

Abstract
Some gene polymorphisms can lead to monogenic diseases, whereas other polymorphisms may confer beneficial traits. A well-characterized example is congenital erythrocytosis—the non-pathogenic hyper-production of red blood cells—that is caused by a truncated erythropoietin receptor. Here we show that Cas9-mediated genome editing in CD34+ human haematopoietic stem and progenitor cells (HSPCs) can recreate the truncated form of the erythropoietin receptor, leading to substantial increases in erythropoietic output. We also show that combining the expression of the cDNA of a truncated erythropoietin receptor with a previously reported genome-editing strategy to fully replace the HBA1 gene with an HBB transgene in HSPCs (to restore normal haemoglobin production in cells with a β-thalassaemia phenotype) gives the edited HSPCs and the healthy red blood cell phenotype a proliferative advantage. Combining knowledge of human genetics with precise genome editing to insert natural human variants into therapeutic cells may facilitate safer and more effective genome-editing therapies for patients with genetic diseases.
Similar content being viewed by others

Main
Many of the initial applications of clinical genome editing have aimed to correct or compensate for disease-causing mutations of monogenic diseases. However, human genetic variation is more nuanced than monogenic diseases as there are also variants that appear to confer positive health benefits. For example, people with bi-allelic deletions in CCR5 show resistance to HIV infection, a variety of polymorphisms cause upregulation of foetal haemoglobin and mutations in PCSK9 cause low cholesterol levels1,2,3. It is important, however, to broadly assess variations that occur in only small numbers of people both for their potential risks and for their potential benefits.
Congenital erythrocytosis (CE) is a rare phenotype in which people have higher than normal levels of red blood cells (RBCs) and consequently elevated haemoglobin. Although there are multiple genetic variants that can lead to this condition, perhaps the best-characterized genotype was first identified in the family of a Finnish Olympic-gold-medal-winning cross-country skier who was found to have levels of haemoglobin >50% higher than normal4. This elevated haemoglobin was attributed to truncations in the erythropoietin receptor (tEPOR) in which the intracellular inhibitory domain to erythropoietin (EPO) signalling is eliminated4,5. This domain contains binding sites for SHP1 that normally leads to downregulation of EPO-dependent JAK2–STAT5 signalling (Supplementary Fig. 1a). Further studies have shown that tEPOR does not create a constitutively active EPOR signalling cascade but rather imparts hypersensitivity to EPO5,6. As a consequence, these kindreds with tEPOR typically present with abnormally low levels of EPO, indicating a new homoeostasis is attained to prevent CE from becoming pathogenic. There have been reports of thrombotic and haemorrhagic events likely due to erythrocytosis but many of these events have a benign clinical course7. More importantly, families with CE have not shown an increased predisposition to cancer, showing that this is not a premalignant genetic condition8.
Although previous studies have investigated the effects of viral-mediated delivery and expression of tEPOR9,10,11, random insertion into the genomes of billions of haematopoietic stem and progenitor cells (HSPCs) in the context of bone marrow (BM) transplant presents a serious safety concern and has resulted in a ‘black box’ warning in the United States for lovotibeglogene autotemcel, a lentiviral gene therapy drug approved for sickle cell disease (SCD)12. In addition, such instances of viral-mediated delivery require expression of tEPOR using a non-native exogenous promoter, which departs from native EPOR regulation and has the potential for unintended consequences, such as pathogenic polycythaemia. Nonetheless, viral-mediated expression studies provide a proof of concept that shows that tEPOR expression can lend a selective advantage to transduced cells and provide a foundation for the utilization of more advanced genome-engineering modalities.
Genome editing is a powerful method that enables the precise changing of nucleotides in the DNA of a cell. There are multiple genome-editing strategies, including nuclease-based insertion–deletion (indel) formation, base editing and prime editing, but the most versatile approach to genome editing is homology-directed repair (HDR). In HDR, a nuclease-induced double-strand break (DSB) is repaired using a donor template. The natural donor template for HDR is the sister chromatid and the natural repair pathway is homologous recombination. By providing a donor template that resembles a sister chromatid with large homology arms flanking the intended cut site, the homologous recombination machinery can use this ‘substitute’ sister chromatid to repair the DSB. HDR editing is the most flexible approach because it can create single nucleotide changes, precisely insert large gene cassettes and even swap out large genomic regions for other gene sequences13,14,15,16. We use all these applications of HDR in this work, including direct creation of the naturally occurring variant found in a human kindred.
One of the challenges in haematopoietic stem cell gene therapy is to achieve sufficient engraftment of the genetically engineered cells to have a beneficial clinical effect without increasing risk. To make this possible, effort is exerted to maximize editing frequencies in HSPCs17,18,19. Even if clinically relevant editing frequencies are achieved, high-morbidity chemotherapeutic regimens are currently required to create niche space in the BM for these edited HSPCs, which can create toxicities, including oncogenic risk20,21,22. In this work we aimed to give edited cells a selective advantage such that low levels of engraftment might still result in a clinical benefit, perhaps enabling less toxic conditioning, through the use of genome editing to recreate the CE phenotype by engineering tEPOR into HSPCs in different ways. We find that when tEPOR is engineered into human HSPCs using genome editing, there is a substantial selective advantage to the derived RBCs. We then show that this selective advantage can be coupled to a therapeutic gene edit to give the cells with the therapeutic edit a selective advantage in RBC development, without affecting the stem and progenitor cells. In this way, we show the power of combining human genetics with precision genome editing to potentially enable safer and more effective genome-editing therapies for patients with serious genetic diseases, particularly those involving RBCs.
Results
Cas9-guided EPOR truncation enhances erythroid proliferation
Truncating mutations in the EPOR gene that cause clinically benign CE4,8 provide a potentially safe avenue to increase erythropoietic output from genome-edited HSPCs. In this study we designed Cas9 single-guide RNAs (sgRNAs)17 (termed EPOR-sg1 and EPOR-sg2) that overlap the location of the originally identified nonsense mutation, EPOR c.1316G>A (p.W439X; Mäntyranta variant)4 (Fig. 1a and Supplementary Fig. 2a). Our hypothesis was that targeting this site in exon 8 with Cas9 would create a spectrum of indels, a subset of which would result in a frameshift of the reading frame and yield premature downstream stop codons in the EPOR gene.

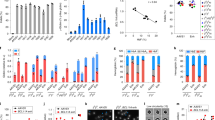
a, Schematic of HSPC editing and model of tEPOR’s effect. Representation of EPOR gene and location of the candidate sgRNA (EPOR-sg1) indicated by a line. Location of c.1316G>A mutation is denoted by the star. Created with BioRender.com. b, Frequency of indels created by EPOR-sg1 in primary human CD34+ HSPCs over the course of erythroid differentiation compared with control HBB sgRNA. Points represent median ± interquartile range. Values represent biologically independent HSPC donors: n = 5 for EPOR-sg1 and n = 1 for control HBB sgRNA. *P = 0.0016 of day 0 versus day 14 by unpaired two-tailed t-test. c, Genome-editing strategy when using an AAV6 DNA repair template to introduce the EPOR c.1316G>A mutation followed by a BGH-poly(A) region and UbC-driven GFP reporter. d, Percentage of GPA+/CD71+ of live single cells on day 14 of differentiation. Bars represent median ± interquartile range. Values represent biologically independent HSPC donors: n = 2–3 for HSPC and n = 3–4 for −EPO and +EPO conditions. *P = 0.0016 of −EPO versus HSPC conditions; **P = 0.003, ***P = 0.0001 of −EPO versus +EPO conditions by unpaired two-tailed t-test. e, Percentage of GFP+ cells of live single cells maintained in RBC media with or without EPO or HSPC media as determined by flow cytometry. Points represent median ± interquartile range. Values represent biologically independent HSPC donors: n = 2 for HSPC condition and n = 3–4 for −EPO and +EPO conditions. *P = 0.04, **P = 0.0006 of day 0 versus day 14 by unpaired two-tailed t-test. f, Fold change in cell count throughout RBC differentiation (for example, if at day 0 starting cell numbers were 1 × 105 cells total, then a fold count change of 1,000 would yield a total cell number of 1 × 108 at day 14). Points represent mean ± s.e.m. Values represent biologically independent HSPC donors: n = 3 for mock and EPOR-sg1 + BGH and n = 2 for EPOR-sg1.
To test this hypothesis, we precomplexed each sgRNA with high-fidelity Cas9 protein23 and delivered these ribonucleoprotein (RNP) complexes to human CD34+ HSPCs. At 2–3 days postediting, we transferred cells into culture medium that promotes erythroid differentiation over the course of 2 weeks (Fig. 1a)24. To determine whether edited HSPCs have a proliferative advantage compared with unedited cells, we collected genomic DNA at day 0, 4, 7, 11 and 14 of RBC differentiation. We then quantified indel frequency by polymerase chain reaction (PCR) amplification followed by Sanger sequencing and decomposition analysis using TIDE25. In the absence of a selective advantage or disadvantage, the percentage of edited alleles in cells at the beginning and end of RBC differentiation will be roughly equivalent—which is what we observe for the editing frequency of the HBB sgRNA used for correction of SCD. However, we observe that the editing frequency of EPOR-targeting sgRNAs increases significantly over the course of erythroid differentiation, to a greater extent in EPOR-sg1 than in EPOR-sg2 (P = 0.0016 for EPOR-sg1 from day 0 to day 14; Fig. 1b and Supplementary Fig. 2b). In addition, we show that the increase in indels for both EPOR-sg1 and EPOR-sg2 is predominantly driven by indels that yield downstream stop codons (Extended Data Fig. 1a,b and Supplementary Fig. 2c,d). This indicates that edited cells, particularly those with premature stop codons, are outcompeting unedited cells because of the EPO hypersensitivity of tEPOR-expressing cells in culture5,6.
As not all indels created by the sgRNAs cause truncations in EPOR, we speculated that we could increase this proliferative effect by using HDR to insert a stop codon at the exact location of the original variant (c.1316G>A). To accomplish this, we designed an adeno-associated virus serotype 6 (AAV6) repair template vector that introduces a stop codon into EPOR at the 439th amino acid (W439X) followed by a BGH-poly(A) tail to terminate transcription. We also included a downstream GFP marker driven by the constitutive human UbC promoter to ensure that each GFP+ allele harbours the intended EPOR-truncating mutation. The entire integration cassette was flanked by 950 bp homology arms that corresponded to the genomic DNA immediately upstream and downstream of the intended Cas9 cut site created by the more effective EPOR-sg1 (Fig. 1c). To determine whether this editing strategy was also able to drive enrichment of genome-edited RBCs, we complexed EPOR-sg1 with Cas9 protein and delivered this by electroporation to human CD34+ HSPCs followed by transduction with an AAV6 DNA repair template. At 2–3 days postediting, we either maintained cells in HSPC media or began erythroid differentiation with 3 U ml−1 of EPO (+EPO), as has been previously described14, or with 0 U ml−1 of EPO (−EPO) to determine whether tEPOR-expressing cells retain EPO sensitivity or became EPO independent during their differentiation. At day 14 of erythroid differentiation, we stained for established RBC markers14 and analysed cells using flow cytometry. We observed no differentiation when cells were kept in HSPC media and efficient RBC differentiation in all treatments with EPO. In the edited conditions in the absence of EPO, we observed moderate differentiation that may indicate the hypersensitivity of tEPOR-expressing cells to trace amounts of EPO in the media, as has been previously observed (Fig. 1d)4. By analysing GFP+ cells over the course of RBC differentiation, in the EPORW439X-edited conditions we observed a significant increase in the frequency of edited cells in only the +EPO conditions (P = 0.0006 comparing day 0 to day 14; Fig. 1e). At the end of differentiation, by looking at both GFP+ cells and frequency of indel formation, we estimated that almost all the RBCs are derived from cells with a truncated EPOR due to either an indel or a UbC-GFP knock-in event (Extended Data Fig. 2a). In addition to the competitive advantage that tEPOR expression gives to edited cells over the course of RBC differentiation, we also observed increased RBC production in both EPOR-sg1 and EPOR-sg1 + UbC-GFP-BGH conditions compared with mock control in the +EPO condition (average 1.45 × 103 total fold increase in mock-edited cells versus 4.71 × 103 in EPOR-sg1 + UbC-GFP-BGH over the 14-day RBC differentiation; Fig. 1f). These increased cell counts were not observed in the −EPO condition or when cells were maintained in HSPC media, indicating an EPO-driven increase in erythroid proliferation in cells expressing tEPOR (Extended Data Fig. 2b). We also assessed whether a gradient of EPO concentrations (0–20 U ml−1) over the course of RBC differentiation yielded varying degrees of enrichment of edited cells (Extended Data Fig. 3a). On day 14, when compared with −EPO, we found minimal differences in GFP+ cells in the 1 U ml−1, 3 U ml−1 and 20 U ml−1 conditions and only a minor reduction in GFP+ cells in the 0.3 U ml−1 EPO condition (Extended Data Fig. 3b,c), indicating that even low levels of EPO are sufficient to impart a selective advantage to edited cells, consistent with previous work on the natural variants5,6. We also observed comparable levels of RBC differentiation at all concentrations except −EPO (Extended Data Fig. 3d).
In terms of the safety of this editing strategy, we show that introduction of these EPOR indels yields RBCs with production of both foetal and adult haemoglobin tetramers following haemoglobin tetramer high-performance liquid chromatography (HPLC) (Supplementary Fig. 3a). We also found similar colony number and lineage distribution from CD34+ HSPCs plated into wells containing methylcellulose media either with or without EPO that were scored for colony-formation ability after 14 days (Supplementary Fig. 3b). As expected, there was a marked decrease in the ability to form erythroid burst-forming unit (BFU-E) colonies in the absence of EPO even if the cells contained the tEPOR. These data reinforce the idea that truncation of EPOR does not alter the lineage bias of HSPCs but rather has an effect only after commitment to the erythroid lineage. In addition, although transient delivery of high-fidelity Cas9 has been shown to be highly specific to the on-target site26, we also evaluated potential off-target effects of the EPOR-sg1–RNP complex in HSPCs. We found that 94% (73 of 78) of candidate off-target sites with scores previously shown to be most informative for identifying sites with potential off-target activity27 resided in intergenic or intronic regions of the genome (Extended Data Fig. 4a,b). We further interrogated potential off-target activity at the five sites that resided in exonic or untranslated regions (UTRs) of genes (Extended Data Fig. 4c) and found no evidence of off-target activity in EPOR-sg1-edited cells when compared with mock-edited cells (Extended Data Fig. 4d,e).
tEPOR at a safe-harbour locus replicates proliferative effect
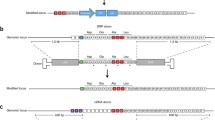
Given the therapeutic utility of transgene integration at safe-harbour sites28, we hypothesized that integration of a tEPOR cDNA at a safe-harbour site may also enable increased erythroid production from edited HSPCs while leaving the endogenous EPOR locus intact. Given the fact that integration at the CCR5 locus is an established method for delivery of therapeutic transgenes in HSPCs29, we developed a custom AAV6-packaged DNA repair template that would facilitate integration of an exogenous human UbC promoter driving expression of tEPOR cDNA followed by a T2A-YFP-BGH reporter (Fig. 2a). Given the strong, constitutive expression of the UbC promoter, this method of insertion is expected to express tEPOR ubiquitously in all haematopoietic cell types, regardless of lineage.

a, Genome-editing strategy to introduce tEPOR-T2A-YFP-BGH-poly(A) cDNA at the CCR5 locus with expression driven by a ubiquitous UbC promoter. b, Percentage of GPA+/CD71+ of live single cells on day 14 of differentiation following introduction of tEPOR at the CCR5 locus. Bars represent median ± interquartile range. Values represent biologically independent HSPC donors: n = 2–3 for HSPC condition and n = 2–4 for −EPO and +EPO conditions. *P = 0.0018 for −EPO versus HSPC conditions, **P < 0.0001 for −EPO versus +EPO conditions by unpaired two-tailed t-test. c, Representative flow cytometry plots of one donor of CCR5-sg3 + tEPOR-edited HSPCs on day 14 of RBC differentiation in the +EPO condition. d, Percentage of YFP+ cells of live single cells as determined by flow cytometry. Points represent mean ± s.e.m. Values represent biologically independent HSPC donors: n = 2 for HSPC condition, n = 3 for −EPO condition and n = 3–4 for +EPO condition. *P = 0.0003 for day 0 versus day 14 by unpaired two-tailed t-test. e, Genome-editing strategy to introduce tEPOR-T2A-YFP cDNA at the HBA1 locus by whole gene replacement to place integration cassette under regulation of the endogenous HBA1 promoter. f, Percentage of GPA+/CD71+ of live single cells on day 14 of differentiation following introduction of tEPOR cDNA at the HBA1 locus. Bars represent median ± 95% confidence interval. Values represent biologically independent HSPC donors: n = 2 for HSPC condition and n = 2–3 for −EPO and +EPO condition. *P = 0.0002 for −EPO to +EPO condition by unpaired two-tailed t-test. g, Representative flow cytometry plots of one donor of HBA1-sg4 + tEPOR-edited HSPCs on day 11 of RBC differentiation in the +EPO condition. h, Percentage of YFP+ cells of live single cells as determined by flow cytometry. Points represent mean ± s.e.m. Values represent biologically independent HSPC donors: n = 2 for HSPC condition and n = 3 for −EPO and +EPO condition. FSC-A, forward scatter area; FITC, fluorescein isothiocyanate.
To test this strategy for tEPOR expression, we edited HSPCs with Cas9 complexed with an established sgRNA targeting exon 2 of CCR5 (CCR5-sg3)29, immediately followed by transduction with our custom DNA repair template. We then performed RBC differentiation postediting and analysed the kinetics of editing frequency, YFP expression and erythroid differentiation using droplet digital PCR (ddPCR) and flow cytometry. As with endogenous EPOR truncation strategies, we observed more efficient erythroid differentiation in all treatments with EPO compared with the −EPO conditions (Fig. 2b). Although flow cytometry confirmed the ubiquitous expression of YFP in edited cells, regardless of presence of CD71 and GPA erythroid markers during differentiation (Fig. 2c), we did observe significant enrichment of YFP-expressing RBCs in the presence of EPO (P < 0.0001 when comparing day 0 with day 14 of RBC differentiation; Fig. 2d). This enrichment was confirmed at the genomic level by ddPCR that showed an increase in the percentage of edited alleles when tEPOR-expressing cells were subjected to erythroid differentiation, increasing by an average of 7.3-fold in the presence of EPO, and enrichment to a limited degree in the −EPO condition (Extended Data Fig. 5a). With this editing strategy, we observed increased RBC production in the CCR5-sg3 + tEPOR condition and no increase in proliferation of CCR5-sg3 alone compared with mock control cells in the +EPO condition. We saw no increased proliferation in the cells maintained in HSPC media or in the −EPO condition (Extended Data Fig. 5b). We then assessed if enrichment of edited cells differed along a gradient of EPO levels during RBC differentiation and again found that there were minimal differences in YFP+ cells in the 1 U ml−1, 3 U ml−1 and 20 U ml−1 conditions and a minor decrease in the 0.3 U ml−1 EPO condition compared with that in the −EPO condition (Extended Data Fig. 5c,d). There were comparable levels of RBC differentiation in all conditions except that of −EPO (Extended Data Fig. 5e).
We again observed that edited and unedited cells had no noticeable difference in lineage commitment or colony-forming ability following a colony-forming unit (CFU) assay (Supplementary Fig. 4a). In addition, we analysed RBC postdifferentiation using haemoglobin tetramer HPLC and found that UbC-mediated expression of tEPOR resulted in both HgbF and HgbA expression but a relative increase in HgbF expression (Supplementary Fig. 4b). These results indicate that expression of tEPOR from a safe-harbour site is an effective means of driving increased RBC production from genome-edited HSPCs.
tEPOR at HBA1 shows erythroid-specific proliferation
Although integration at a safe-harbour locus effectively increased erythropoietic output from edited HSPCs, there is the concern that constitutive expression in all cell types could disrupt stemness or lead to other unintended effects. As an alternative, we can introduce promoterless transgenes into endogenous genes for the integration cassette to be regulated by endogenous expression machinery. For instance, in previous work we designed a genome-editing strategy to fully replace the HBA1 gene with an HBB transgene to correct β-thalassaemia14. We found that because α-globin is produced by duplicate genes, HBA1 may serve as a safe-harbour site to deliver custom payloads with strong erythroid-specific expression.
We therefore hypothesized that integration of the tEPOR cDNA at the HBA1 locus could further enhance production of edited RBCs while avoiding potential complications with ubiquitous transgene expression because of the specificity of expression of HBA1 in the RBC lineage. To test this hypothesis, we designed a custom integration cassette (also packaged in AAV6) to use with an established sgRNA (HBA1-sg4) to introduce a promoterless tEPOR cDNA followed by a T2A-YFP reporter under expression of endogenous HBA1 regulatory machinery (Fig. 2e). Following editing, we observed efficient erythroid differentiation in all treatments with EPO and little to no differentiation in the absence of EPO (Fig. 2f). We found that this integration strategy indeed yielded RBC-specific expression of YFP, which was only detectable as cells gained CD71 and GPA erythroid markers (Fig. 2g). Over the course of RBC differentiation, we observed dramatic enrichment of YFP+ cells exclusively in the +EPO condition (Fig. 2h). Although we observed a mild degree of enrichment of edited alleles using ddPCR in the absence of EPO, this effect was more pronounced in the presence of EPO, eliciting an average 4.4-fold increase in the percentage of edited alleles over the course of RBC differentiation (Extended Data Fig. 6a). We again cultured edited cells in a gradient of EPO during RBC differentiation and observed minimal differences in YFP+ cell enrichment in all conditions containing EPO (Extended Data Fig. 6b,c). There were also comparable levels of RBC differentiation in all conditions except that of −EPO (Extended Data Fig. 6d).
Again we found that cells edited with tEPOR at HBA1 showed similar colony-forming ability as mock-edited cells (Supplementary Fig. 5a). In addition, we found that tEPOR-expressing cells produce ratios of human haemoglobin that are similar to unedited RBCs (Supplementary Fig. 5b).
HBB-tEPOR increases production of thalassaemia-corrected RBCs
As the above results show that tEPOR expression yields increased RBC production from edited HSPCs, we then sought to couple this selective advantage with a therapeutic gene edit. We chose to combine tEPOR with our previous β-thalassaemia correction approach to simultaneously correct the disease and increase production of clinically meaningful RBCs from these corrected HSPCs. One way this can be accomplished is by creating a bicistronic cassette that links expression of the therapeutic full-length HBB transgene with a tEPOR cDNA. Previous studies have found that the type of linker domain used can have a great bearing on transgene expression and protein function—particularly when function is dependent on formation of protein complexes, as is the case with the globin genes14. Therefore, we designed and tested a variety of AAV6 repair template vectors linking the two genes using standard T2A peptides, optimized T2A peptides with furin cleavage sites30 (referred to as FuT2A) and internal ribosome entry sites (referred to as IRES) and by driving tEPOR expression from a separate exogenous promoter—human PGK1 (referred to as PGK). To evaluate the different vectors, we edited healthy donor HSPCs as previously described at HBA1 using the bicistronic AAV6 repair templates and evaluated their ability to differentiate and enrich for edited RBCs over the course of erythroid differentiation (Fig. 3a). Although efficient RBC differentiation was achieved in all editing conditions (Fig. 3b), we found that all four bicistronic cassettes drove >2-fold enrichment of edited alleles (range, 2.1-fold to 3.5-fold; P = 0.003 for PGK-tEPOR, P = 0.0055 for tEPOR-T2A, P = 0.0259 for tEPOR-FuT2A and P = 0.0003 for IRES-tEPOR when comparing day 0 to day 14; Fig. 3c,d). We note that allele-targeting frequencies of 60% translate into >80% of the cells having at least one allele targeted (cell-targeting frequency) and thus we would not expect to see much more enrichment than we observed.

a, Design of HBB (control) and HBB-tEPOR (bicistronic) AAV6 donor cassettes targeted to the HBA1 locus by whole gene replacement. b, Percentage of GPA+/CD71+ of CD34−/CD45− cells on day 14 as determined by flow cytometry. Points are shown as median ± 95% confidence interval. Values represent biologically independent HSPC donors: n = 2 for HBB and n = 3 for all other vectors. c, Percentage of edited alleles for control (HBB) and bicistronic HBB-tEPOR in cord-blood-derived CD34+ cells over the course of RBC differentiation. Points are shown as median ± 95% confidence interval. Values represent biologically independent HSPC donors: n = 2 for HBB, n = 3 for all other vectors. *P = 0.003 for PGK-tEPOR, P = 0.0055 for tEPOR-T2A, P = 0.0259 for tEPOR-FuT2A, P = 0.0003 for IRES-tEPOR (day 0 versus day 14) by unpaired two-tailed t-test. d, Fold change in edited alleles from the beginning (day 0) to end (day 14) of RBC differentiation.The dashed line represents no fold change. Bars represent median ± 95% confidence interval. e, Percentage of edited alleles for control (HBB) and bicistronic HBB-tEPOR vectors in cells from patients with SCD over the course of RBC differentiation. Points are shown as median ± 95% confidence interval. Values represent biologically independent HSPC donors: n = 3 for PGK-tEPOR and n = 4 for all other vectors. *P = 0.0061 for PGK-tEPOR, *P = 0.011 for tEPOR-T2A, *P = 0.0016 for tEPOR-FuT2A, *P = 0.0153 for IRES-tEPOR (day 0 versus day 14) by unpaired two-tailed t-test. f, Fold change in edited alleles from the beginning (day 0) to end (day 14) of RBC differentiation. The dashed line represents no fold change. Bars represent median ± 95% confidence interval.
To ensure this strategy was also effective in patient-derived cells, we tested these bicistronic vectors in HSPCs derived from patients with SCD, this time comparing them with a therapeutic full-length HBB transgene14. All the constructs are knocked in to the HBA1 locus without disrupting the endogenous HBB locus expressing HgbS. We found that vectors did not disrupt erythroid differentiation when compared with mock-edited cells in the same donor, although their ability to differentiate was likely impacted by the variable quality of the frozen patient samples (Extended Data Fig. 7a). Again, we observed >2-fold enrichment of edited alleles for all four bicistronic vectors (range, 2.0-fold to 3.1-fold), but no change in editing frequency for the original β-thalassaemia correction vector (P = 0.0061 for PGK-tEPOR, P = 0.011 for tEPOR-T2A, P = 0.0016 for tEPOR-FuT2A, P = 0.0153 for IRES-tEPOR when comparing day 0 with day 14; Fig. 3e,f). We estimate that at the end of the differentiation almost all the cells have at least one allele with the HBB-tEPOR knock-in and thus no biological drive for further enrichment. When we analysed differentiated RBCs for haemoglobin tetramers by HPLC, we found that the T2A vectors showed almost no HgbA expression (consistent with a previous observation that addition of a T2A can disrupt HBB protein function31). In contrast, we found that PGK-tEPOR and IRES-tEPOR vectors showed an improvement in HgbA production relative to the HBB-only edited cells (Extended Data Fig. 7b). There was no change in the HgbF expression in these samples.
Multiplexed tEPOR and HBB editing increases β-globin mRNA
In lieu of coupling the tEPOR cDNA and therapeutic edit at the same locus, an alternative strategy would be to multiplex two editing events at different loci to simultaneously truncate the endogenous EPOR and introduce the original β-thalassaemia correction vector at HBA1. This strategy may have the additional advantage that the endogenous EPOR truncation will more reliably recapitulate CE. We hypothesized that we could simultaneously deliver Cas9 separately precomplexed with EPOR-truncating EPOR-sg1 and HBA1-sg4 gRNA and then transduce HSPCs with both the β-thalassaemia correction vector and the W439X EPOR-targeting vector. As homology arms of each vector are specific for each site—HBB for the HBA1 locus and W439X for the EPOR locus—integration for each vector will occur only at the intended locus. This strategy may allow simultaneous correction of β-thalassaemia and increased erythropoietic output from corrected HSPCs. For this to be maximally effective, the two editing events must be present in the same cell. Therefore, during editing we used a DNA-PKcs inhibitor to increase the frequency of template integration at each locus32,33. Following editing, we determined whether this multiplexed editing strategy increases the frequency of corrected RBCs over the course of erythroid differentiation.
To model the clinical setting where both edited and unedited HSPCs would occupy the patient’s BM, we introduced unedited cells at various concentrations at the start of erythroid differentiation (Fig. 4a). Importantly, none of the multiplexed conditions disrupted erythroid differentiation compared with single-edited HBB or mock conditions (Fig. 4b). In all the multiplexed conditions, we observed an increase in the frequency of corrected alleles over the course of RBC differentiation (P = 0.0332 for HBB + tEPOR 100%, P = 0.0086 for HBB + tEPOR 30% and P = 0.0122 for HBB + tEPOR 10% when comparing day 0 with day 14; Fig. 4c). In fact, in both the 30% and 10% multiplexed conditions, we achieved a higher frequency of edited alleles by the conclusion of erythroid differentiation compared with single-edited conditions with unedited cells introduced at an equivalent concentration (P = 0.0113 for HBB versus HBB + tEPOR 30% at day 14 and P = 0.008 for HBB versus HBB + tEPOR 10% at day 14; Fig. 4d). We confirmed truncation at the EPOR locus measured by GFP+ cells at day 14 of RBC differentiation (Extended Data Fig. 8a). We also observed a corresponding increase in HBB mRNA expression from the HBA1 locus in the 30% and 10% multiplexed edited conditions on day 14 of RBC differentiation compared with single-edited conditions (Fig. 4e), indicating an improved ability for multiplexed editing to increase therapeutic potential of this β-thalassaemia correction strategy. When we measured the colony-forming ability of the edited cells, we found that differentiation into the various lineages was similar between mock cells and cells edited with HBB alone or HBB + tEPOR; however, we did observe a decrease in total colonies produced in both edited conditions as would be expected from the increased amount of AAV used to target two loci (Extended Data Fig. 8b,c)34,35.

a, Schematic of multiplexed editing strategy with spike-in of unedited cells at the start of erythroid differentiation to model HSPC transplantation. b, Percentage of GPA+/CD71+ of CD34–/CD45– cells on day 14 of RBC differentiation as determined by flow cytometry. Points are shown as median ± interquartile range. n = 3 biologically independent HSPC donors. c, Percentage of edited alleles at HBA1 in all multiplexed editing or spike-in conditions throughout RBC differentiation. Points are shown as median ± 95% confidence interval. n = 3 biologically independent HSPC donors. *P = 0.0332 for HBB + tEPOR 100%, *P = 0.0086 for HBB + tEPOR 30%, *P = 0.0122 for HBB + tEPOR 10% (day 0 versus day 14) by unpaired two-tailed t-test; **P = 0.0113 for HBB versus HBB + tEPOR 30% at day 14, **P = 0.008 for HBB versus HBB + tEPOR 10% at day 14 by unpaired two-tailed t-test. d, Fold increase in edited alleles on day 14 of differentiation of multiplexed conditions versus HBB only. The dashed line represents no fold change. Bars represent median ± 95% confidence interval. *P = 0.0315, **P = 0.0123 by unpaired two-tailed t-test. e, mRNA expression of integrated HBB at HBA1 locus normalized to HBB expression from mock. GPA mRNA expression was used as a reference. n = 3 biologically independent HSPC donors. Bars represent median ± 95% confidence interval.
Discussion
Insights from clinical genetics have typically implicated new genes and pathways in disease. Here we sought to use human genetics to develop new strategies to treat disease. We used the precision of genome editing to capitalize on a previously characterized disorder called CE, which leads to EPO hypersensitivity and hyper-production of erythrocytes, without causing pathology4. As previously shown with variants identified in human genetics, such as those found in CCR5, PCSK9 and the γ-globin promoter region, we hypothesized that we could use CRISPR to introduce this natural EPOR variant (tEPOR) to increase erythropoietic output from edited HSPCs. Previous work has highlighted the challenge of achieving long-term correction of disease following delivery of gene therapy or genome-editing correction strategies2,36,37. Although many efforts are underway to improve editing and engraftment frequencies, we hypothesized that we could develop a strategy to increase production of the clinically relevant cell type—the RBC—from edited HSPCs. If successful, then lower editing and engraftment frequencies could yield sufficient production of RBCs to achieve therapeutic benefit and thus be curative for patients. Previous work has shown that introducing the tEPOR variant using lentiviral delivery enhanced the efficacy of small hairpin RNA knockdown of BCL11A in upregulating HgbF, confirming the beneficial function of this variant9. However, our work deploys the precision of genome editing to generate tEPOR, which may have broad utility across a spectrum of blood disorders.
We explored multiple genome-editing strategies to create the tEPOR variant, either through truncation of the endogenous EPOR gene or integration of a tEPOR cDNA at safe-harbour loci. We found that HSPCs expressing tEPOR consistently showed increased erythropoiesis but otherwise normal, EPO-dependent production of haemoglobin. To increase RBC production of genome-edited HSPCs in the context of disease correction, we combined the tEPOR cassette with a previously described β-thalassaemia strategy in which the HBB gene replaces the HBA1 gene using HDR-based genome editing14. This allowed us to simultaneously introduce an HBB transgene to restore normal haemoglobin production and to increase erythropoietic output from edited HSPCs. To show the flexibility of the various tEPOR-introduction strategies, we also developed an alternative, multiplexed, site-specific genome-editing strategy to pair the original β-thalassaemia correction strategy with introduction of the EPOR truncation at the endogenous locus. Both strategies led to enrichment of genome-edited RBCs over the course of erythroid differentiation compared with the traditional β-thalassaemia correction strategy. As we found the effects to be EPO dependent, it is possible the selective advantage in vivo may be even more pronounced because patients suffering from the haemoglobinopathies display elevated EPO levels due to their severe anaemia38.
In terms of safety, the strategy to use CRISPR to introduce natural variants has the benefit of having been already ‘tested’ in vivo in humans. However, it must be noted that several of the genome-editing strategies introduce tEPOR under non-native regulation that could alter the normal function of tEPOR. In considering this possibility, we note that although every cell in patients with CE harbours an EPOR truncation, therapeutic deployment of the genome-editing strategy will result in the introduction of tEPOR in only a subset of HSPCs resident in the BM. Therefore, any aberrant effects of non-native tEPOR expression (such as bias away from lymphoid or other cell types) are unlikely to lead to cytopenia given the large number of unedited HSPCs remaining in the BM post-transplant. Furthermore, by increasing erythropoietic output from edited HSPCs, we believe this work could enable the reduction or elimination of high-morbidity myeloablation regimens that are currently required to attain therapeutic levels of edited HSPCs. Expression of tEPOR could therefore be integrated into any treatment for blood disorders that involve transplantation of HSPCs. For example, even in an allogeneic haematopoietic stem cell transplant for RBC disorders, a truncation in the natural EPOR could be created using indel-based genome editing to give the derived transplanted erythroid progenitors a selective advantage. This strategy could thereby enable less toxic myeloablative conditioning to be effectively used where mixed chimerism might be the result.
Taken together, these results show the power of combining knowledge from human genetics with the precision of CRISPR genome-editing technology to introduce clinically meaningful variants. As human genome sequencing becomes more commonplace and clinically routine39,40, it is likely that a greater number of variants of unknown significance will be discovered and characterized. We therefore believe that the strategy defined in this work—using CRISPR to introduce natural human variants—may be deployed to amplify the therapeutic potential of current and future cell therapies.
Methods
AAV6 vector design, production and purification
AAV6 vector plasmids were cloned into the pAAV-MCS plasmid (Agilent Technologies) comprising inverted terminal repeats derived from AAV2. Gibson Assembly Master Mix (New England Biolabs) was used for the creation of all DNA repair vectors as per the manufacturer’s instructions. AAV6 vector was produced and purified with little variation from previously described processes41. 293T cells (Life Technologies) were seeded in five dishes (15 cm2) with 13 × 106–15 × 106 cells per plate at 24-h pretransfection. Each dish was then transfected with a standard polyethylenimine (PEI) transfection of 6 μg inverted-terminal-repeat-containing plasmid and 22 μg pDGM6 (gift from David Russell, University of Washington), which holds the AAV6 cap, AAV2 rep and Ad5 helper genes. After a 48–72-h incubation, cells were collected and vectors were purified using the AAVpro purification kit (catalogue number 6666; Takara Bio) per the manufacturer’s instructions and then stored at −80 °C until further use. AAV6 vectors were titred using ddPCR to measure the number of vector genomes as previously described42.
In vitro culture of CD34+ HSPCs
Human CD34+ HSPCs were cultured in conditions as previously described13,43,44,45,46. CD34+ HSPCs were isolated from cord blood (provided by Stanford Binns Program for Cord Blood Research) or sourced from plerixafor- and/or G-CSF-mobilized peripheral blood (AllCells and STEMCELL Technologies). Frozen plerixafor- and/or G-CSF-mobilized peripheral blood of patients with SCD were provided by Dr Vivien Sheehan at Emory University. CD34+ HSPCs were cultured at 1 × 105–5 × 105 cells ml−1 in StemSpan Serum-Free Expansion Medium II (STEMCELL Technologies) or Good Manufacturing Practice Stem Cell Growth Medium (SCGM; CellGenix) supplemented with a human cytokine (PeproTech) cocktail: stem cell factor (100 ng ml−1), thrombopoietin (100 ng ml−1), Fms-like tyrosine kinase 3 ligand (100 ng ml−1), interleukin-6 (100 ng ml−1), streptomycin (20 mg ml−1), penicillin (20 U ml−1) and 35 nM of UM171 (catalogue number A89505; APExBIO). The cell incubator conditions were 37 °C, 5% CO2 and 5% O2.
Electroporation-aided transduction of cells
The synthetic chemically modified sgRNAs used to edit CD34+ HSPCs were purchased from Synthego or TriLink Biotechnologies and were purified by HPLC. These modifications comprise 2′-O-methyl-3′-phosphorothioate at the three terminal nucleotides of the 5′ and 3′ ends described previously17. The target sequences for the gRNAs were as follows.
EPOR gRNA (EPOR-sg1)
5′-AGCTCAGGGCACAGTGTCCA-3′
EPOR gRNA (EPOR-sg2)
5′-GCTCCCAGCTCTTGCGTCCA-3′
CCR5 gRNA (CCR5-sg3)
5′-GCAGCATAGTGAGCCCAGAA-3′
HBA1 gRNA (HBA1-sg4)
5′-GGCAAGAAGCATGGCCACCG-3′
The HiFi Cas9 protein was purchased from Integrated DNA Technologies (IDT) or Aldevron. Before electroporation, RNPs were complexed at a Cas9/sgRNA molar ratio of 1:2.5 at 25 °C for 10–20 min. Next, CD34+ cells were resuspended in P3 buffer (Lonza) with complexed RNPs and subsequently electroporated using the Lonza 4D-Nucleofector and 4D-Nucleofector X Unit (program DZ-100). Electroporated cells were then plated at 1 × 105–5 × 105 cells ml−1 in the previously described cytokine-supplemented media. Immediately after electroporation, AAV6 was dispensed onto cells at 2.5 × 103–5 × 103 vector genomes per cell based on titre determined by ddPCR. For multiplex editing experiments, in addition to the steps described above, cells were incubated with 0.5 μM of the DNA-PKcs inhibitor AZD7648 (catalogue number S8843; Selleck Chemicals) for 24 h, as previously described32,33.
Allelic modification analysis using ddPCR
Edited HSPCs were collected within 2–3 days postelectroporation and at each media change throughout erythrocyte differentiation and then analysed for modification frequencies of the alleles of interest. To quantify editing frequencies, we created custom ddPCR primers and probes to quantify HDR alleles (using in–out PCR and probe corresponding to the expected integration event) compared with an established genomic DNA reference (REF) at the CCRL2 locus14. QuickExtract DNA extraction solution (catalogue number QE09050; Biosearch Technologies) was used to collect genomic DNA input, which was then digested using BamHI-HF or HindIII-HF as per the manufacturer’s instructions (New England Biolabs). The percentage of targeted alleles within a cell population was measured with a Bio-Rad QX200 ddPCR machine and QuantaSoft software (v.1.7; Bio-Rad) using the following reaction mixture: 1–4 μl genomic DNA input, 10 μl of ddPCR Supermix for Probes (no dUTP; Bio-Rad), primer and probes (1:3.6 ratio; IDT), and volume up to 20 μl with H2O. ddPCR droplets were then generated following the manufacturer’s instructions (Bio-Rad): 20 μl of ddPCR reaction, 70 μl of droplet generation oil and 40 μl of droplet sample. Thermocycler (Bio-Rad) settings were as follows: 98 °C (10 min), 94 °C (30 s), 55.7–60 °C (30 s), 72 °C (2 min), return to step 2 for 40–50 cycles and then 98 °C (10 min). Analysis of droplet samples was then performed using the QX200 Droplet Digital PCR System (Bio-Rad). We next divided the copies per microlitre for HDR (%): HDR/REF. The following primers and probes were used in the ddPCR reaction.
CCR5 (for tEPOR-YFP construct)
Forward primer (FP): 5′-GGGAGGATTGGGAAGACA-3′
Reverse primer (RP): 5′-AGGTGTTCAGGAGAAGGACA-3′
Probe: 5′-6-FAM/AGCAGGCATGCTGGGGATGCGGTGG/3IABkFQ-3′
HBA1 (for tEPOR-YFP construct)
FP: 5′-AGTCCAAGCTGAGCAAAGA-3′
RP: 5′-ATCACAAACGCAGGCAGAG-3′
Probe: 5′-6-FAM/CGAGAAGCGCGATCACATGGTCCTGC/3IABkFQ-3′
HBA1 (for HBB construct and tEPOR-HBB constructs)
FP: 5′-GTGGCTGGTGTGGCTAATG-3′
RP: 5′-CAGAAAGCCAGCCAGTTCTT-3′
Probe: 5′-6-FAM/CCTGGCCCACAAGTATCACT/3IABkFQ-3′
HBA1 (for HBB-tEPOR constructs)
FP: 5′-TCTGCTGCCAGCTTTGAGTA-3′
RP: 5′-GCTGGAGTGGGACTTCTCTG-3′
Probe: 5′-6-FAM/ACTATCCTGGACCCCAGCTC/3IABkFQ-3′
CCRL2 (reference)
FP: 5′-GCTGTATGAATCCAGGTCC-3′
RP: 5′-CCTCCTGGCTGAGAAAAAG-3′
Probe: 5′-HEX/TGTTTCCTC/ZEN/CAGGATAAGGCAGCTGT/3IABkFQ-3′
Indel analysis using TIDE software
Within 2–4 days postelectroporation, HSPCs were collected with QuickExtract DNA extraction solution (catalogue number QE09050; Biosearch Technologies) to collect genomic DNA. The following primer sequences were used to amplify the respective cut sites at the EPOR locus:
FP: 5′-CAGCTGTGGCTGTACCAGAA-3′
RP: 5′-CAGCCTGGTGTCCTAAGAGC-3′
Sanger sequencing of the respective samples was then used as input for indel frequency analysis relative to a mock, unedited sample using TIDE as previously described25.
In vitro differentiation of CD34+ HSPCs into erythrocytes
Following editing, HSPCs derived from healthy individuals or patients with SCD were cultured for 2–3 days as described above. Subsequently, a 14-day in vitro differentiation was performed in supplemented SFEMII medium as previously described24,47. SFEMII base medium was supplemented with 100 U ml−1 penicillin–streptomycin, 10 ng ml−1 SCF (PeproTech), 1 ng ml−1 IL-3 (PeproTech), 3 U ml−1 EPO (eBiosciences), 200 μg ml−1 transferrin (Sigma-Aldrich), 3% human serum (heat-inactivated; Sigma-Aldrich or Thermo Fisher Scientific), 2% human plasma (isolated from umbilical cord blood provided by the Stanford Binns Cord Blood Program), 10 μg ml−1 insulin (Sigma-Aldrich) and 3 U ml−1 heparin (Sigma-Aldrich). Cells were cultured in the first phase of medium for 7 days at 1 × 105 cells ml−1. In the second phase of medium, days 7–10, cells were maintained at 1 × 105 cells ml−1 and IL-3 was removed from the culture. In the third phase of medium, days 11–14, cells were cultured at 1 × 106 cells ml−1, with a transferrin increase to 1 mg ml−1.
Immunophenotyping of differentiated erythrocytes
Differentiated erythrocytes were analysed by flow cytometry on day 14 for erythrocyte lineage-specific markers using a FACS Aria II (BD Biosciences). Edited and unedited cells were analysed using the following antibodies: hCD45-V450 (HI30; BD Biosciences), CD34-APC (561; BioLegend), CD71-PE-Cy7 (OKT9; Affymetrix) and CD235a-PE (GPA) (GA-R2; BD Biosciences). In addition to cell-specific markers, cells were also stained with Ghost Dye Red 780 (Tonbo Biosciences) to measure viability.
Haemoglobin tetramer analysis
Frozen pellets of approximately 1 × 106 in vitro-differentiated erythrocytes were thawed and lysed in 30 µl of RIPA buffer with 1× Halt Protease Inhibitor Cocktail (Thermo Fisher Scientific) for 5 min on ice. The mixture was vigorously vortexed and cell debris was removed by centrifugation at 13,000 r.p.m. for 10 min at 4 °C. HPLC analysis of haemoglobins in their native form was performed on a cation-exchange PolyCAT A column (35 mm2 × 4.6 mm2, 3 µm, 1,500 Å; PolyLC) using a Perkin-Elmer Flexar HPLC system at room temperature and detection at 415 nm. Mobile phase A consisted of 20 mM Bis-Tris and 2 mM KCN at pH 6.94, adjusted with HCl. Mobile phase B consisted of 20 mM Bis-Tris, 2 mM KCN and 200 mM NaCl at pH 6.55. Haemolysate was diluted in buffer A before injection of 20 µl onto the column with 8% buffer B and eluted at a flow rate of 2 ml min−1 with a gradient made to 40% B in 6 min, increased to 100% B in 1.5 min, returned to 8% B in 1 min and equilibrated for 3.5 min. Quantification of the area under the curve of the peaks was performed with TotalChrom software (Perkin-Elmer) and raw values were exported to GraphPad Prism 9 for plotting and further analysis.
mRNA analysis
After differentiation of HSPCs into erythrocytes, cells were collected and RNA was extracted using the RNeasy Plus Mini Kit (Qiagen). Subsequently, cDNA was made from approximately 100 ng of RNA using the iScript Reverse Transcription Supermix for quantitative PCR with reverse transcription (Bio-Rad). Expression levels of the β-globin transgene and α-globin mRNA were quantified with a Bio-Rad QX200 ddPCR machine and QuantaSoft software (v.1.7; Bio-Rad) using the following primers and 6-FAM/ZEN/IBFQ-labelled hydrolysis probes, purchased as custom-designed PrimeTime qPCR Assays from IDT.
HBB and HBB-tEPOR into HBA1
FP: 5′-GGTCCCCACAGACTCAGAGA-3′
RP: 5′-CAGCATCAGGAGTGGACAGA-3′
Probe: 5′-6-FAM/AACCCACCATGGTGCATCTG/3IABkFQ-3′
To normalize for RNA input, levels of the RBC-specific reference gene GPA were determined in each sample using the following primers and HEX/ZEN/IBFQ-labelled hydrolysis probes, purchased as custom-designed PrimeTime qPCR Assays from IDT.
GPA (reference)
FP: 5′-ATATGCAGCCACTCCTAGAGCTC-3′
RP: 5′-CTGGTTCAGAGAAATGATGGGCA-3′
Probe: 5′-HEX/AGGAAACCGGAGAAAGGGTA/3IABkFQ-3′
ddPCR reactions were created using the respective primers and probes and droplets were generated as described above. Thermocycler (Bio-Rad) settings were as follows: 98 °C (10 min), 94 °C (30 s), 54 °C (30 s), 72 °C (30 s), return to step 2 for 50 cycles and then 98 °C (10 min). Analysis of droplet samples was done using the QX200 Droplet Digital PCR System (Bio-Rad). To determine relative expression levels, the numbers of HBB transgene copies per millilitre were divided by the numbers of GPA copies ml−1.
Methylcellulose CFU assay
At 2–3 days postelectroporation, HSPCs were plated in SmartDish 6 well plates (catalogue number 27370; STEMCELL Technologies) containing MethoCult H4434 Classic or MethoCult H4434 Classic without EPO (catalogue numbers 04444 and 04544; STEMCELL Technologies). After 14 days, the wells were imaged using the STEMvision Hematopoietic Colony Counter (STEMCELL Technologies). Colonies were counted and scored to determine the number of CFU-GEMM (colony-forming unit-granulocyte, erythroid, macrophage, megakaryocyte), CFU-GM (colony-forming unit-granulocyte, macrophage), BFU-E (burst-forming unit-erythroid) and CFU-E (colony-forming unit-erythroid) colonies.
Quantification of editing efficiency at evaluated off-target sites
Potential sgRNA off-target sites were predicted using the CRISPR Off-target Sites with Mismatches, Insertions and Deletions (COSMID) online tool48. Sites were ranked according to score and duplicate predictions at the same location were removed. All sites with a score ≤5.5 were included in the analysis and the 5 sites in exonic or untranslated regions were further analysed. PCR amplification of these sites was performed using genomic DNA from mock-edited and RNP-edited cells. The following primers were used with Illumina adaptors (FP adaptor, 5′-ACACTCTTTCCCTACACGACGCTCTTCCGATCT-3′; RP adaptor, 5′-GACTGGAGTTCAGACGTGTGCTCTTCCGATCT-3′).
EPOR-OT1
FP: 5′-GAGCGGGCTACAGAGCTAGA-3′
RP: 5′-TGGCAGAAAGTAAGGGGATG-3′
EPOR-OT2
FP: 5′-ACTTGTGGAGCCACAGTTTG-3′
RP: 5′-AATGCCCTTGAGATGAATGC-3′
EPOR-OT3
FP: 5′-TCACACACCCGTAGCCATAA-3′
RP: 5′-AGAATGCTCTTTGCAGTAGCC-3′
EPOR-OT4
FP: 5′-CTCAAAACTTCACCCAGGCT-3′
RP: 5′-GGTCTGTCATTGAATGCCTT-3′
EPOR-OT5
FP: 5′-CAACCCTGATGGGTCTGC-3′
RP: 5′-CCACAGCTGGCTGACCTT-3′
Following amplification, PCR products were purified by gel electrophoresis and subsequent extraction using the GeneJet Gel Extraction Kit (catalogue number FERK0692; Thermo Fisher Scientific). Purified samples were submitted for library preparation and sequencing by Amplicon-EZ next-generation sequencing (Azenta Life Sciences), ensuring a yield of over 100,000 reads per sample. Amplicons, flanked by Illumina partial adaptor sequences, which encompassed the programmed DSBs for CRISPR–Cas9, underwent sequencing using Illumina chemistry. FastQC (v.0.11.8, default parameters; http://www.bioinformatics.babraham.ac.uk/projects/fastqc/) was used to assess the quality of raw reads. Subsequently, paired-end reads were aligned to the specified off-target regions using CRISPResso2 (v.2.2.14; fastq.gz files were used as input)49.
Statistical analysis
GraphPad Prism 9 software was used for all statistical analysis.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The main data supporting the results in this study are available within the article and its Supplementary Information. High-throughput-sequencing data generated for off-target analysis are available through the NCBI Sequence Read Archive database via the accession number PRJNA1102034. Sequences of the gRNA and ddPCR primers and probes are provided in Methods. Source data for the figures are provided with this paper. All data generated in this study are available from the corresponding authors on reasonable request.
References
Hütter, G. et al. Long-term control of HIV by CCR5 Delta32/Delta32 stem-cell transplantation. N. Engl. J. Med. 360, 692–698 (2009).
Frangoul, H. et al. CRISPR-Cas9 gene editing for sickle cell disease and β-thalassemia. N. Engl. J. Med. 384, 252–260 (2021).
Fitzgerald, K. et al. A highly durable RNAi therapeutic inhibitor of PCSK9. N. Engl. J. Med. 376, 41–51 (2017).
de la Chapelle, A., Träskelin, A. L. & Juvonen, E. Truncated erythropoietin receptor causes dominantly inherited benign human erythrocytosis. Proc. Natl Acad. Sci. USA 90, 4495–4499 (1993).
Sokol, L. et al. Primary familial polycythemia: a frameshift mutation in the erythropoietin receptor gene and increased sensitivity of erythroid progenitors to erythropoietin. Blood 86, 15–22 (1995).
Juvonen, E., Ikkala, E., Fyhrquist, F. & Ruutu, T. Autosomal dominant erythrocytosis caused by increased sensitivity to erythropoietin. Blood 78, 3066–3069 (1991).
Huang, L. J., Shen, Y. M. & Bulut, G. B. Advances in understanding the pathogenesis of primary familial and congenital polycythaemia. Br. J. Haematol. 148, 844–852 (2010).
Arcasoy, M. O., Degar, B. A., Harris, K. W. & Forget, B. G. Familial erythrocytosis associated with a short deletion in the erythropoietin receptor gene. Blood 89, 4628–4635 (1997).
Uchida, N. et al. Sustained fetal hemoglobin induction in vivo is achieved by BCL11A interference and coexpressed truncated erythropoietin receptor. Sci. Transl. Med. 13, eabb0411 (2021).
Negre, O. et al. Correction of murine β-thalassemia after minimal lentiviral gene transfer and homeostatic in vivo erythroid expansion. Blood 117, 5321–5331 (2011).
Kirby, S. L., Cook, D. N., Walton, W. & Smithies, O. Proliferation of multipotent hematopoietic cells controlled by a truncated erythropoietin receptor transgene. Proc. Natl Acad. Sci. USA 93, 9402–9407 (1996).
Hacein-Bey-Abina, S. et al. Insertional oncogenesis in 4 patients after retrovirus-mediated gene therapy of SCID-X1. J. Clin. Invest. 118, 3132–3142 (2008).
Dever, D. P. et al. CRISPR/Cas9 β-globin gene targeting in human haematopoietic stem cells. Nature 539, 384–389 (2016).
Cromer, M. K. et al. Gene replacement of α-globin with β-globin restores hemoglobin balance in β-thalassemia-derived hematopoietic stem and progenitor cells. Nat. Med. 27, 677–687 (2021).
Vaidyanathan, S. et al. High-efficiency, selection-free gene repair in airway stem cells from cystic fibrosis patients rescues CFTR function in differentiated epithelia. Cell Stem Cell 26, 161–171.e4 (2020).
Choi, E. & Koo, T. CRISPR technologies for the treatment of Duchenne muscular dystrophy. Mol. Ther. 29, 3179–3191 (2021).
Hendel, A. et al. Chemically modified guide RNAs enhance CRISPR-Cas genome editing in human primary cells. Nat. Biotechnol. 33, 985–989 (2015).
Shin, J. J. et al. Controlled cycling and quiescence enables efficient HDR in engraftment-enriched adult hematopoietic stem and progenitor cells. Cell Rep. 32, 108093 (2020).
Baik, R. et al. Transient inhibition of 53BP1 increases the frequency of targeted integration in human hematopoietic stem and progenitor cells. Nat. Commun. 15, 111 (2024).
Chakraborty, N. G. et al. Myeloablative chemotherapy with autologous peripheral blood stem cell transplantation for metastatic breast cancer: immunologic consequences affecting clinical outcome. Bone Marrow Transplant. 24, 837–843 (1999).
Bhatia, S. Long-term health impacts of hematopoietic stem cell transplantation inform recommendations for follow-up. Expert Rev. Hematol. 4, 437–454 (2011).
Inamoto, Y. et al. Secondary solid cancer screening following hematopoietic cell transplantation. Bone Marrow Transplant. 50, 1013–1023 (2015).
Vakulskas, C. A. et al. A high-fidelity Cas9 mutant delivered as a ribonucleoprotein complex enables efficient gene editing in human hematopoietic stem and progenitor cells. Nat. Med. 24, 1216–1224 (2018).
Dulmovits, B. M. et al. Pomalidomide reverses γ-globin silencing through the transcriptional reprogramming of adult hematopoietic progenitors. Blood 127, 1481–1492 (2016).
Brinkman, E. K., Chen, T., Amendola, M. & van Steensel, B. Easy quantitative assessment of genome editing by sequence trace decomposition. Nucleic Acids Res. 42, e168 (2014).
Cromer, M. K. et al. Ultra-deep sequencing validates safety of CRISPR/Cas9 genome editing in human hematopoietic stem and progenitor cells. Nat. Commun. 13, 4724 (2022).
Cromer, M. K. et al. Comparative analysis of CRISPR off-target discovery tools following ex vivo editing of CD34+ hematopoietic stem and progenitor cells. Mol. Ther. 31, 1074–1087 (2023).
Papapetrou, E. P. & Schambach, A. Gene insertion into genomic safe harbors for human gene therapy. Mol. Ther. 24, 678–684 (2016).
Gomez-Ospina, N. et al. Human genome-edited hematopoietic stem cells phenotypically correct Mucopolysaccharidosis type I. Nat. Commun. 10, 4045 (2019).
Fang, J. et al. An antibody delivery system for regulated expression of therapeutic levels of monoclonal antibodies in vivo. Mol. Ther. 15, 1153–1159 (2007).
Reinhardt, A., Kagawa, H. & Woltjen, K. N-terminal amino acids determine KLF4 protein stability in 2A peptide-linked polycistronic reprogramming constructs. Stem Cell Reports 14, 520–527 (2020).
Selvaraj, S. et al. High-efficiency transgene integration by homology-directed repair in human primary cells using DNA-PKcs inhibition. Nat. Biotechnol. https://doi.org/10.1038/s41587-023-01888-4 (2023).
Wimberger, S. et al. Simultaneous inhibition of DNA-PK and Polϴ improves integration efficiency and precision of genome editing. Nat. Commun. 14, 4761 (2023).
Schiroli, G. et al. Precise gene editing preserves hematopoietic stem cell function following transient p53-mediated DNA damage response. Cell Stem Cell 24, 551–565.e8 (2019).
Ferrari, S. et al. Choice of template delivery mitigates the genotoxic risk and adverse impact of editing in human hematopoietic stem cells. Cell Stem Cell 29, 1428–1444.e9 (2022).
Kanter, J. et al. Biologic and clinical efficacy of LentiGlobin for sickle cell disease. N. Engl. J. Med. 386, 617–628 (2022).
George, L. A. et al. Hemophilia B gene therapy with a high-specific-activity factor IX variant. N. Engl. J. Med. 377, 2215–2227 (2017).
Nişli, G., Kavakli, K., Aydinok, Y., Oztop, S. & Cetingül, N. Serum erythropoietin levels in patients with beta thalassemia major and intermedia. Pediatr. Hematol. Oncol. 14, 161–167 (1997).
Halldorsson, B. V. et al. The sequences of 150,119 genomes in the UK Biobank. Nature 607, 732–740 (2022).
Gudbjartsson, D. F. et al. Large-scale whole-genome sequencing of the Icelandic population. Nat. Genet. 47, 435–444 (2015).
Khan, I. F., Hirata, R. K. & Russell, D. W. AAV-mediated gene targeting methods for human cells. Nat. Protoc. 6, 482–501 (2011).
Aurnhammer, C. et al. Universal real-time PCR for the detection and quantification of adeno-associated virus serotype 2-derived inverted terminal repeat sequences. Hum. Gene Ther. Methods 23, 18–28 (2012).
Bak, R. O. & Porteus, M. H. CRISPR-mediated integration of large gene cassettes using AAV donor vectors. Cell Rep. 20, 750–756 (2017).
Bak, R. O., Dever, D. P. & Porteus, M. H. CRISPR/Cas9 genome editing in human hematopoietic stem cells. Nat. Protoc. 13, 358–376 (2018).
Charlesworth, C. T. et al. Priming human repopulating hematopoietic stem and progenitor cells for Cas9/sgRNA gene targeting. Mol. Ther. Nucleic Acids 12, 89–104 (2018).
Cromer, M. K. et al. Global transcriptional response to CRISPR/Cas9-AAV6-based genome editing in CD34+ hematopoietic stem and progenitor cells. Mol. Ther. 26, 2431–2442 (2018).
Hu, J. et al. Isolation and functional characterization of human erythroblasts at distinct stages: implications for understanding of normal and disordered erythropoiesis in vivo. Blood 121, 3246–3253 (2013).
Cradick, T. J., Qiu, P., Lee, C. M., Fine, E. J. & Bao, G. COSMID: a web-based tool for identifying and validating CRISPR/Cas off-target sites. Mol. Ther. Nucleic Acids 3, e214 (2014).
Clement, K. et al. CRISPResso2 provides accurate and rapid genome editing sequence analysis. Nat. Biotechnol. 37, 224–226 (2019).
Acknowledgements
We thank the following funding sources that made this work possible: American Society of Hematology Junior Faculty Scholar Award to M.K.C.; Stanford Medical Scholars Research Program, the American Society of Hematology Minority Medical Student Award Program and the Stanford Medical Scientist Training Program to S.E.L.; and American Society of Hematology Minority Medical Student Award Program to J.C. We thank the Stanford Binns Program for Cord Blood Research for providing CD34+ HSPCs and the FACS Core Facility at the Stanford Institute for Stem Cell Biology and Regenerative Medicine for access to FACS machines.
Author information
Authors and Affiliations
Contributions
S.E.L., J.C., J.P.H., K.R.M., C.T.C., E.S. and M.K.C. designed and performed experiments and analysed data. S.S. and K.J. designed experiments and analysed data. V.A.S. provided reagents for experiments. M.H.P. and M.K.C. supervised the project. S.E.L., J.C. and M.K.C. wrote the article with input from other authors.
Corresponding authors
Ethics declarations
Competing interests
M.H.P. serves on the scientific advisory board of Allogene Tx and is an advisor to Versant Ventures. M.H.P., M.K.C. and J.C. have equity in Graphite Bio. M.H.P. has equity in CRISPR Tx and serves on the board of directors of Graphite Bio and Kamau Therapeutics. J.C., M.K.C. and M.H.P. have filed provisional patent number PCT/US2022/077951. All other authors declare no competing interests.
Peer review
Peer review information
Nature Biomedical Engineering thanks Van Trung Chu and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Indel analysis of HSPCs edited with most effective EPOR sgRNA (sg1) over course of erythroid differentiation.
a, Frequency of five most common indels found in one HSPC donor targeted with EPOR-sg1 over the course of RBC differentiation. b, Fold enrichment of five most common indels over course of RBC differentiation in one HSPC donor targeted with EPOR-sg1.
Extended Data Fig. 2 Almost all cells edited at EPOR locus contain tEPOR at end of differentiation and show increased erythroid proliferation.
a, Plot of percentage of GFP+ cells of live single cells maintained in RBC media +EPO (from data shown in Fig. 1e) overlaid with percentage of alleles containing indels in EPOR in same three biological donors. Points represent median ± 95% confidence interval. b, Cell count fold change in mock and edited cells maintained in RBC media +/- EPO or HSPC media. Points represent mean ± SEM. Values represent biologically independent HSPC donors N = 3 for Mock and EPOR-sg1+ BGH and N = 2 for sg1. Data for +EPO condition same as in Fig. 1f.
Extended Data Fig. 3 Enrichment of edited cells and RBC differentiation is only minorly affected by different concentration of EPO in media.
a, Schematic depicting HSPC editing and subsequent RBC differentiation using different levels of EPO cytokine. b, Percentage of GFP+ cells of live single cells maintained in RBC media with 0 U/mL – 20 U/mL EPO from one biological HSPC donor. c, Fold increase of GFP+ cells at each concentration of EPO compared to 0 U/mL EPO at day 14 of RBC differentiation. Bars represent median ± 95% confidence interval. N = 2 biological HSPC donors. d, Percentage of GPA+/CD71+ of live single cells on day 14 of differentiation. Bars represent median ± 95% confidence interval. Values represent N = 2 biologically independent HSPC donors.
Extended Data Fig. 4 Off-target analysis of EPOR-sg1 by in silico prediction and targeted next-generation sequencing (NGS).
a, Complete list of potential off-target sites predicted by COSMID with a score ≤5.5. b, Summary of off-target sites classified by region in genome. c, Detailed summary of 5 candidate off-target sites found in an exon or UTR of the genome. Sequence nucleotide mismatches highlighted in red, inserted nucleotides underlined, and missing nucleotides indicated by the ‘^’ symbol. PAM sequence highlighted in blue. d, Sequencing read depth of mock-edited and EPOR-sg1 edited sample sent for each off-target site. N = 1 biological HSPC donor. e, Frequency of indels detected in each sample at each off-target site. N = 1 biological HSPC donor.
Extended Data Fig. 5 Safe harbour integration of tEPOR leads to increased erythroid proliferation and editing frequencies with little impact at different concentrations of EPO.
a, Fold change in edited alleles at CCR5 over course of RBC differentiation +/- EPO or maintained in HSPC media measured by ddPCR. Points shown as median ± interquartile range. N = 3-4 biologically independent HSPC donors. b, Cell count fold change in mock and edited cells maintained in RBC media +/- EPO or HSPC media. Points represent mean ± SEM. Values represent biologically independent HSPC donors N = 3 for Mock and CCR5-sg3 + tEPOR and N = 2 for CCR5-sg3. c, Percentage of YFP+ cells of live single cells throughout RBC differentiation with 0 U/mL – 20 U/mL EPO from one biological HSPC donor. d, Fold increase of YFP+ cells at each concentration of EPO compared to 0 U/mL EPO at day 14 of differentiation. Bars represent median ± 95% confidence interval. N = 2 biological HSPC donors. e, Percentage of GPA+/CD71+ of live single cells on day 14 of differentiation. Bars represent median ± 95% confidence interval. Values represent N = 2 biologically independent HSPC donors.
Extended Data Fig. 6 Erythroid specific expression of tEPOR leads to increased editing frequencies and is not affected by EPO concentration.
a, Fold change in edited alleles at HBA1 over course of RBC differentiation +/- EPO measured by ddPCR. Points shown as median ± 95% CI. N = 3 biologically independent HSPC donors. b, Percentage of YFP+ cells of live single cells throughout RBC differentiation with 0 U/mL – 20 U/mL EPO in one biological HSPC donor. c, Fold increase of YFP+ cells at each EPO concentration compared to 0 U/mL EPO at day 14 of RBC differentiation. Bars represent median ± 95% confidence interval. N = 2 biological HSPC donors. d, Percentage of GPA+/CD71+ of live single cells on day 14 of differentiation. Bars represent median ± 95% confidence interval. Values represent N = 2 biologically independent HSPC donors.
Extended Data Fig. 7 Haemoglobin tetramer analysis of tEPOR-edited SCD patient HSPCs following RBC differentiation.
a, Percentage of GPA+/CD71+ of CD34−/CD45−cells on day 14 of RBC differentiation as determined by flow cytometry. Points shown as median ± 95% confidence interval. N = 4 biologically independent HSPC donors. b, Ratio of adult haemoglobin to sickle haemoglobin from HPLC analysis of haemoglobin tetramers from differentiated SCD patient HSPCs. Bars shown as mean ± SD. N = 4 biologically independent HSPC donors.
Extended Data Fig. 8 Multiplexed editing shows maintenance of EPOR truncation at end of RBC differentiation and does not disrupt HSPC lineage formation.
a, Percentage of GFP+ cells of live single cells on day 14 of RBC differentiation as determined by flow cytometry. Points represent median ± interquartile range. Values represent N = 3 biologically independent HSPC donors. b, CFU assay of mock, single edited, and multiplex edited HSPCs. Bars represent percent of total colonies: CFU-GEMM (multi-potential granulocyte, erythroid, macrophage, megakaryocyte progenitor cells), CFU-GM (colony forming unit-granulocytes and monocytes), and BFU-E (erythroid burst forming units). N = 1. c, Number of total colonies in CFU assay produced per 500 plated cells in each condition.
Supplementary information
Source data
Source Data Figs. 1–4 and Extended Data Figs. 1–8
Source data and statistics.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Luna, S.E., Camarena, J., Hampton, J.P. et al. Enhancement of erythropoietic output by Cas9-mediated insertion of a natural variant in haematopoietic stem and progenitor cells. Nat. Biomed. Eng (2024). https://doi.org/10.1038/s41551-024-01222-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41551-024-01222-6
- Springer Nature Limited