 134.7, 132.6, 130.8, 129.4, 128.4, 127.1, 123.1, 122.3, 106.9, 106.4, 70.1, 69.4, 30.1, 29.7, 29.1, 29.0, 27.1, 26.5; HRMS (ESI): m/z 1,125.6226 [M–H+]. Calcd. for C76H85O8+: 1,125.6244.
X-ray crystallography
X-ray data were collected on a Bruker D8 QUEST diffractometer, equipped with an Incoatec IμS Cu microsource (λ = 1.5418 Å) and a PHOTON III detector operating in the shutterless mode. Crystals were mounted on a MiTeGen crystal mount using inert polyfluoroether oil and the analysis was carried out under an Oxford Cryosystems open-flow N2 Cryostream operating at 180(2) K. The control and processing software was Bruker APEX4 (v. 2022.1-1)60. The diffraction images were integrated using SAINT in APEX4, and a multiscan correction was applied using SADABS. The final unit-cell parameters were refined against all reflections. Structures were solved using SHELXT61 and refined using SHELX62. All of the crystal structures include CHCl3 solvent molecules, and the crystals were generally prone to solvent loss and degradation on removal from the mother liquour. In some cases, the SQUEEZE algorithm within PLATON was applied to complete the refinement63. Supplementary Table 1 provides summary details of the data collection and structure/refinement parameters.
Computational details
Computations were performed using (time-dependent) density functional theory as implemented in ORCA 5.0 (ref. 64). Ground- and excited-state structure optimizations, Hessians and vibronic coupling parameters were computed at the ωB97X-D3/def2-SVP level of theory65,66. Vertical excitations are reported at the LC-BLYP/def2-TZVP level using a range separation parameter of μ = 0.1 a.u. chosen to experimentally reproduce the observed singlet and triplet excitation energies67. Absorption and emission spectra were computed using a path integral approach68 as implemented in the ORCA ESD module, employing ωB97X-D3/def2-SVP Hessians along with LC-BLYP/def2-TZVP excitation energies. A linear vibronic coupling model69,70,71 was used for interpreting the spectral lineshapes. Here the Huang–Rhys factor for normal mode i was computed as
where κi is the vibronic coupling parameter and ωi is the wavenumber for mode i, both inserted in atomic units and computed with respect to the ground-state modes. These computations were carried out using the linear vibronic coupling functionality of SHARC70. Natural transition orbitals72 were computed using the TheoDORE program73. Molecular volumes were calculated via a marching tetrahedron model74.
Electrochemical measurements
Cyclic voltammetry was carried out using a PalmSens EmStat4S at a scan rate of 100 mV s−1. Solutions were prepared in dry, degassed THF with 0.1 M n-Bu4NPF6 as the supporting electrolyte. All the experiments were run under argon with a glassy carbon working electrode, Ag/AgCl as the wire quasi-reference and a Pt wire as the counter electrode. The potentials were internally referenced to the half-potential of an fcH/fcH+ redox couple.
Photophysical measurements
Solution absorption spectra were measured using a Shimadzu UV-1800 instrument. Molar extinction coefficients were determined from triplicate runs using a method intended to minimize weighing and dilution error. For each run, >3 mg of the compound was accurately weighed into a 25 ml volumetric flask to make a stock solution. Then, 100 µl of the stock solution was titrated five times into a 1-cm-path-length cuvette (2 ml starting blank solvent volume), measuring the absorption spectra after each addition. Fluorescence and time-correlated single-photon-counting experiments were carried out on Edinburgh Instruments FS5. Solution PL quantum yield measurements were carried out in an integrating sphere on Edinburgh Instruments FS5.
For organic films for photophysics, glass substrates (steady-state PL and absorption, PL quantum efficiency and transient PL) and quartz substrates (TA) were prepared, and they were cleaned with acetone and isopropyl alcohol with sonification for 10 min before loading them into an evaporator (Angstrom Engineering). Then, 50-nm- and 100-nm-thick films were formed on glass and quartz substrates, respectively. The thermal evaporating process was conducted in a vacuum chamber under <5 × 10−7 mbar. Steady-state PL spectra were measured by an Edinburgh Instruments fluorescence spectrometer (FLS980) with a monochromated xenon arc lamp at λEx = 330 nm under nitrogen flow. FLS980 with an integrating sphere under nitrogen flow was used to measure the PL quantum efficiency, and the films were excited by a 330 nm laser. Transient PL was recorded by using an Andor electrically gated intensified charge-coupled device (iCCD) with 330 nm laser excitation; the decay kinetics were obtained from the integration of the total spectrum at each time.
To determine the emitter triplet energies, the samples were prepared via drop casting onto a sapphire substrate from a mixture of the compound at 1 wt% to ZEONEX in toluene. The films were then dried in a vacuum oven at room temperature for 1 h to remove any trace solvent. The time-resolved PL spectra were recorded using nanosecond gated luminescence and lifetime measurement setup (from 400 ps to 1 s). The sample was loaded into a Janis Research VNF-100 cryostat, which was placed under a vacuum and kept at room temperature. The excitation pulses were provided by an Ekspla Nd:YAG laser at the third harmonic of 355 nm and the emission was collected after passing through a spectrograph on a Stanford Computer Optics iCCD camera to produce the time-resolved emission spectra.
For photostability measurements, the samples were prepared in degassed toluene and irradiated with a 400 nm upconverted pump laser. The PL intensity was monitored as a function of time with an iCCD camera.
TA spectroscopy was performed on a setup powered using a commercially available Ti:sapphire amplifier (Spectra Physics Solstice Ace). The amplifier operates at 1 kHz and generates 100 fs pulses centred at 800 nm with an output of 7 W. For the ultrafast TA measurements, a portion of the laser fundamental was frequency doubled using a 1-mm-thick β-barium borate crystal for sample excitation at 400 nm, whereas the third harmonic (355 nm) of an electronically triggered, Q-switched Nd:YVO4 laser (Innolas Picolo 25) provided the ~1 ns pump pulses for the nanosecond–microsecond (1 ns to 100 μs) TA measurements. The probe was provided by a broadband visible (525–775 nm) non-collinear optical parametric amplifier. The probe pulses are collected with a Si dual-line array detector (Hamamatsu S8381-1024Q), driven and read by a custom-built board from Entwicklungsbüro Stresing. For determining the triplet-excited-state absorption of the terminal emitters, the probe was instead generated by a LEUKOS Disco 1 ultraviolet low-timing-jitter supercontinuum laser (STM-1-UV), which was then electronically delayed relative to the femtosecond 400 nm excitation by an electronic delay generator (Stanford Research Systems DG645).
Organic light-emitting devices
For the fabrication of OLED devices, indium-tin-oxide-coated substrates (~15 Ω cm–2) were cleaned with acetone and isopropyl alcohol, and then O2 plasma treatment was applied to align the energy level with a hole-transporting layer. All the layers, including the organic layers and a LiF/aluminium cathode, were thermally deposited in a high vacuum (~10−7 torr). The performance of the OLED devices was measured by a Keithley 2635 source meter and a calibrated Si photodiode. The EL spectra were recorded by an Ocean Optics Flame spectrometer.
Data availability
The main data supporting the findings of this study are available within this Article and its Supplementary Information. Further data are available from the corresponding author on reasonable request. The X-ray crystallographic coordinates for structures reported in this study have been deposited at the Cambridge Crystallographic Data Centre (CCDC) under deposition numbers 2236336, 2236337, 2236338, 2236339 and 2282946.
References
Monkman, A. Why do we still need a stable long lifetime deep blue OLED emitter? ACS Appl. Mater. Interfaces 14, 20463–20467 (2022).
Chan, C. Y. et al. Stable pure-blue hyperfluorescence organic light-emitting diodes with high-efficiency and narrow emission. Nat. Photon. 15, 203–207 (2021).
Nakanotani, H. et al. High-efficiency organic light-emitting diodes with fluorescent emitters. Nat. Commun. 5, 4016 (2014).
Cheng, S. H. et al. Hyperfluorescence™: excel the performance, create the future. J. Soc. Inf. Display 30, 514–522 (2021).
Braveenth, R. et al. Achieving narrow FWHM and high EQE over 38% in blue OLEDs using rigid heteroatom-based deep blue TADF sensitized host. Adv. Funct. Mater. 31, 2105805 (2021).
Adachi, J., Endo, A., Kakizoe, H. & Oyamada, T. Hyperfluorescence™; a game changing technology of OLED display. Proc. Int. Disp. Work. 27, 399–401 (2021).
Jeon, S. O. et al. High-efficiency, long-lifetime deep-blue organic light-emitting diodes. Nat. Photon. 15, 208–215 (2021).
Rayappa Naveen, K. et al. Deep blue diboron embedded multi-resonance thermally activated delayed fluorescence emitters for narrowband organic light emitting diodes. Chem. Eng. J. 432, 134381 (2022).
Wei, J. et al. Indolo[3,2,1-jk]carbazole embedded multiple-resonance fluorophors for narrowband deep-blue electroluminescence with EQE≈34.7 % and CIEy≈0.085. Angew. Chem. Int. Ed. 60, 12269–12273 (2021).
Tanaka, H. et al. Hypsochromic shift of multiple-resonance-induced thermally activated delayed fluorescence by oxygen atom incorporation. Angew. Chem. Int. Ed. 60, 17910–17914 (2021).
Oda, S. et al. One-shot synthesis of expanded heterohelicene exhibiting narrowband thermally activated delayed fluorescence. J. Am. Chem. Soc. 144, 106–112 (2022).
Kondo, Y. et al. Narrowband deep-blue organic light-emitting diode featuring an organoboron-based emitter. Nat. Photon. 13, 678–682 (2019).
Lo, K.-W., Tong, G. S. M., Cheng, G., Low, K.-H. & Che, C.-M. Dinuclear PtII complexes with strong blue phosphorescence for operational stable organic light‐emitting diodes with EQE up to 23% at 1000 cd m‐2. Angew. Chem. Int. Ed. 61, e202115515 (2022).
Stavrou, K., Danos, A., Hama, T., Hatakeyama, T. & Monkman, A. Hot vibrational states in a high-performance multiple resonance emitter and the effect of excimer quenching on organic light-emitting diodes. ACS Appl. Mater. Interfaces 13, 8643–8655 (2021).
Huang, T., Wang, Q., Meng, G., Duan, L. & Zhang, D. Accelerating radiative decay in blue through‐space charge transfer emitters by minimizing the face‐to‐face donor‐acceptor distances. Angew. Chem. Int. Ed. 61, e202200059 (2022).
Cho, H. H., Romanov, A. S., Bochmann, M., Greenham, N. C. & Credgington, D. Matrix-free hyperfluorescent organic light-emitting diodes based on carbene–metal–amides. Adv. Opt. Mater. 9, 2001965 (2021).
Zhang, D. et al. High-efficiency fluorescent organic light-emitting devices using sensitizing hosts with a small singlet-triplet exchange energy. Adv. Mater. 26, 5050–5055 (2014).
Stavrou, K. et al. Key requirements for ultraefficient sensitization in hyperfluorescence organic light-emitting diodes. Nat. Photon. https://doi.org/10.1038/s41566-024-01395-1 (2024).
Gottardi, S., Barbry, M., Coehoorn, R. & Van Eersel, H. Efficiency loss processes in hyperfluorescent OLEDs: a kinetic Monte Carlo study. Appl. Phys. Lett. 114, 73301 (2019).
Haase, N. et al. Are the rates of Dexter transfer in TADF hyperfluorescence systems optically accessible? Mater. Horiz. 8, 1805–1815 (2021).
Lee, Y. T. et al. Tailor-made multi-resonance terminal emitters toward narrowband, high-efficiency, and stable hyperfluorescence organic light-emitting diodes. Adv. Opt. Mater. 10, 2200682 (2022).
Han, S. H., Jeong, J. H., Yoo, J. W. & Lee, J. Y. Ideal blue thermally activated delayed fluorescence emission assisted by a thermally activated delayed fluorescence assistant dopant through a fast reverse intersystem crossing mediated cascade energy transfer process. J. Mater. Chem. C 7, 3082–3089 (2019).
Madayanad Suresh, S., Hall, D., Beljonne, D., Olivier, Y. & Zysman-Colman, E. Multiresonant thermally activated delayed fluorescence emitters based on heteroatom-doped nanographenes: recent advances and prospects for organic light-emitting diodes. Adv. Funct. Mater. 30, 1908677 (2020).
Han, S. H. & Lee, J. Y. Spatial separation of sensitizer and fluorescent emitter for high quantum efficiency in hyperfluorescent organic light-emitting diodes. J. Mater. Chem. C 6, 1504–1508 (2018).
Jang, J. S., Han, S. H., Choi, H. W., Yook, K. S. & Lee, J. Y. Molecular design of sensitizer to suppress efficiency loss mechanism in hyper-fluorescent organic light-emitting diodes. Org. Electron. 59, 236–242 (2018).
Niebel, C. et al. Dibenzo[2,3:5,6]pyrrolizino[1,7-bc]indolo[1,2,3-lm]carbazole: a new electron donor. New J. Chem. 34, 1243–1246 (2010).
Liu, T. et al. Ultrapure blue organic light-emitting diodes exhibiting 13 nm full width at half-maximum. J. Mater. Chem. C 10, 7799–7802 (2022).
Zeng, X. et al. Nitrogen‐embedded multi‐resonance heteroaromatics with prolonged homogeneous hexatomic rings. Angew. Chem. 134, e202117181 (2022).
Zhang, Q. et al. Nearly 100% internal quantum efficiency in undoped electroluminescent devices employing pure organic emitters. Adv. Mater. 27, 2096–2100 (2015).
Congrave, D. G. et al. Suppressing aggregation induced quenching in anthracene based conjugated polymers. Polym. Chem. 12, 1830–1836 (2021).
Royakkers, J. et al. Molecular encapsulation of naphthalene diimide (NDI) based π-conjugated polymers: a tool for understanding photoluminescence. Angew. Chem. Int. Ed. 60, 25005–25012 (2021).
Royakkers, J. et al. Doubly encapsulated perylene diimides: effect of molecular encapsulation on photophysical properties. J. Org. Chem. 85, 207–214 (2020).
Royakkers, J. et al. Suppressing solid-state quenching in red-emitting conjugated polymers. Chem. Mater. 32, 10140–10145 (2020).
Leventis, A. et al. Highly luminescent encapsulated narrow bandgap polymers based on diketopyrrolopyrrole. J. Am. Chem. Soc. 140, 1622–1626 (2018).
Fujiwara, Y. et al. Double alkylene-strapped diphenylanthracene as a photostable and intense solid-state blue-emitting material. J. Org. Chem. 78, 2206–2212 (2013).
Zhang, D., Song, X., Cai, M. & Duan, L. Blocking energy-loss pathways for ideal fluorescent organic light-emitting diodes with thermally activated delayed fluorescent sensitizers. Adv. Mater. 30, 1705250 (2018).
Jakoby, M., Richards, B. S., Lemmer, U. & Howard, I. A. Investigations of singlet and triplet diffusion in thermally activated delayed-fluorescence emitters: implications for hyperfluorescence. Phys. Rev. B 100, 45303 (2019).
Xie, W. et al. Blocking the energy loss of Dexter energy transfer in hyperfluorescence OLEDs via one-step phenyl-fluorene substitution of TADF assistant host. Adv. Opt. Mater. 10, 2200665 (2022).
Rodriguez, L.-I., Lackner, A., Pflumm, C. & Mekic, A. Materials for organic electroluminescent devices. Taiwan Patent WO2020053150A1 (2020).
Zhang, Y. et al. Sterically wrapped multiple resonance fluorophors for suppression of concentration quenching and spectrum broadening. Angew. Chem. Int. Ed. 61, e202113206 (2022).
Lee, I. H., Song, W., Lee, J. Y. & Hwang, S. H. High efficiency blue fluorescent organic light-emitting diodes using a conventional blue fluorescent emitter. J. Mater. Chem. C 3, 8834–8838 (2015).
Song, W., Lee, I. & Lee, J. Y. Host engineering for high quantum efficiency blue and white fluorescent organic light-emitting diodes. Adv. Mater. 27, 4358–4363 (2015).
Song, W. & Yook, K. S. Hyperfluorescence-based full fluorescent white organic light-emitting diodes. J. Ind. Eng. Chem. 61, 445–448 (2018).
Cheng, Z. et al. Achieving efficient blue delayed electrofluorescence by shielding acceptors with carbazole units. ACS Appl. Mater. Interfaces 11, 28096–28105 (2019).
Tasaki, S. et al. Realization of ultra-high-efficient fluorescent blue OLED. J. Soc. Inf. Display 30, 441–451 (2022).
Lim, H., Woo, S. J., Ha, Y. H., Kim, Y. H. & Kim, J. J. Breaking the efficiency limit of deep-blue fluorescent OLEDs based on anthracene derivatives. Adv. Mater. 34, 2100161 (2022).
Ahn, Y. et al. The positional effect of arylamines on pyrene core in a blue fluorescent dopant significantly affecting the performance of organic light emitting diodes. Dyes Pigm. 205, 110505 (2022).
Qu, Y.-K. et al. Steric modulation of spiro structure for highly efficient multiple resonance emitters. Angew. Chem. Int. Ed. 61, e202201886 (2022).
Cheon, H. J., Shin, Y., Park, N., Lee, J. & Kim, Y. Boron‐based multi‐resonance TADF emitter with suppressed intermolecular interaction and isomer formation for efficient pure blue OLEDs. Small 18, 2107574 (2022).
Jiang, P. et al. Quenching-resistant multiresonance TADF emitter realizes 40% external quantum efficiency in narrowband electroluminescence at high doping level. Adv. Mater. 34, 2106954 (2022).
Meng, G. et al. Highly efficient and stable deep-blue OLEDs based on narrowband emitters featuring orthogonal spiro-configured indolo[3,2,1-de]acridine structure. Chem. Sci. 13, 5622–5630 (2022).
Cui, L. S. et al. Fast spin-flip enables efficient and stable organic electroluminescence from charge-transfer states. Nat. Photon. 14, 636–642 (2020).
Han, J. et al. Narrowband blue emission with insensitivity to the doping concentration from an oxygen-bridged triarylboron-based TADF emitter: nondoped OLEDs with a high external quantum efficiency up to 21.4%. Chem. Sci. 13, 3402–3408 (2022).
Xia, G. et al. A TADF emitter featuring linearly arranged spiro-donor and spiro-acceptor groups: efficient nondoped and doped deep-blue OLEDs with CIEy <0.1. Angew. Chem. Int. Ed. 60, 9598–9603 (2021).
Shi, Y. Z. et al. Recent progress in thermally activated delayed fluorescence emitters for nondoped organic light-emitting diodes. Chem. Sci. 13, 3625–3651 (2022).
Gu, R., Van Snick, S., Robeyns, K., Van Meervelt, L. & Dehaen, W. A facile and general method for the synthesis of 6,12-diaryl-5,11-dihydroindolo[3,2-b]carbazoles. Org. Biomol. Chem. 7, 380–385 (2009).
Stoltenberg, D., Lüthje, S., Winkelmann, O., Näther, C. & Lüning, U. Tetraols as templates for the synthesis of large endo-functionalized macrocycles. Eur. J. Org. Chem. 4, 5845–5859 (2011).
Reck, L. M., Haberhauer, G. & Lüning, U. Enantiopure chiral concave 1,10-phenanthrolines. Eur. J. Org. Chem. 2016, 1119–1131 (2016)
Wisser, F. M. et al. Molecular porous photosystems tailored for long-term photocatalytic CO2 reduction. Angew. Chem. Int. Ed. 59, 5116–5122 (2020).
APEX4, SAINT and SADABS (Bruker AXS Inc., 2022).
Sheldrick, G. M. SHELXT—integrated space-group and crystal-structure determination. Acta Cryst. A71, 3–8 (2015).
Sheldrick, G. M. Crystal structure refinement with SHELXL. Acta Cryst. C71, 3–8 (2015).
Spek, A. L. PLATON SQUEEZE: a tool for the calculation of the disordered solvent contribution to the calculated structure factors. Acta Cryst. C71, 9–18 (2015).
Neese, F. Software update: the ORCA program system—version 5.0. WIREs Comput. Mol. Sci. 12, e1606 (2022).
Weigend, F. & Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: design and assessment of accuracy. Phys. Chem. Chem. Phys. 7, 3297–3305 (2005).
Chai, J. D. A. & Head-Gordon, M. Systematic optimization of long-range corrected hybrid density functionals. J. Chem. Phys. 128, 84106 (2008).
Tawada, Y., Tsuneda, T., Yanagisawa, S., Yanai, T. & Hirao, K. A long-range-corrected time-dependent density functional theory. J. Chem. Phys. 120, 8425–8433 (2004).
De Souza, B., Neese, F. & Izsák, R. On the theoretical prediction of fluorescence rates from first principles using the path integral approach. J. Chem. Phys. 148, 34104 (2018).
Köuppel, H., Domcke, W. & Cederbaum, L. S. Multimode molecular dynamics beyond the Born-Oppenheimer approximation. Adv. Chem. Phys. 57, 59–246 (1984).
Mai, S., Marquetand, P. & González, L. Nonadiabatic dynamics: the SHARC approach. WIREs Comput. Mol. Sci. 8, e1370 (2018).
Plasser, F., Gómez, S., Menger, M. F. S. J., Mai, S. & González, L. Highly efficient surface hopping dynamics using a linear vibronic coupling model. Phys. Chem. Chem. Phys. 21, 57–69 (2019).
Martin, R. L. Natural transition orbitals. J. Chem. Phys. 118, 4775–4777 (2003).
Plasser, F. TheoDORE: a toolbox for a detailed and automated analysis of electronic excited state computations. J. Chem. Phys. 152, 84108 (2020).
Lu, T. & Chen, F. Quantitative analysis of molecular surface based on improved Marching Tetrahedra algorithm. J. Mol. Graph. Model. 38, 314–323 (2012).
Acknowledgements
P. L. D. Santos and D. Credgington are acknowledged for their contribution to important discussion at the beginning of this project. A. P. Monkman is acknowledged for the use of facilities at Durham University for the time-resolved PL measurements. H.-H.C. acknowledges George and Lilian Schiff Foundation for PhD studentship funding. D.G.C. acknowledges the Herchel Smith fund for an early career fellowship. A.J.G. thanks the Leverhulme Trust for an Early Career Fellowship (ECF-2022-445). V.R.-G. acknowledges the Faraday Institution Degradation Project (grant no. FIRG001 and FIRG024). J.Y. acknowledges support from a UK Research and Innovation (UKRI) Frontier Grant (no. EP/X029900/1), awarded via the European Research Council Starting Grant 2021 scheme. This work was supported by the Engineering and Physical Sciences Research Council (EPSRC, grant nos. EP/M005143/1 and EP/S003126/1). H.-H.C. and R.H.F. acknowledge the European Research Council for European Union’s Horizon 2020 research and innovation programme grant agreement no. 101020167.
Author information
Authors and Affiliations
Contributions
H.-H.C. and D.G.C. contributed equally and are considered joint first authors, ordering is alphabetical. All authors contributed to the data analysis, discussion and revision of the manuscript. H.-H.C. fabricated and measured the OLEDs and performed the majority of the solid-state photophysics. D.G.C. and H.-H.C. wrote the manuscript. D.G.C. coordinated the project; designed and synthesized the molecules; and contributed to the solution photophysics, electrochemistry and density functional theory calculations. A.J.G. performed the TA spectroscopy. S.M. contributed to the synthesis and solution photophysics. H.E.F. and V.R.-G. performed the thermal analysis. J.Y. performed the single-crystal PL experiments. R.C. performed the photostability experiments. W.Z. performed and advised in the density functional theory calculations. M.K.E. performed the triplet energy experiments. J.R. advised in the molecular design. O.M. contributed to the data analysis. A.D.B. measured and solved the single-crystal X-ray structures. F.P. performed the majority of the computational chemistry. J.M.F. contributed to the computational chemistry. C.P.G. supervised H.E.F. and V.R.-G. A.R. supervised J.Y. R.H.F. supervised H.-H.C. and R.C. N.C.G. supervised H.-H.C. H.B. supervised D.G.C., H.E.F., W.Z. and O.M. D.G.C. and H.B. conceived the research.
Corresponding authors
Ethics declarations
Competing interests
All authors declare no competing interests.
Peer review
Peer review information
Nature Materials thanks Chihaya Adachi, Zuo-Quan Jiang and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Synthesis of NB-1.
Scheme for the synthesis of NB-1 via sequential Suzuki cross-coupling, Grubbs metathesis and catalytic hydrogenation chemistry.
Extended Data Fig. 2 Synthesis of NB-2.
Scheme for the synthesis of NB-2 via sequential Suzuki cross-coupling, de-benzylation and nucleophilic substitution chemistry.
Extended Data Fig. 3 Synthesis of NB-3.
Scheme for the synthesis of NB-2 via acid-catalysed condensation and Cu-catalysed intramolecular Ullmann chemistry.
Extended Data Fig. 4 Synthesis of En-Per.
Scheme for the synthesis of En-Per via chemistry analogous to that employed in the synthesis of NB-2.complete.
Supplementary information
Supplementary Information
Supplementary Figs. 1–138, Notes 1–7, Tables 1–22 and Discussion.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Cho, HH., Congrave, D.G., Gillett, A.J. et al. Suppression of Dexter transfer by covalent encapsulation for efficient matrix-free narrowband deep blue hyperfluorescent OLEDs. Nat. Mater. 23, 519–526 (2024). https://doi.org/10.1038/s41563-024-01812-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41563-024-01812-4
- Springer Nature Limited
This article is cited by
-
Efficient blue emitter with a hoop
Nature Materials (2024)