
Abstract
Magnetic transition metals (mTM = Cr, Mn, Fe, Co, and Ni) and their complex compounds (oxides, hydroxides, and oxyhydroxides) are highly important material platforms for diverse technologies, where electrochemical phase diagrams with respect to electrode potential and solution pH can be used to effectively understand their corrosion and oxidation behaviors in relevant aqueous environments. Many previous decades-old mTM–Pourbaix diagrams are inconsistent with various direct electrochemical observations, because experimental complexities associated with extracting reliable free energies of formation (ΔfG) lead to inaccuracies in the data used for modeling. Here, we develop a high-throughput simulation approach based on density-functional theory (DFT), which quickly screens structures and compounds using efficient DFT methods and calculates accurate ΔfG values, using high-level exchange-correlation functions to obtain ab initio Pourbaix diagrams in comprehensive and close agreement with various important electrochemical, geological, and biomagnetic observations reported over the last few decades. We also analyze the microscopic mechanisms governing the chemical trends among the ΔfG values and Pourbaix diagrams to further understand the electrochemical behaviors of mTM-based materials. Last, we provide probability profiles at variable electrode potential and solution pH to show quantitatively the likely coexistence of multiple-phase areas and diffuse phase boundaries.
Similar content being viewed by others


Introduction
Magnetic transition metals (mTM = Cr, Mn, Fe, Co, and Ni) are among the most important elements for human civilization. Numerous mTM alloys and compounds have been applied broadly and frequently throughout history. Various conventional mTM-based structural alloys (e.g., Fe and Ni alloys) are widely used in many low- and high-temperature fields, including civilian tools, construction frameworks, biocompatible alloys,1,2,3 gas turbines,4,5,6 and nuclear-power equipment,7,8,9 owing to their excellent mechanical properties, environmental benignity, and oxidation and corrosion resistances. Superior mechanical properties are also present in mTM-based high-entropy alloys,10,11,12 and their corrosion resistance is under intensive investigation due to its importance.13,14 A variety of mTM metals, oxides, and (oxy)hydroxides are superior materials for photonic and electrochemical catalyses (e.g., water splitting and pollutant decomposition).15,16,17,18 In addition, electrode materials based on mTM (hydr)oxides are utilized in electrochemical capacitors19,20,21 and rechargable lithium/sodium-ion batteries.22,23,24 Last, mTM oxides find promising application in nonvolatile resistive random access memory.25,26
Electrochemical stability is among the most critical factors determining the applications of materials in biological, marine, and civilian fields. The stabilities of mTM metals and their compounds against continuous corrosion and oxidation are one of the prerequisites for systems-level integration of new materials in aqueous and humid environments. On the other hand, the synthesis and optimization of many functional materials (e.g., metal–organic frameworks,27 TiO2,28,29 and TM (hydr)oxides18) in aqueous solutions require accurate knowledge about the electrochemical behaviors of related compounds. The electrochemical stabilities of materials can be effectively understood and predicted from a Pourbaix diagram30—thermodynamic phase maps indicating the equilibrium phases of a material system spanning a space defined by electrode potential and solution pH.
Simulating a Pourbaix diagram requires the free energies of formation (ΔfG) of all the involved species (the metal, its compounds, and the associated aqueous ions). Although the experimental ΔfG (or chemical potentials) for the most common aqueous ions (e.g., X2+ and X3+, X = Cr, Mn, Fe, Co, and Ni) usually have a relatively small uncertainty (~0.02 eV/ion), those for many solid compounds may have large uncertainties (e.g., δ ~ 1.6 eV per formula unit (f.u.) for Co3O4) or are not available (e.g., ΔfG of Cr3O4 is undetermined). Supplementary Information (part D) contains our collected ΔfG data of mTM compounds and their aqueous ions from various databases. In addition, there are many unavoidable technical or physical limitations imposed by experiment, for example, fierce combustion, defect contamination, uncontrollable/unmeasurable hydration, and solution filtering, which can result in large uncertainties (inaccuracies) in the extracted experimental ΔfG values.31 Indeed, the Ti and Ni Pourbaix diagrams simulated using experimental ΔfG values are found to be inconsistent with various direct electrochemical phenomena,31,32 which have been ascribed to the inaccuracies in the free energies of formation. Furthermore, some of the important compounds presenting potential and pH-dependent stabilities may exhibit structures that are poorly or difficult to characterize (e.g., NiOOH31,33) or remain to be determined by experiment (e.g., Cr3O4, Cr(OH)2, FeO2, Co2O3, Co(OH)2, and Ni3O434). Alternatively, state-of-the-art density-functional theory (DFT) can be used as a reliable approach to assess the most stable structures and determine accurate thermodynamic energies. Such structures and thermodynamic energies can then be utilized to obtain reliable Pourbaix diagrams that completely account for various electrochemical observations.31,32,33
Here, we develop a high-throughput ab initio approach to rapidly assess many possible structures and magnetic states of mTM compounds, using efficient DFT methods, and then accurately calculate ΔfG data for the most stable phases, using expensive high-level DFT methods to produce reliable mTM–Pourbaix diagrams. We compare our DFT Pourbaix diagrams with directly observed electrochemical, geological, and biomagnetic phenomena on mTM-based metals and compounds to comprehensively demonstrate the high accuracy of our fully ab initio scheme. In addition, we reveal many chemical trends appearing across both the ΔfG data and Pourbaix diagrams. Finally, we compute condition-dependent probability profiles for the mTM metals, compounds, and aqueous ions to further establish structure–electrochemical property relationships.
Results
Thermodynamic principles
The electronic formation energy (Δfεe) of a mTM compound (XnOmHl) is calculated as
where εe is the total electronic energy obtained from DFT, and the elemental X (=mTM), O2 molecule, and H2 molecule are the reference species. At finite temperatures, the total free energy (Gtot) of a solid or molecular gas is expressed as
where GT is the temperature-dependent component that consists of contributions from atomic vibrations (including zero-point energy), electronic excitation (exclusively in metals), and molecular rotation and translation in the diatomic O2 and H2 gases. Here, we use the standard state O2 and H2 gases at 298.15 K and 1.0 bar as the reference species, and their GT values are obtained by summing their zero-point vibrational energies (calculated herein from DFT) and free-energy drops from 0 to 298.15 K (measured in experiment35). The GT values of the mTM compounds are derived from the phonon spectra calculated from DFT.
The standard free energy of formation (ΔfG) of a mTM compound is calculated as
where the thermal correction ΔGT is obtained in a similar manner as given by Eq. (1), with the εe values therein replaced by the corresponding GT values here. The standard chemical potential of a solid (μs) equals its ΔfG value, i.e., μs = ΔfG.
For aqueous ions, the concentration-dependent chemical potential of an aqueous ion I (μI) is calculated from its standard chemical potential (μ0, at 298.15 K, 1.0 M, and 1.0 bar, pH = 0) using
where, R is the gas constant (8.314 J⋅mol−1⋅K−1), μ0 is obtained from the experimental databases, and [I] is the aqueous-ion activity that is approximated to be its concentration. In aqueous environments, the relative electrochemical stabilities between different species (e.g., metals, oxides, hydroxides, oxyhydroxides, and aqueous ions) are described by their chemical potentials of reaction (Δμ), which are calculated using the reaction paths that connect all of the considered species (see Supplementary Information, part E).
High-throughput approach
Our high-throughput DFT approach is based on the following considerations: first, efficient DFT methods can be used to quickly determine the most stable structures among all the possible structures. Second, precise but expensive DFT methods utilizing higher-level density functionals can be used to calculate accurate ΔfG values of these stable structures. Finally, the obtained ΔfG values can be used to construct Pourbaix diagrams. This simulation scheme is implemented as a high-throughput ab initio workflow, Fig. 1, consisting of five major parts described next. Details of the DFT methods utilized in this work are described in the Methods section, and include the use of the LDA, GGA (PBE and PBEsol), metaGGA (RTPSS and MS2), and hybrid (HSE06) functionals.

The workflow of the high-throughput ab initio approach utilized herein to generate accurate Pourbaix diagrams (see the section High-throughput approach for details). This workflow can also be directly used to simulate the Pourbaix diagrams of materials other than the mTM-based ones, and for nonmagnetic materials (e.g., Ti metal and oxides32), the calculations of different magnetic states are not required
The first major part is “Fast DFT Structural Screening”, which consists of five steps (boxes) described below: the boxes labeled “Structure database” and “Collect possible structures in Cr, Mn, Fe, Co, Ni compounds” in Fig. 1 show that the many possible structures for the mTM oxides, hydroxides, and oxyhydroxides are obtained from the Inorganic Crystal Structure Database34 as well as the literature.33 There are 26 different compound structures collected (Supplementary Table S1), and the cations in these structures are successively substituted by the 5 mTM elements to sample the structure-composition space, resulting in 130 (=5 × 26) structures in total.
As indicated by the “Set various initial magnetic states for each structure” box, at least three initial magnetic configurations (e.g., nonmagnetic, ferromagnetic, and antiferromagnetic) are considered to determine the ground-state magnetic structure for each phase. In some compounds (e.g., X3O4 and XOOH), the finally calculated magnetic configurations may be ferrimagnetic or another complex order, for which we still simply use FM or AFM to conveniently indicate the magnetic coupling character between the neighboring cations therein. In some complex structures (e.g., defective X3O4 and layered X2O3), there may be at least two or three inequivalent AFM configurations requiring sampling. Thus, there are ≈4 magnetic configurations for each structure on average, resulting in a total of ~520 structure-magnetism configurations to consider.
The next step in the screening scheme uses the efficient PBEsol functional first to fully optimize the lattice constants and atomic positions of all 520 structure-magnetism configurations, which are followed by further optimizations at the MS2 level (“Calculate PBEsol & MS2 energies,” Fig. 1). Thus, there are about 1,040 DFT structural optimizations in the structural screening step. At a specific chemical composition, we find that both PBEsol and MS2 give the same relative stabilities among different polymorphs (Supplementary Table S1).
The final step in the screening procedure is to “Select the most stable structures” with the lowest electronic energy for each composition. These phases are then used as inputs for additional more advanced DFT calculations to obtain higher-accuracy electronic energies and vibrational free energies. We are conservative in our down-selection process, and also include one to four metastable configurations for the next step, which further justifies that our assignment of the relative stabilities among polymorphs from PBEsol and MS2 is the same as those from the higher-level hybrid functional HSE06.
Furthermore, we note that for transition-metal systems with localized 3d orbitals, the electronic exchange potentials in conventional DFT methods (e.g., LDA and GGA) may need improvement due to the delocalization error therein.33,36,37 An efficient alternative to the computationally expensive hybrid functional with exact electronic exchange (e.g., HSE06 used here) is the so-called DFT plus Hubbard U (DFT + U) method with a static mean-field on-site Hartree–Fock approximation,36,37 which has an efficiency equivalent to that of a GGA functional and is quite useful for large-scale computation of lattice and thermodynamic energies of complex materials. The DFT + U method, however, usually requires the experimental compound free energies of formation, ΔfG values, as the energetic references to fit both the tunable parameter U and the ad hoc energetic correction to the O2 molecule,38,39,40,41 where the fitted U for a transition-metal cation may also depend on its coordination number and anion type.41
In simulating electrochemical phase diagrams, the DFT + U method additionally requires ad hoc energetic corrections to the important aqueous ions to reproduce the dissolution energies of compounds, as derived from experimental characterizations.42,43,44 Therefore, when using the DFT + U method, it will involve complicated ad hoc numerical processing, and the quality of the simulated electrochemical phase diagrams will also rely on the accuracy and availability of the experimental energies. For these reasons, we do not consider the DFT + U method here, which was frequently used in earlier simulations of Pourbaix diagrams and are only as accurate as the experimental energies they reproduce.42,43,44 This aspect makes assessment of calculated Pourbaix diagrams to direct measurements important as we show here.
The second major part “Accurate DFT Free Energies” consists of three boxes as described here. We use the most stable structures to recalculate the electronic formation energies Δfεe using HSE06 (“Calculate Δfεe using HSE06”). Phonon spectra for these phases are computed from DFT, using the efficient PBEsol functional (“Calculate ΔGvib using PBEsol”), which is sufficiently accurate to obtain reliable vibrational free energies (Gvib) and vibrational formation free energies (ΔGvib) of solids.32,45 In addition, other DFT methods (i.e., LDA, PBE, and RTPSS) are also used to calculate the functional-dependent Δfεe values (Supplementary Tables S2–S6). Accurate ΔfG values at standard conditions (298.15 K and 1.0 bar) are derived using the HSE06 Δfεe and PBEsol ΔGvib data (from Eq. (3)). The obtained ΔfG values are then assigned as the standard chemical potentials (μs) of the mTM compounds (“Calculate μs = ΔfG”). The aforementioned DFT ΔfG data appear in Supplementary Tables S7 and S8. The experimental values are given in Supplementary Tables S9–S14.
The third major part “Experimental Data of Aqueous Ions” consists of three steps. Rather than computing the chemical potentials of the aqueous ions involved in the electrochemical phase diagrams, we first “List relevant aqueous ions” and then tabulate their standard chemical potentials (μ0) by collecting them from various experimental databases (“Collect experimental standard chemical potentials (μ0)’’, Fig. 1). Based on the collected μ0 values (Supplementary Tables S9–S14), the μI values at any specified [I] are calculated using Eq. (4) (“Calculate μI at specified [I]”).
The fourth major part “Reaction Thermochemical Formula” consists of two boxes. Apart from the calculated and collected chemical potentials, the relevant formula and numerical solvers are required to model the electrochemical thermodynamics. Regarding the former aspect, we list the reaction paths that connect all of the solids and aqueous species (“List relevant reaction paths”), based on which the dependencies of the chemical potentials of reaction (Δμ) on electrode potential V (with respect to the standard hydrogen electrode, i.e., VSHE) and solution pH are formulated (“Formulate Δμ(V, pH) using Nernst equation”). All Δμ(V, pH) formula for the considered reaction paths are available in Supplementary Information (part E).
The fifth major part is the “Final Numerical Modelling” consisting of three steps. As indicated by the box of “Generate a discrete phase space spanned by V and pH”, prior to the diagram modelings, a dense descrete numerical grid (400 × 400) is used to precisely describe the complete range of phase space of interest. Here, we focus on VSHE ∈ [−2, 3] V and pH ∈ [−2, 16]. Next, the calculated μs and μI values for all of the mTM species (i.e., metals, oxides, hydroxides, oxyhydroxides, and aqueous ions) are used as inputs into the Δμ(V, pH) formula, and their relative Δμ values at each numerical grid point are calculated (“Calculate Δμ(V, pH) within the specified phase space”). From this assessment at each grid point, the most stable chemical form is identified. Last, a Pourbaix diagram is generated after scanning the complete numerical grid.
Although only mTM-based materials are studied here, this high-throughput ab initio approach (as depicted in Fig. 1) can be directly applied to simulate the ab initio Pourbaix diagrams of many materials other than the mTM-based compounds. In addition, for many nonmagnetic materials, (e.g., Ti metal and oxides32), it is unnecessary to perform the complex magnetic-state screening step required in this work.
Data presentation
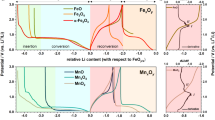
The calculated electronic formation energies (Δfεe) and free energies of formation at the standard condition (ΔfG at 298.15 K and 1.0 bar) for the mTM oxides, hydroxides, and oxyhydroxides are shown in Fig. 2. The ΔfG values per atom (Fig. 2b) indicate the relative stabilities among the component compounds in a sample with a fixed global composition. The ΔfG values per cation (Fig. 2c) can be used to derive the relative stabilities among the compounds when in contact with a reactive environment, e.g., an O2 (and/or H2) atmosphere or an aqueous solution. Indeed, the DFT-calculated ΔfG values per cation are used in the simulation of the mTM–Pourbaix diagrams (Fig. 3), which are also compared with the diagrams simulated using the experimental (Expt) ΔfG values.

The DFT–HSE06-calculated (a) electronic formation energies (\(\Delta _f\varepsilon _e\)) and (b, c) free energies of formation (\(\Delta _fG\) per atom and cation) for mTM compounds (X = Cr, Mn, Fe, Co, and Ni) at the standard condition. The nominal valences of the mTM cations are indicated on the upper axis of panel (a)

The experimental (Expt) Pourbaix diagrams (\([I] = 10^{ - 6}\) M) and DFT Pourbaix diagrams (\([I] = 10^{ - 6}\) and 10−2 M). The two inclined parallel blue dashed lines indicate the electrode potentials for the oxidation (\(2{\mathrm{H}}_2{\mathrm{O}} - 4e^ - \to {\mathrm{O}}_2 + 4{\mathrm{H}}^ +\), upper) and reduction (\(2{\mathrm{H}}_2{\mathrm{O}} + 2e^ - \to {\mathrm{H}}_2 + 2{\mathrm{OH}}^ -\), lower) of water. The phase domains for some secondary phases (e.g., Cr2O3, Mn2O3, Fe2O3, FeO, Co(OH)2, and Ni(OH)2 as labeled in brackets) are also indicated by their red dotted boundaries
In addition, we will use the mTM compound and aqueous-ion ΔfG chemical trends (Figs 2c, 4) to explain the chemical trends in the Pourbaix diagrams (Fig. 3). Detailed DFT and Expt ΔfG data can be found in Supplementary Information (part A–D). We also consider any possible precipitation of metastable phases at various electrochemical conditions by analyzing probability profiles for all of the mTM species at variable pH and VSHE (Fig. 5). These data are also useful for understanding the synthesis, characterization, and application of the related materials in aqueous environments.


Probability profiles for the mTM compounds and species with respect to (left column) pH (\(V_{{\mathrm{SHE}}} = 0\) V) and (right column) \(V_{{\mathrm{SHE}}}\) (pH = 7)
Thermodynamic energies
The Δfεe and ΔfG per atom data can indicate the intrinsic stabilities of materials, and be readily explained using microscopic electronic structure-based models. They are also widely used to describe the thermodynamic stabilities of numerous alloys.46,47 The observable difference of 0.1 –0.3 eV per atom between any Δfεe (Fig. 2a) and its counterpart ΔfG (Fig. 2b) indicates the considerable destabilizing thermal effects. Such thermal effects, however, do not alter any chemical trends, i.e., the relative stabilities among different compositions of a mTM or between different mTM compounds at the same O (and H) compositions. Nonetheless, inclusion of the thermal effects is required for the precise simulation of thermodynamic and electrochemical phase diagrams.
Figure 2a, b shows that Δfεe (and ΔfG) generally increase with increasing the number of 3d electrons (n3d: Cr < Mn < Fe < Co < Ni). MnO and Mn(OH)2, however, are an exception to this trend; these two compounds are lower in energy than CrO and Cr(OH)2, respectively. We ascribe the general decrease in compound stability to the variation of the 3d-orbital physics47,48,49 as follows: first, the 3d orbitals become more localized and lower in energy with increasing nuclear charge; therefore, both the intra-atomic orbital hybridization and interatomic electron transfer become less energetically favorable, resulting in the decreased strength of the covalent–ionic mTM–O bonds,33 i.e., decreased stability. Second, the 3d orbitals are close to half filling for Cr and Mn. The further addition of electrons will lead to the pairing of spins to form spin singlets, which also makes the mTM atoms less sensitive to changes in bonding.
Interestingly, MnO and Mn(OH)2 (Fig. 2b), as well as the aqueous Mn2+ ion (Fig. 4d) have unexpectedly low ΔfG values, which we again ascribe to the orbital character of the Mn(II) cation. For an atom with half-filled d orbitals (i.e., with the largest number of unpaired 3d electrons), the electronic structure is especially sensitive to the coordination environment, leading to coordination flexibility and low stability. This is further supported by another abnormal behavior of elemental Mn: it exhibits a distorted BCC* structure (α phase34), which is not anticipated from the structural trend (HCP–BCC–HCP–FCC) for the TM elements in the same 3d row.47,49
Figure 2a, b also shows that upon going from Cr to Ni, the nominal cation valence of the most stable oxide decreases from +3 (Cr2O3), to +2.67 (Fe3O4 and Mn3O4) and +2.0 (CoO and NiO). We attribute this behavior to the increased mTM electronegativity, i.e., the less-favored interatomic electron transfer mentioned above. The relative stability of the mTM oxyhydroxides (XOOH) with respect to hydroxides (X(OH)2) also decreases from Cr to Ni, due to the same increased mTM electronegativity.
The aforementioned chemical trends for mTM compounds can help to predict and understand various thermodynamic and electrochemical phenomena, and provide much insights for the design, synthesis, and application of related materials. To fully quantitatively understand the electronic mechanisms underlying these chemical trends, an in-depth and comprehensive investigation into the orbital properties (e.g., energy level, occupation, hybridization, and bonding) of the TM atoms in the various coordination environments is required. We do not address these issues further, but rather focus on compound stability in aqueous environments in the form of electrochemical phase diagrams.
In realistic situations, the compounds are always in contact with a reactive environment, e.g., in an O2 (plus H2) atmosphere. Under such conditions, the oxidation of a metal (X) is described by the reaction path
whereas in an aqueous solution, the dissolution of a compound is described by
In this later scenario, the ΔfG values are specified per cation, Fig. 2c, rather than per atom to assess the relative stabilities among all of the involved mTM species (metal, compounds, and aqueous ions). Then the ΔfG values per cation, electrode potential, and solution pH should be simultaneously considered to clearly understand the Pourbaix diagrams presented in the following section.
Pourbaix diagrams: general trends
The mTM–Pourbaix diagrams at a moderate [I] of 10−6 mol/L (10−6 M) simulated using Expt and DFT ΔfG values are compared in Fig. 3 (left and center columns). In electrochemical experiments, [I] is usually not controlled and characterized; thus, the DFT Pourbaix diagrams at [I] = 10−2 M are also provided in Fig. 3 (right column). The Pourbaix diagrams at [I] = 10−2 M are also useful for the experimental synthesis of related compounds through solution precipitation, where a relatively high [I] should be required (DFT Pourbaix diagrams constructed for a broader range of [I], from 10−8 to 10−2 M, are also provided in Supplementary Fig. S2). In addition, some compounds such as Cr2O3, Mn2O3, Fe2O3, FeO, Cr(OH)2, and Ni(OH)2 with secondary electrochemical stabilities find importance in numerous realistic applications; for that reason, we also calculate their phase domains by excluding the more stable phases in the DFT Pourbaix diagrams found in the center and right panels of Fig. 3.
In a Pourbaix diagram, the domains consisting of the metal, its compounds, and aqueous ions are called immunity, passivation, and corrosion domains,30 respectively. We find in the DFT Pourbaix diagrams that the relative stabilities (i.e., phase domains) of the passivating compounds increase with increasing [I] in Fig. 3a–e (center and right columns), due to the decreased stabilities of the aqueous ions (by Eq. (4)). The critical pH value at the left (right) passivation-domain boundary generally decreases by ~2 (increases by ~4) with increasing [I] by 104 times. The phase areas at low electrode potentials (VSHE) are always occupied by mTM metals, and the mTM compounds and aqueous ions with more oxidized mTM are stabilized with increasing VSHE. For example, there is a phase transition of Co → CoO/Co(OH)2 → Co3O4 → CoOOH → CoO2 (Fig. 3d). This electrochemical trend is ascribed to the behavior of the positively (negatively) charged electrode, which extracts electrons from (introduces electrons into) the materials, making them more oxidized (reduced).
In the Pourbaix diagrams of Cr, Mn, and Fe (Fig. 3a–c), the high-VSHE areas are occupied by complex aqueous ions with highly oxidized mTM (e.g., [XO4]2−). However, these aqueous ions are absent in the Pourbaix diagrams of Co and Ni (Fig. 3d, e), because the data for [CoO4]2− and [NiO4]2− ions are unavailable and thus are not considered in our simulations. This lack of data could be related to the decreased aqueous-ion stabilities upon going from Cr to Ni (Supplementary Table S14), making it challenging or even impossible to detect the [CoO4]2− and [NiO4]2− ions at high voltages in electrochemical experiments. Nonetheless, the most common aqueous ions (i.e., X2+ and X3+) can be used to understand the energetic trend for aqueous ions from Cr to Ni. Figure 4d depicts their ΔfG values, revealing an obvious general increasing trend, except for the abnormal dip at Mn2+. This energetic trend for aqueous ions and the special behavior of Mn2+ are similar to those described above for the solid-state compounds and thus are likely governed by the same electronic mechanisms.
Upon moving from Fe to Co, and Ni, the electrochemical phase domains of XO and X(OH)2 expand toward higher VSHE (Fig. 3c–e). This trend occurs because the stabilities of other compounds (e.g., X2O3 and XOOH) decrease much faster than those of XO and X(OH)2 (Fig. 4a–c). The appearance of MnO and Mn(OH)2 in the Mn Pourbaix diagrams (Fig. 3b) arises from their unexpected low ΔfG values (see Figs 2a, 4a). In the DFT mTM–Pourbaix diagrams, the nominal cation charge of the most favored mTM compound at VSHE ~ 0 generally increases, e.g., from CrOOH to Mn3O4, Fe3O4, CoO, and NiO, due to the different destabilizing rates for the compounds with different cation charges (Fig. 4a–c).
In the following sections, we demonstrate the advancement of our high-throughput ab initio method in simulating accurate mTM–Pourbaix diagrams ranging from Cr to Ni, which in some cases have been lacking for over 50 years. Assessments are made in detail with various electrochemical phenomena directly observed in recent decades. An explicit comparison of the improvement enabled by our approach is provided for the Ni Pourbaix diagram, which is shown together with many electrochemical observation results in Supplementary Fig. S3. As described earlier, the mTM-based materials have been widely used in numerous fields (e.g., structural materials, catalysts, electrode materials, and electronic devices), where the materials always closely contact with different aqueous environments during their synthesis and exploitation. Thus, precisely knowing their electrochemical phase stabilities can be highly helpful for designing materials, optimizing the synthesis and application conditions, and controlling material phases and properties.
Cr Pourbaix diagrams
The experimental (Expt) and our DFT Cr Pourbaix diagrams exhibit quite similar phase domains for Cr2O3 (Fig. 3a, left and center panels), owing to the closeness in the free energies of formation (Fig. 4c). This good theory–experiment agreement is ascribed to the high thermodynamic stability (large ΔfG) of Cr2O3, which serves to suppress defect generation during the combustion process in thermodynamic experiments used for estimating ΔfG (as discussed in the Introduction). Thus, the contaminating effect of defects is largely minimized in the experimental ΔfG of Cr2O3.
Cr2O3 is a ubiquitous oxide readily formed on various alloys, e.g., steels,50 under atmospheric conditions. However, it is well known that Cr2O3 never forms on Cr in aqueous solutions, but its hydrous counterparts (CrOOH and Cr(OH)3) appear as the passivating compounds, as detected in several electrochemical experiments.51,52,53 Cr(OH)3 is a highly hydrated material consisting of molecular Cr(OH)3 units, and its structure still has not been well characterized. For these reasons, we do not considered it explicitly in Fig. 3a. The reason Cr(OH)3 will form initially on Cr metal in solutions is probably due to its higher kinetic activity, whereas CrOOH will gradually grow beneath the outer Cr(OH)3 layer,53 indicating the higher electrochemical stability of CrOOH. Therefore, CrOOH should be the stable phase in aqueous solutions, and both Cr2O3 and Cr(OH)3 should appear as metastable phases. This assessment is consistent with our DFT Cr Pourbaix diagrams (Fig. 3a, center and right panels), whereas the experimental Cr Pourbaix diagram reveals a much smaller phase domain over which Cr2O3 is stable (Fig. 3a, left panel).
CrOOH precipitates are widely observed in solutions at pH ≳ 3,53,54,55,56,57 which is consistent with the boundary between CrOOH and Cr3+ at pH 2 ~ 3.5 and VSHE ~ 0 V in the DFT diagrams (Fig. 3a, center and right panels). The hydrous Cr2O3 (e.g., CrOOH) formed in aqueous solutions transforms into the anhydrous Cr2O3 only upon heating at temperatures ≳700 K.55,57 In addition, the formation of anhydrous Cr2O3 (not CrOOH) underneath an outer Cr(OH)3 layer has been observed on 254 MO stainless steels (Fe–20%, Cr–18%, and Ni–6% Mo, in wt.%).58 These corrosion processes likely originate from the kinetic and/or thermodynamic effects of the other alloying elements on the electrochemical stabilities of Cr2O3 and CrOOH. Such interactions still require further detailed experimental and theoretical investigations to understand the microscopic mechanisms governing the appearance of these phases.
Mn Pourbaix diagrams
Mn oxides (e.g., Mn3O4, Mn2O3, and MnO2) have promising application in water electrolysis, due to their favorable catalytic reactivity under electrochemical conditions, and these oxides always coexist in experimentally synthesized samples.59,60,61,62,63 This is well explained by the calculated ΔfG values per cation (Fig. 2c), and their small differences indicate the similar thermochemical stabilities of these oxides in contact with a dry/aqueous environment. However, Mn (hydr)oxides present serious adverse dissolution problems in aqueous environments,20 which should be due to their low electrochemical stabilities, as indicated by their relatively small or absent phase domains in both the experimental and DFT Mn Pourbaix diagrams (Fig. 3b). In contrast, the layered MnO2 compound is readily stabilized by intercalation with the alkaline cations (e.g., Li, K, and Ca) from aqueous solutions.59,60,64
An early experiment using X-ray diffraction65 observed in solutions with [I] ≲ 10−2 M that (1) at T ~ 298 K, only Mn3O4 is present at pH 8.5–9; (2) at T ≳ 310 K, Mn3O4 appears at pH down to 7; (3) at T ∈ (283, 290) K, Mn3O4 and MnOOH coexist at pH 8.0–8.5; and (4) at T ~ 273.6 K, only MnOOH occurs at pH 8.5~9.0. These observations were later confirmed using multiple probes, including high-resolution X-ray diffraction, Raman spectroscopy, and X-ray photoelectron spectroscopy.66 In those experiments, MnOOH or/and Mn(OH)2 was found to initially precipitate and then transform into Mn3O4 in solutions with [I] ≲ 10−4 M and pH 10. It should be noted that precisely distinguishing MnOOH from Mn(OH)2 in experiment is nontrivial, due to their similar layered structures and the uncontrollable degree of hydrogenation.33 The initial formation of MnOOH/Mn(OH)2 is ascribed to the high kinetic activity that may be due their layered structures—the oxidation of metals in solutions always initiates with the adsorption of OH radicals.67,68,69,70
We can conclude from these electrochemical observations that MnOOH/Mn(OH)2 is an intermediate precipitate, and it will spontaneously convert into more stable Mn3O4 at temperatures ≳283 K, while this kinetic transition will be deactivated at lower temperatures (<283 K).65 Indeed, the Mn3O4 domain appears as a major field in the DFT Mn Pourbaix diagrams (Fig. 3b, center and right panels). Furthermore, Mn3O4 is observed to be stable in solutions with pH ≳ 8.5 and [I] 10−2–10−4 M,65,66 which also strongly supports the DFT-determined phase boundary at pH 8.5~9.5 (VSHE ~ 0 V). In addition, an operando X-ray absorption near-edge structure (XANES) spectroscopy63 has recently been performed and observed the oxidation of Mn3O4 at 0.2–0.4 V in a solution with pH = 14. This result is also consistent with the upper phase boundary of Mn3O4 at 0.3–0.4 V in the DFT-simulated Mn Pourbaix diagrams.
In the DFT Mn Pourbaix diagrams, Mn(OH)2 only has a negligibly small phase domain at pH ~ 11.5 when [I] is as high as 10−2 M, and MnOOH is totally absent. These aspects are consistent with the experimentally observed intermediate roles played by Mn(OH)2 and MnOOH. According to Pourbaix diagrams obtained using experimental free energies of formation, however, stable Mn(OH)2 is expected to persist at pH values >8.7 in a solution with [I] ≲ 10−2 M. In addition, MnOOH also presents an observable region of phase stability at low anodic potentials ([I] = 10−6 M, Fig. 3b, left panel). At a phase boundary, there is an inevitable thermodynamic blurring (δpH ~ 1 at room temperature, discussed later in the section Probability analysis) due to the nonzero probability for a metastable phase at a finite temperature. Thus, Mn(OH)2 precipitates are expected to appear at a pH lower than the phase boundary in the experimental Pourbaix diagram (at pH < 8.7). These derived phenomena from the experimental Pourbaix diagrams are clearly inconsistent with the above experimental observations.
Fe Pourbaix diagrams
The most frequently observed Fe oxides are Fe3O4 and Fe2O3, owing to their close thermodynamic stabilities (Fig. 2c) and the fact that the less stable FeO phase is only possible under highly reducing conditions, e.g., high H2 concentration.50,71 The major discrepancy between the experimental and DFT Fe Pourbaix diagrams appears as a difference in the relative stabilities between Fe3O4 (magnetite) and Fe2O3 (hematite) as shown in Fig. 3c (left and center panels). In addition, FeO is only observable in the DFT diagrams (Fig. 3c, center and right panels).
Fe3O4 forms rapidly in various electrochemical experiments; however, any direct formation of Fe2O3 has not been observed in the Fe3O4 products.72,73,74 Fe2O3 is only obtained by further oxidizing Fe3O4 using additional aeration plus heating.72,74 In aqueous solutions, however, the formation of Fe2O3 may be facilitated by other alloying elements or related oxides (e.g., stainless steel with Cr, Ni, and Mo58). It is the small difference in the ΔfG values of these phases (Fig. 2c) that results in their readily variable relative stabilities in different environments. These electrochemical observations are more consistent with the DFT Fe Pourbaix diagrams (Fig. 3c, center and right panels), where Fe3O4 is more stable than Fe2O3 at VSHE ~ 0 V. Furthermore, solid Fe3O4 is reported to be stable at pH ≳ 3.5,72,73,75,76 which is also in good agreement with its dissolution boundary at pH 4.5–3.0 in the DFT diagrams. In contrast, the experimental Pourbaix diagrams show that the dissolution boundary for the stable oxide (Fe2O3) resides at pH 6.5–5.0, which is inconsistent with the observed electrochemical boundary.
We also note that although Fe2O3 is more stable in the experimental diagram (Fig. 3c, left panel), it has a phase domain that is quite close to that of the metastable Fe2O3 in the DFT diagram (Fig. 3c, center panel, dotted lines). This similarity indicates that the experimental ΔfG of Fe2O3 is likely quite accurate, while that of Fe3O4 is somewhat underestimated. The inaccurate experimental ΔfG of Fe3O4 may be ascribed to the uncontrollable defect concentration in defective Fe3O4 spinel samples during combustion heat measurements.
Furthermore, regarding the relative electrochemical stabilities between Fe3O4 and Fe2O3, available geological and biomagnetic evidences also strongly support our DFT results. During the long-term geological evolution of ultramafic rocks driven by aqueous fluid, magnetite (Fe3O4) rather than hematite (Fe2O3) minerals formed from primary olivine minerals.77 It should be the electrochemically stable Fe3O4 in iron ore (i.e., lodestone, a magnetite mineral) that is pervasive. Indeed, magnetite nanocrystals have also been widely found in magnetotactic bacteria78,79 and under the upper-beak skin of homing pigeons.80 These evidences clearly indicate the higher electrochemical stability of Fe3O4 than that of Fe2O3, as revealed by our DFT Fe Pourbaix diagrams. Our assessment here motivates additional measurements to quantify the experimental accuracy of ΔfG(Fe3O4).
During the electrochemical oxidation of pure Fe with increasing electrode potential (scan rate 0.04 V/s) in solutions with pH = 14,69,70,75 the reaction starts with the adsorption of OH on Fe surface at VSHE ~ −0.9 V. Next, the formation of FeO (and/or Fe(OH)2) occurs at ≈−0.7 V and the further oxidation of the outer (inner) FeO layer into Fe3O4 and/or Fe2O3 occurs at ≈−0.5 V (≈−0.2 V). From our DFT Pourbaix diagrams for Fe (Fig. 3c, center and right panels), we find that the Fe–FeO boundary at pH 14 resides at VSHE ~ −1.04 V, which is a little lower than the observed initial-oxidation potential of ~−0.7 V. This small difference (by ≈0.3 V) in oxidation potential is reasonable, because any effective kinetic factor will tend to slow the oxidation process in experiment. The different oxidation potentials for the inner and outer FeO layers themselves are indicative of a non-thermodynamic factor at play. In addition, the DFT FeO–Fe3O4 and FeO–Fe2O3 boundaries (at pH 14) reside at −0.99 and −0.5 V, respectively, which are reasonably lower than the observed oxidation potentials of FeO at −0.2 ~ −0.5 V. In these early voltammetric experiments, it was either not possible or exceedingly difficult to distinguish between FeO and Fe(OH)2 (Fe3O4 and Fe2O3). For this reason, we suggest that additional in situ characterization of the samples be performed, e.g., using X-ray diffraction (XRD), X-ray photoelectron spectroscopy (XPS), or Raman spectroscopy, to better differentiate the phases present.
In addition to the consistency between our DFT Fe Pourbaix diagrams and various electrochemical/geological observations, the knowledge gained from our DFT diagrams can also help correctly understand the oxide formation on Fe samples with corrosion-resistant polymer coatings.81,82 The Bragg peaks observed in the XRD patterns on such corroded samples were ascribed to the Fe substrate, Fe2O3, and FeOOH;82 however, our analysis indicates that Fe3O4 should form as the stable oxide under electrochemical conditions. To assess the interpretation in refs. 81,82, we examined the XRD patterns for standard Fe, Fe2O3, Fe3O4, and FeOOH samples83,84,85 in detail and found that for the corroded Fe samples with polymer coatings,82 the XRD peaks, can be ascribed solely to Fe and Fe3O4. When the polymer composition changes,81 however, XRD peaks consistent with the formation of FeOOH appear. The FeOOH phase that appears may be retained after the adsorption of OH (described above). It remains unknown whether there exist any additional chemical or voltaic-cell effects at the Fe/Fe3O4–polymer interface that alter the stability of Fe compounds (e.g., FeOOH). Therefore, our DFT Fe Pourbaix diagrams are useful for reinterpreting experimental results and should motivate further experimental and theoretical studies on the corrosion mechanisms for Fe-based materials.
Co Pourbaix diagrams
Based on various measurements using cyclic voltammetry, ellipsometry, X-ray photoelectron spectroscopy, and Mössbauer spectroscopy on the oxidation of Co metal in solutions with pH 10 –14.4,68,86,87,88,89 it is known that (1) Co(OH)2 initially forms on a Co surface; (2) CoO may grow underneath Co(OH)2, resulting in a sandwich heterostructure of Co/CoO/Co(OH)2; and (3) CoO and Co(OH)2 will transform into Co3O4 and/or CoOOH at VSHE at ≳0.3 V. Given these experimental observations, we now assess the experimental and DFT-simulated Co Pourbaix diagrams.
In the DFT Co Pourbaix diagrams (Fig. 3d, center and right panels), CoO and Co(OH)2 are stable against dissolution into Co2+ in alkaline solutions, which is consistent with their formations on Co surface at pH ≳ 10 in experiment. In the experimental Co Pourbaix diagram, however, the phase domain of CoO (and Co(OH)2) is significantly undestimated to be within pH ∈ (10.3, 11.0), which is inconsistent with the observed passivation domain at pH values >~10. In the observed Co/CoO/Co(OH)2 sandwich structure, the initial formation of Co(OH)2 should be ascribed to the higher kinetic activity for its formation, and the later growth of CoO underneath Co(OH)2 indicates that CoO is thermodynamically more stable than Co(OH)2 in solution. These conclusions strongly support our DFT electrochemical results, where Co(OH)2 is a metastable phase relative to CoO. According to the DFT Co Pourbaix diagrams, a VSHE ≳ −0.1 V is required to activate the CoO–Co3O4/CoOOH transition at pH ≈ 14, which is close to the experimental value (≳0.3 V). The deviation of ~0.4 V is reasonable when considering the possible influence of kinetic effects.
In a recent experiment,90 in situ Raman spectroscopy was used to characterize a Co electrode with deposited Co3O4, which was then immersed into a solution at pH ~ 13. Here, it was found that Co3O4 coexists with CoOOH at an initial 0.16 V, and a Co3O4–CoOOH transition occurred upon increasing the anodic potential. This experimental observation is also consistent with the boundary at VSHE ~ 0 V in our DFT Co Pourbaix diagrams. In another experimental measurement using extended X-ray absorption fine-structure (EXAFS) spectroscopy,91 CoOOH was observed to be stable at VSHE ≈ 0.75 V in a solution at pH 7, which is consistent with the stability of CoOOH at >0.3 V in the DFT Pourbaix diagram.
In Yeo’s measurement,90 there is no evidence of Co(IV) (e.g., CoO2) up to 0.86 V. In an earlier measurement, however, using Mössbauer spectroscopy,92 stable CoO2 (with intercalated Fe) was observed at VSHE ≳ 1.1 V and pH 8.5, which was further confirmed by Kanan’s EXAFS measurement,91 i.e., a stable Co(IV) state exists at VSHE ≳ 1.25 V and pH 7. These measured VSHE values for stable CoO2 are obviously lower than the DFT ones by about 1.0 V, which may be due to the stabilizing effects of some aqueous ions (e.g., Fe and Na) that can readily intercalate into the layered CoO2 structure. To that end, the effects of electrolyte composition and possible intercalation for CoO2 are interesting topics for future experimental and theoretical studies.
Ni Pourbaix diagrams
NiO and Ni(OH)2 are especially important for Ni-based corrosion-resistant alloys, electrodes, and catalysts, and there are numerous experimental observations reported that can be used to assess the DFT Ni Pourbaix diagrams presented here. In the DFT Ni Pourbaix diagrams (Fig. 3e, center and right panels), NiO exhibits a slightly larger phase domain than the metastable Ni(OH)2, which explains their ubiquitous experimentally observed coexistence in solutions.31,67,93,94,95,96 Similar to the situation described above for Co, initially, Ni surfaces will be passivated by Ni(OH)2, likely due to the higher kinetic activity for its formation, which is followed by the growth of NiO underneath,94,95,96 indicating the higher thermodynamic stability of NiO as our DFT results show. In addition, NiO and/or Ni(OH)2 are stable experimentally against dissolution at pH ≳ 4,31,67,93,94,95,96,97,98,99,100,101,102,103,104,105,106 which is consistent with their dissolution boundaries in the DFT Ni Pourbaix diagrams at pH 3–5 and 4–6, respectively. However, in the experimental Ni Pourbaix diagram (Fig. 3e, left panel), their phase stabilities are highly underestimated, with Ni(OH)2 and NiO only stable over the pH range from 9 to 12.
In various alkaline solutions at pH 13–15, the Ni(OH)2–NiOOH transition is observed at VSHE 0.5~1.0 V, which is highly consistent with their phase boundary at ~0.75 V in the DFT Ni Pourbaix diagrams. In contrast, the experimental Ni Pourbaix diagram (Fig. 3e, left panel), shows that both Ni(OH)2 and NiOOH are unstable at pH of ~14, and the upper boundary of Ni(OH)2 resides only at ≲0.3 V.
Probability analysis
To better reveal more of the electrochemical subtleties of the mTMs, their compounds, and aqueous ions, we calculate probabilities with respect to VSHE and pH as31
where kB is Boltzmann constant, i and j index the species, and Δμ depends on VSHE, pH, and [I] (fixed at 10−2 M here). Two types of electrochemical conditions are considered here: first variable pH at fixed VSHE (=0 V) and second, variable VSHE at fixed pH (=7) as shown in Fig. 5. The calculated probabilities for the Cr, Mn, Fe, Co, and Ni species within these two conditions at values ⩾10−6 would indicate possible precipitation of the metastable species. It should be noted that a decrease in probability by one order of magnitude corresponds to an increase in Δμ by about 0.06 eV. Two observations from these probability profiles can be discerned that may be important for the mTM compounds under various electrochemical conditions: phase-boundary blurring and coexistence of multiple (stable and metastable) phases.
First, at finite temperatures (e.g., 298.15 K), the probability P of a stable phase exponentially decreases from 1 down to 0 upon traversing a phase boundary from one stable domain to another. Rather than an abrupt transition, finite temperature effects result in a diffuse crossover or “thermodynamic blurring” of the phase boundary (Fig. 5). This indicates that a detectable precipitation of a metal/compound may occur at an electrochemical condition beyond its domain of stability. If P = 1% is used as an approximate cutoff criterion, then the mTM species generally exhibit the aforementioned phase-boundary blurring effects (δ) of the order δpH ≈ 1 and δVSHE ≈ 0.1 V. The thermodynamic blurring in VSHE is much smaller, because the electrode potential more significantly affects the reaction thermodynamics, especially when species with different cation-charge states are involved. CoO, Co3O4, and CoOOH have exceptionally large δpH values (≳2), because they are quite close in electrochemical stability (Fig. 4a–c), resulting in their comparable probabilities within a relatively large pH range (Fig. 5g).
Second, within a phase domain or at a phase boundary, many secondary metastable phases with observable probabilities can be found (Fig. 5). Here, we list the domains with significant phase competition: (1) Cr2O3 in the CrOOH domain; (2) MnO, Mn2O3, and MnO2 in the domain (and at the domain boundaries) of Mn3O4; (3) FeO, Fe(OH)2, and Fe2O3 in the domain (and at the domain boundaries) of Fe3O4; (4) Co(OH)2 and Co3O4 in the domains (and at the domain boundaries) of CoO and CoOOH; (5) Ni(OH)2 in the NiO domain; (6) Ni3O4 and Ni2O3 at the NiO–NiOOH boundary.
Discussion
In summary, we proposed a first-principles high-throughput method to calculate reliable Pourbaix diagrams, which have largely motivated us to understand various electrochemical behaviors of mTMs (Cr, Mn, Fe, Co, and Ni) and their complex compounds (e.g., oxides, hydroxides, and oxyhydroxides). In our approach, a large number of structure-magnetism configurations are screened using efficient DFT methods, and precise ΔfG values are calculated using high-level DFT methods for down-selected configurations. Many chemical trends in the calculated ΔfG values and Pourbaix diagrams were also uncovered. Last, we calculated the probability profiles for mTM metals, their compounds, and aqueous ions at different conditions, which has advanced our understanding of electrochemical transformations.
The high accuracy of our DFT Pourbaix diagrams was comprehensively supported by various electrochemical phenomena observed over the past several decades, which justifies the scheme implemented and motivates a careful reassessment of some experimental energies used in the construction of electrochemical phase diagrams. We anticipate the wide applications of these DFT Pourbaix diagrams in the design, characterization, synthesis, and application of various mTM-based materials in many future technological fields. Furthermore, there may be many factors other than the bulk thermodynamics determining the formations of mTM compounds in realistic (complex) situations, e.g., kinetic activity, hydration, size effects, and substrate-induced phenomena. We anticipate that the information obtained from our ab initio Pourbaix diagrams and these thermodynamic probability profiles will be especially helpful to qualitatively and quantitatively determine the influence of such additional factors in the future.
Methods
There exist various density functionals to approximate the exact electronic exchange-correlation interaction, and according to Jacob’s ladder for DFT,107 they can be categorized into four tiers: (1) local-density approximations (LDA), (2) generalized-gradient approximations (GGA), (3) metaGGA functionals, which additionally include electronic kinetic energy to improve the description of nonlocal electronic exchange, and (4) hybrid functionals, which partially incorporate the exact nonlocal electronic exchange. The rank-order increasing accuracy of these DFT methods is LDA, GGA, and metaGGA, followed by hybrids at the expense of computational efficiency in a decreasing rank-order LDA, GGA, metaGGA, and hybrids.
The increased accuracy from the semilocal LDA and GGA functionals to the nonlocal metaGGA and hybrid functionals is mainly ascribed to the better approximated exchange potentials, e.g., the enhanced nonlocal nature and remedied self-interaction error.33,108,109,110,111,112,113,114 To simultaneously exploit both the accuracy and efficiency of DFT, different approximations for the electronic exchange and correlation to DFT can be used to solve different aspects of these complex problems. In addition, by comparing the results from different DFT methods, it is possible to build insight into both the performance of these methods and the underlying electronic interactions in a material.
The DFT calculations for the structures and electronic energies of magnetic transition metals and their compounds are performed using the VASP code,115 where projector-augmented wave (PAW) pseudopotentials116 are used to describe the electronic wavefunctions and potentials. In the PAW pseudopotentials for the mTM-based materials with localized 3d orbitals, we include the aspherical (ASPH) gradient corrections to the PAW spheres to reduce the large energetic errors that may occur (e.g., δ ≲ 1.0 eV/f.u. in NiO). The cutoff energy for the plane-wave expansion is 600 eV. The reciprocal k grid is \(\gtrsim{\frac{20}{a_0}} \times {\frac{20}{b_0}} \times {\frac{20}{c_0}}\) for the mTM compounds (a0, b0, and c0 are the lattice constants scaled by unit of angstrom) and 12 × 12 × 12 for the mTMs. Phonon spectra are calculated using the PHONOPY code,117 where the small-displacement method118 is implemented. The energy and atomic-force convergence thresholds for the self-consistent DFT calculations are 10−7 eV and 10−3 eV/Å, respectively.
In this work, the LDA functional,119,120 GGA functionals (PBE121 and PBEsol122), metaGGA functionals (RTPSS123 and MS2109), and hybrid functional (HSE06113) are considered. In HSE06, 25% PBE electronic exchange is replaced by a screened nonlocal Fock exchange (screening length ~10 Å). Due to the two-body electronic exchange potential in HSE06, two sets of reciprocal grids (e.g., k and k* grids) are required in the calculations, and the second k* grid is set to be approximately \(\frac{{10}}{{a_0}} \times \frac{{10}}{{b_0}} \times \frac{{10}}{{c_0}}\).
Data availability
The authors declare that the data supporting the findings of this study are available within the paper and its Supplementary Information files. The simulation output files are available upon reasonable request. They are not publicly available due to the very large file sizes. Parameters of the input files are described in the computational methods. Atomic structures for the simulation cells are available upon request.
Code availability
The Vienna Ab Initio Simulation Package (VASP) is a proprietary software available for purchase at https://www.vasp.at/. Data processing scripts written to process output files and create figures are available upon request.
References
Hoar, T. P., Mears, D. C. & Evans, U. R. Corrosion-resistant alloys in chloride solutions: materials for surgical implants. Proc. R. Soc. Lond. Ser. A. Math. Phys. Sci. 294, 486–510 (1966).
Pourbaix, M. Electrochemical corrosion of metallic biomaterials. Biomaterials 5, 122–134 (1984).
Jacobs, J. J., Gilbert, J. L. & Urban, R. M. Current concepts review–corrosion of metal orthopaedic implants. J. Bone Jt. Surg. 80, 268–282 (1998).
Eliaz, N., Shemesh, G. & Latanision, R. M. Hot corrosion in gas turbine components. Eng. Fail. Anal. 9, 31–43 (2002).
Padture, N. P., Gell, M. & Jordan, E. H. Thermal barrier coatings for gas-turbine engine applications. Science 296, 280–284 (2002).
Clarke, D. R. & Levi, C. G. Materials design for the next generation thermal barrier coatings. Ann. Rev. Mater. Res. 33, 383–417 (2003).
Zinkle, S. J. & Snead, L. L. Design ingradiation resistance in materials for fusion energy. Annu. Rev. Mater. Res. 44, 241–267 (2014).
Rodriguez, D. & Chidambaram, D. Accelerated estimation of corrosion rate in supercritical and ultra-supercritical water. npj Mater. Degrad. 1, 14 (2017).
Carranza, R. M. & Rodríguez, M. A. Crevice corrosion of nickel-based alloys considered as engineering barriers of geological repositories. npj Mater. Degrad. 1, 9 (2017).
Yeh, J.-W. et al. Nanostructured high-entropy alloys with multiple principal elements: novel alloy design concepts and outcomes. Adv. Eng. Mater. 6, 299–303 (2004).
Zhang, Y. et al. Microstructures and properties of high-entropy alloys. Prog. Mater. Sci. 61, 1–93 (2014a).
Li, Z. & Raabe, D. Strong and ductile non-equiatomic high-entropy alloys: design, processing, microstructure, and mechanical properties. JOM 69, 2099–2106 (2017).
Qiu, Y., Thomas, S., Gibson, M. A., Fraser, H. L. & Birbilis, N. Corrosion of high entropy alloys. npj Mater. Degrad. 1, 15 (2017).
Shi, Y., Yang, B. & Liaw, P. K. Corrosion-resistant high-entropy alloys: a review. Metals 7, 43 (2017).
Sivula, K., Le Formal, F. & Grätzel, M. Solar water splitting: progress using hematite (α-Fe2O3) photoelectrodes. ChemSusChem. 4, (432–449 (2011).
Lin, H., Zhang, Y., Wang, G. & Li, J.-B. Cobalt-based layered double hydroxides as oxygen evolving electrocatalysts in neutral electrolyte. Front. Mater. Sci. 6, 142–148 (2012).
Subbaraman, R. et al. Trends in activity for the water electrolyser reactions on 3d M (Ni, Co,Fe, Mn) hydr (oxy) oxide catalysts. Nat. Mater. 11, 550–557 (2012).
Burke, M. S., Enman, L. J., Batchellor, A. S., Zou, S. & Boettcher, S. W. Oxygen evolution reaction electrocatalysis on transition metal oxides and (oxy)hydroxides: activity trends and design principles. Chem. Mater. 27, 7549–7558 (2015).
Deng, W., Ji, X., Chen, Q. & Banks, C. E. Electrochemical capacitors utilising transition metaloxides: an update of recent developments. RSC Adv. 1, 1171–1178 (2011).
Wang, G., Zhang, L. & Zhang, J. A review of electrode materials for electrochemical supercapacitors. Chem. Soc. Rev. 41, 797–828 (2012).
Cheng, J. P., Zhang, J. & Liu, F. Recent development of metal hydroxidesas electrode material of electrochemical capacitors. RSC Adv. 4, 38893–38917 (2014).
Fergus, J. W. Recent developments in cathode materials for lithium ion batteries. J. Power Sources 195, 939–954 (2010).
Kim, S.-W., Seo, D.-H., Ma, X., Ceder, G. & Kang, K. Electrode materials for rechargeable sodium-ion batteries: potential alternatives to current lithium-ion batteries. Adv. Energy Mater. 2, 710–721 (2012).
Yuan, C., Wu, H. B., Xie, Y. & Lou, X. W. Mixed transition-metal oxides: design, synthesis, and energy-related applications. Angew. Chem. Int. Ed. 53, 1488–1504 (2014).
Waser, R. & Aono, M. Nanoionics-based resistive switching memories. Nat. Mater. 6, 833–840 (2007).
Sawa, A. Resistive switching in transition metal oxides. Mater. Today 11, 28–36 (2008).
Devic, T. & Serre, C. High valence 3p and transition metal based MOFs. Chem. Soc. Rev. 43, 6097–6115 (2014).
Kumar, S. G. & Devi, L. G. Review on modified TiO2 photocatalysis under UV/Visible light: selected results and related mechanisms on interfacial charge carrier transfer dynamics. J. Phys. Chem. A 115, 13211–13241 (2011).
Macwan, D. P., Dave, P. N. & Chaturvedi, S. A review on nano-TiO2 sol–gel type syntheses and its applications. J. Mater. Sci. 46, 3669–3686 (2011).
Pourbaix, M. ATLAS of Electrochemical Equilibria in Aqueous Solutions. (Pergamon Press, Oxford, 1966).
Huang, L.-F., Hutchison, M. J., Santucci, R. J. Jr., Scully, J. R. & Rondinelli, J. M. Improved electrochemical phase diagrams from theory and experiment: the Ni-water system and its complex compounds. J. Phys. Chem. C. 121, 9782–9789 (2017).
Huang, L.-F. & Rondinelli, J. M. Electrochemical phase diagrams for Ti oxides from density functional calculations. Phys. Rev. B 92, 245126 (2015).
Huang, L.-F. & Rondinelli, J. M. Electrochemical phase diagrams of Ni from ab initio simulations: role of exchange interactions on accuracy. J. Phys.: Condens. Matter 29, 475501 (2017).
Belsky, A., Hellenbrandt, M., Karen, V. L. & Luksch, P. New developments in the inorganic crystal structure database (ICSD): accessibility in support of materials research and design. Acta Crystallogr. Sect. B 58, 364–369 (2002).
Chase, M. W. NIST-JANAF Thermochemical Tables. 4th edn (American Institute of Physics, New York, 1998).
Anisimov, V. I., Aryasetiawan, F. & Lichtenstein, A. I. First-principles calculations of the electronic structure and spectra of strongly correlated systems: the LDA + U method. J. Phys.: Condens. Matter 9, 767 (1997).
Campo, V. L. Jr. & Cococcioni, M. Extended DFT + U + V method with on-site and inter-site electronic interactions. J. Phys.: Condens. Matter 22, 055602 (2010).
Wang, L., Maxisch, T. & Ceder, G. Oxidation energies of transition metaloxides within the GGA + U framework. Phys. Rev. B 73, 195107 (2006).
Jain, A. et al. Formation enthalpies by mixing GGA and GGA + U calculations. Phys. Rev. B 84, 045115 (2011).
Lutfalla, S., Shapovalov, V. & Bell, A. T. Calibration of the DFT/GGA+U method for determination of reduction energies for transition and rare earth metal oxides of Ti, V, Mo, and Ce. J. Chem. Theory Comput. 7, 2218–2223 (2011).
Aykol, M. & Wolverton, C. Local environment dependent GGA + U method for accurate thermochemistry of transition metal compounds. Phys. Rev. B 90, 115105 (2014).
Persson, K. A., Waldwick, B., Lazic, P. & Ceder, G. Prediction of solid-aqueous equilibria: scheme to combine first-principles calculations of solids with experimental aqueous states. Phys. Rev. B 85, 235438 (2012).
Zeng, Z. et al. Towards first principles-based prediction of highly accurate electrochemical pourbaix diagrams. J. Phys. Chem. C. 119, 18177–18187 (2015).
Huang, L.-F., Scully, J. R. & Rondinelli, J. M. Modeling corrosion with first-principles electrochemical phase diagrams. Annu. Rev. Mater. Res. 49 (2019). https://doi.org/10.1146/annurev-matsci-070218-010105.
Huang, L.-F., Lu, X.-Z., Tennessen, E. & Rondinelli, J. M. An efficient ab-initio quasiharmonic approach for the thermodynamics of solids. Comput. Mater. Sci. 120, 84–93 (2016).
Müller, S. Bulk and surface ordering phenomena in binary metal alloys. J. Phys.: Condens. Matter 15, R1429 (2003).
Huang, L.-F. et al. From electronic structure to phase diagrams: a bottom-up approach to understand the stability of titanium-transition metal alloys. Acta Mater. 113, 311–319 (2016).
Landrum, G. A. & Dronskowski, R. The orbital origins of magnetism: from atoms to molecules to ferromagnetic alloys. Angew. Chem. Int. Ed. 39, 1560–1585 (2000).
Huang, L.-F., Grabowski, B., McEniry, E., Trinkle, D. R. & Neugebauer, J. Importance of coordination number and bond length in titanium revealed by electronic structure investigations. Phys. Status Solidi B 252, 1907–1924 (2015).
Young, D. J. High Temperature Oxidation and Corrosion of Metals, Vol. 1 (Elsevier, Amsterdam, 2008).
Piazza, C. S. & Di Quarto, Fo Photocurrent spectroscopic investigations of passive films on chromium. J. Electrochem. Soc. 137, 2411–2417 (1990).
Maurice, V., Yang, W. P. & Marcus, P. XPS and STM investigation of the passive film formed on Cr(110) single‐crystal surfaces. J. Electrochem. Soc. 141, 3016–3027 (1994).
Kim, J., Cho, E. & Kwon, H. Photo-electrochemical analysis of passive film formed on (cr in ph 8.5) buffer solution. Electrochim. Acta 47, 415–421 (2001).
Olazabal, M. A., Nikolaidis, N. P., Suib, S. A. & Madariaga, J. M. Precipitation equilibria of the chromium(vi)/iron(iii) system and spectrospcopic characterization of the precipitates. Environ. Sci. Technol. 31, 2898–2902 (1997).
Abd El-sadek, M. S. & Babu, S. M. A controlled approach for synthesizing CdTe@CrOOH (core-shell) composite nanoparticles. Curr. Appl. Phys. 11, 926–932 (2011).
Collins, R. N., Clark, M. W. & Payne, T. E. Solid phases responsible for MnII, CrIII, CoII, Ni, CuII and Zn immobilization by a modified bauxite refinery residue (red mud) at pH 7.5. Chem. Eng. J. 236, 419–429 (2014).
Pardo, P., Calatayud, J. M. & Alarcón, J. Chromium oxide nanoparticles with controlled size prepared from hydrothermal chromium oxyhydroxide precursors. Ceram. Int. 43, 2756–2764 (2017).
Liu, C. T. & Wu, J. K. Influence of pH on the passivation behavior of 254 SMO stainless steel in 3.5 NaCl solution. Corros. Sci. 49, 2198–2209 (2007).
Jiao, F. & Frei, H. Nanostructured cobalt and manganese oxide clusters as efficient water oxidation catalysts. Energy Environ. Sci. 3, 1018–1027 (2010).
Najafpour, M. M., Haghighi, B., Sedigh, D. J. & Ghobadi, M. Z. Conversions of Mn oxides to nanolayered Mn oxide in electrochemical water oxidation at near neutral pH, all to a better catalyst: catalyst evolution. Dalton Trans. 42, 16683–16686 (2013).
Ramírez, A. et al. Evaluation of MnOx, Mn2O3, and Mn3O4 electrodeposited films for the oxygen evolution reaction of water. J. Phys. Chem. C. 118, 14073–14081 (2014b).
Gorlin, Y. et al. Understanding interactions between manganese oxide and gold that lead to enhanced activity for electrocatalytic water oxidation. J. Am. Chem. Soc. 136, 4920–4926 (2014c).
Frydendal, R. et al. Operando investigation of Au-MnOx thin films with improved activity for the oxygen evolution reaction. Electrochim. Acta 230, 22–28 (2017a).
Takashima, T., Hashimoto, K. & Nakamura, R. Mechanisms of pH-dependent activity for water oxidation to molecular oxygen by MnO2 electrocatalysts. J. Am. Chem. Soc. 134, 1519–1527 (2012).
Hem, J. D. Rates of manganese oxidation in aqueous systems. Geochim. Cosmochim. Acta 45, 1369–1374 (1981).
Jung, H. & Jun, Y.-S. Ionic strength-controlled mn(hydr)oxide nanoparticle nucleation on quartz: effect of aqueous Mn(OH)2. Environ. Sci. Technol. 50, 105–113 (2016).
Lyons, M. E. G. & Brandon, M. P. The oxygen evolution reaction on passive oxide covered transition metal electrodes in aqueous alkaline solution. part I - nickel. Int. J. Electrochem. Sci. 3, 1386–1424 (2008).
Lyons, M. E. G. & Brandon, M. P. The oxygen evolution reaction on passive oxide covered transition metal electrodes in alkaline solution part II - cobalt. Int. J. Electrochem. Sci. 3, 1425–1462 (2008).
Lyons, M. E. G. & Brandon, M. P. The oxygen evolution reaction on passive oxide covered transition metal electrodes in alkaline solution. part III - iron. Int. J. Electrochem. Sci. 3, 1463–1503 (2008).
Lyons, M. E. G. & Brandon, M. P. A comparative study of the oxygen evolution reaction on oxidised nickel, cobalt and iron electrodes in base. J. Electroanal. Chem. 641, 119–130 (2010).
Cornell, R. M. & Schwertmann, U. The Iron Oxides: Structure, Properties, Reactions, Occurrences and Uses (John Wiley & Sons, Weinheim, 2003).
Kang, Y. S., Risbud, S., Rabolt, J. F. & Stroeve, P. Synthesis and characterization of nanometer-size Fe3O4 and γ-Fe2O3 particles. Chem. Mater. 8, 2209–2211 (1996).
Jolivet, J.-P., Chanéac, C. & Tronc, E. Iron oxide chemistry. From molecular clusters to extended solid networks. Chem. Commun. 481–483 (2004).
Petcharoen, K. & Sirivat, A. Synthesis and characterization of magnetite nanoparticles via the chemical co-precipitation method. Mater. Sci. Eng. B 177, 421–427 (2012).
Burke, L. D. & Lyons, M. E. G. The formation andstability of hydrous oxide films on iron under potential cycling conditions in aqueous solution at high pH. J. Electroanal. Chem. 198, 347–368 (1986).
Lee, J., Isobe, T. & Senna, M. Preparation of ultrafine Fe3O4 particles by precipitation in the presence of PVA at high pH. J. Colloid Interface Sci. 177, 490–494 (1996).
Beinlich, A. et al. Peridotite weathering is the missing ingredient of Earth’s continental crust composition. Nat. Commun. 9, 634 (2018a).
Frankel, R. B., Papaefthymiou, G. C., Blakemore, R. P. & O’Brien, W. Fe3O4 precipitation in magnetotactic bacteria. Biochim. et. Biophys. Acta 763, 147–159 (1983).
Mandernack, K. W., Bazylinski, D. A., Shanks, W. C. & Bullen, T. D. Oxygen and iron isotope studies of magnetite produced by magnetotactic bacteria. Science 285, 1892–1896 (1999).
Hanzlik, M. et al. Superparamagnetic magnetite in the upper beak tissue of homing pigeons. Biometals 13, 325–331 (2000).
Chen, C. et al. Achieving high performance corrosion and wear resistant epoxy coatings via incorporation of noncovalent functionalized graphene. Carbon 114, 356–366 (2017b).
Cui, M. et al. Processable poly(2-butylaniline)/hexagonal boron nitride nanohybrids for synergetic anticorrosive reinforcement of epoxy coating. Corros. Sci. 131, 187–198 (2018b).
Han, R. et al. 1D magnetic materials of Fe3O4 and Fe with high performance of microwave absorption fabricated by electrospinning method. Sci. Rep. 4, 7493 (2014d).
Wei, Y. et al. Facile synthesisof self-assembled ultrathin α-FeOOH nanorod/graphene oxide composites for supercapacitors. J. Colloid Interface Sci. 504, 593–602 (2017c).
Song, H., Xia, L., Jia, X. & Yang, W. Polyhedral α-Fe2O3 crystals at RGO nanocomposites: synthesis, characterization, and application in gassensing. J. Alloy. Compd. 732, 191–200 (2018).
Meier, H. G., Vilche, J. R. & Arvía, A. J. The electrochemical behaviour of cobalt in alkaline solutions part II. the potentiodynamic response of Co(OH)2 electrodes. J. Electroanal. Chem. 138, 367–379 (1982).
Ohtsuka, T. & Sato, N. Anodic oxide film on cobalt in weakly alkaline solution. J. Electroanal. Chem. 147, 167–179 (1983).
Foelske, A. & Strehblow, H.-H. Structure and composition of electrochemically prepared oxide layers on Co in alkaline solutions studied by XPS. Surf. Interface Anal. 34, 125–129 (2002).
Bewick, A., Gutiérrez, C. & Larramona, G. An in-situ IR spectroscopic study of the anodic oxide film on cobalt in alkaline solutions. J. Electroanal. Chem. 333, 165–175 (1992).
Yeo, B. S. & Bell, A. T. Enhanced activity of gold-supported cobalt oxide for the electrochemical evolution of oxygen. J. Am. Chem. Soc. 133, 5587–5593 (2011).
Kanan, M. W. et al. Structure and valency of a cobalt−phosphate water oxidation catalyst determined by in situ x-ray spectroscopy. J. Am. Chem. Soc. 132, 13692–13701 (2010).
Simmons, G. W., Vertes, A., Varsanyi, M. L. & Leidheiser, H. Emission Mössbauer studies of anodically formed CoO2. J. Electrochem. Soc. 126, 187–189 (1979).
Lo, Y. L. & Hwang, B. J. In situ Raman studies on cathodically deposited nickel hydroxide films and electroless Ni-P electrodes in 1 M KOH solution. Langmuir 14, 944–950 (1998).
Hoppe, H.-W. & Strehblow, H.-H. XPS and UPS examinations of passive layers on Ni and Fe53Ni alloys. Corros. Sci. 31, 167–177 (1990).
Druska, P., Strehblow, H. H. & Golledge, S. A surface analytical examination of passive layers on CuNi alloys: part I. alkaline solution. Corros. Sci. 38, 835–851 (1996).
Druska, P. & Strehblow, H.-H. Surface analytical examination of passive layers on Cu-Ni alloys Part II. acidic solutions. Corros. Sci. 38, 1369–1383 (1996).
Schrebler Guzmán, R. S., Vilche, J. R. & Arvía, A. J. The kinetics and mechanism of the nickel electrode III. The potentiodynamic response of nickel electrodes in alkaline solutions in the potential region of Ni(OH)2 formation. Corros. Sci. 18, 765–778 (1978).
Melendres, C. A. & Xu, S. In situ laser Raman spectroscopic study of anodic corrosion films on nickel and cobalt. J. Electrochem. Soc. 131, 2239–2243 (1984).
Lillard, R. S. & Scully, J. R. Electrochemical passivation of ordered NiAl. J. Electrochem. Soc. 145, 2024–2032 (1998).
Han, S. Y. et al. The growth mechanism of nickel oxide thin films by room-temperature chemical bath deposition. J. Electrochem. Soc. 153, C382–C386 (2006).
Yeo, B. S. & Bell, A. T. In situ Raman study of nickel oxide and gold-supported nickel oxide catalysts for the electrochemical evolution of oxygen. J. Phys. Chem. C. 116, 8394–8400 (2012).
Trotochaud, L., Young, S. L., Ranney, J. K. & Boettcher, S. W. Nickel-iron oxyhydroxide oxygen-evolution electrocatalysts: the role of intentional and incidental iron incorporation. J. Am. Chem. Soc. 136, 6744–6753 (2014).
Klaus, S., Cai, Y., Louie, M. W., Trotochaud, L. & Bell, A. T. Effects of Fe electrolyte impurities on Ni(OH)2/NiOOH structure and oxygen evolution activity. J. Phys. Chem. C. 119, 7243–7254 (2015).
Chen, J. S., Gui, Y. & Blackwood, D. J. A versatile ionic liquid-assisted approach to synthesize hierarchical structures ofβ-Ni(OH)2 nanosheets for high performance pseudocapacitor. Electrochim. Acta 188, 863–870 (2016).
Abbas, S. A. & Jung, K. D. Preparation of mesoporous microspheres of nio with high surface area and analysis on their pseudocapacitive behavior. Electrochim. Acta 193, 145–153 (2016).
Jung, S. C. et al. Synthesis of nanostructured β-ni(oh)2 by electrochemical dissolution–precipitation and its application as a water oxidation catalyst. Nanotechnology 27, 275401 (2016b).
Perdew, J. P. & Schmidt, K. Jacob’s ladder of density functional approximations for the exchange-correlation energy. AIP Conf. Proc. 577, 1–20 (2001).
Sun, J. et al. Semilocal and hybrid meta-generalized gradient approximations based on the understanding of the kinetic-energy-density dependence. J. Chem. Phys. 138, 044113 (2013a).
Sun, J. et al. Density functionals that recognize covalent, metallic, and weak bonds. Phys. Rev. Lett. 111, 106401 (2013b).
Heyd, J., Scuseria, G. E. & Ernzerhof, M. Hybrid functionals based on a screened coulomb potential. J. Chem. Phys. 118, 8207–8215 (2003).
Heyd, J. & Scuseria, G. E. Assessment and validation of a screened coulomb hybrid density functional. J. Chem. Phys. 120, 7274–7280 (2004a).
Heyd, J. & Scuseria, G. E. Efficient hybrid density functional calculations in solids: assessment of theheyd–scuseria–ernzerhof screened coulomb hybrid functional. J. Chem. Phys. 121, 1187–1192 (2004b).
Heyd, J., Scuseria, G. E. & Ernzerhof, M. Erratum: “hybrid functionals based on a screened coulomb potential” [J. Chem. Phys. 118, 8207 (2003)]. J. Chem. Phys. 124, 219906 (2006).
Vydrov, O. A., Heyd, J., Krukau, A. V. & Scuseria, G. E. Importance of short-range versus long-range hartree-fock exchange for the performance of hybrid density functionals. J. Chem. Phys. 125, 074106 (2006).
Hafner, J. Ab-initio simulations of materials using VASP: density-functional theory and beyond. J. Comput. Chem. 29, 2044–2078 (2008).
Blöchl, P. E. Projector augmented-wave method. Phys. Rev. B 50, 17953–17979 (1994).
Togo, A. & Tanaka, I. First principles phonon calculations in materials science. Scr. Mater. 108, 1–5 (2015).
Parlinski, K., Li, Z. Q. & Kawazoe, Y. First-principles determination of the soft mode in cubic ZrO2. Phys. Rev. Lett. 78, 4063–4066 (1997).
Ceperley, D. M. & Alder, B. J. Ground state of the electron gas by a stochastic method. Phys. Rev. Lett. 45, 566–569 (1980).
Perdew, J. P. & Zunger, A. Self-interaction correction to density-functional approximations for many-electron systems. Phys. Rev. B 23, 5048–5079 (1981).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1996).
Perdew, J. P. et al. Restoring the density-gradient expansion for exchange in solids and surfaces. Phys. Rev. Lett. 100, 136406 (2008).
Perdew, J. P., Ruzsinszky, A., Csonka, G. I., Constantin, L. A. & Sun, J. Workhorse semilocal density functional for condensed matter physics and quantum chemistry. Phys. Rev. Lett. 103, 026403 (2009).
Samsonov, G. V. The Oxide Handbook. (IFI/Plenum, New York, 1973).
Burgess, J. Metal Ions in Solution. (Ellis Horwood Limited, Chichester, 1978).
Bard, A. J., Parsons, R. & Jordan, J. Standard Potentials in Aqueous Solution. (Marcel Dekker, New York, 1985).
Kubaschewski, O., Alcock, C. B. & Spencer, P. J. Materials Thermochemistry. (Pergamon Press, Oxford, 1993).
Beverskog, B. & Puigdomenech, I. Revised pourbaix diagrams for iron at 25–300 °c. Corros. Sci. 38, 2121–2135 (1996).
Beverskog, B. & Puigdomenech, I. Revised pourbaix diagrams for nickel at 25–300 °C. Corros. Sci. 39, 969–980 (1997).
Beverskog, B. & Puigdomenech, I. Revised pourbaix diagrams for chromium at 25–300 °c. Corros. Sci. 39, 43–57 (1997).
Chivot, J., Mendoza, L., Mansour, C., Pauporté, T. & Cassir, M. New insight in the behavior of Co-H2O system at 25–150 °C, based on revised pourbaix diagrams. Corros. Sci. 50, 62–69 (2008).
Acknowledgements
L.-F.H. and J.M.R. were supported by the Office of Naval Research under Award no. N00014-16-1-2280. Calculations were performed using the QUEST HPC Facility at Northwestern University, the HPCMP facilities at the Navy DSRC, the Extreme Science and Engineering Discovery Environment (XSEDE) supported by the National Science Foundation (NSF) under award number ACI-1548562, and the Center for Nanoscale Materials (Carbon Cluster). Use of the Center for Nanoscale Materials, an Office of Science user facility, was supported by the U.S. Department of Energy, Office of Science, and Office of Basic Energy Sciences, under Contract No. DE-AC02-06CH11357.
Author information
Authors and Affiliations
Contributions
L.-F.H. and J.M.R. designed the theoretical method, performed ab initio calculations, analyzed the results, and wrote the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Huang, LF., Rondinelli, J.M. Reliable electrochemical phase diagrams of magnetic transition metals and related compounds from high-throughput ab initio calculations. npj Mater Degrad 3, 26 (2019). https://doi.org/10.1038/s41529-019-0088-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41529-019-0088-z
- Springer Nature Limited
This article is cited by
-
Electrochemical properties of magnetite nanoparticles supported on carbon paste electrode in various electrolyte solutions: A study by cyclic voltammetry
Journal of Applied Electrochemistry (2024)
-
Elemental partitioning and corrosion resistance of Ni–Cr alloys revealed by accurate ab-initio thermodynamic and electrochemical calculations
npj Materials Degradation (2023)
-
A perspective on corrosion of multi-principal element alloys
npj Materials Degradation (2021)
-
Predicting aqueous stability of solid with computed Pourbaix diagram using SCAN functional
npj Computational Materials (2020)