
Abstract
The nexus between classic ferroelectricity and the structure of perovskite materials hinges on the concept of lattice disorder. Although the ordered perovskites display short-range displacements of the central cations around their equilibrium points, the lattice disorder dynamically unfolds to generate a myriad of distorted rhombohedral lattices characterized by the hopping of the central cations across <111> directions. It is discovered that the lattice disorder correlates with the emergence of minimum configuration energy <100> pathways for the central cations, resulting in spatially modulated ultrafast polarization nanocluster arrangements that are stabilized by the electric charge defects in the material. Through high-resolution phonon dispersion analyses encompassing molecular dynamics (MD) and density functional theory (DFT) simulations, we provide unequivocal evidence linking the hopping of central cations to the development of diffuse soft phonon modes observed throughout the phase transitions of the perovskite. Through massive MD simulations, we unveil the impact of lattice disorder on the structures of domain walls at finite-temperature vis-à-vis collective activation and deactivation of <100> pathways. Furthermore, our simulations demonstrate the development of hierarchical morphotropic phase boundary (MPB) nanostructures under the combined influence of externally applied pressure and stress relaxation, characterized by sudden emergence of zig-zagged monoclinic arrangements that involve dual <111> shifts of the central cations. These findings have implications for tailoring MPBs in thin-film structures and for the light-induced mobilization of DWs. Avenues are finally uncovered to the exploration of lattice disorder through gradual shear strain application.
Similar content being viewed by others


Introduction
With the advent of advanced material characterization methods and computational simulations, evidence has become increasingly available in that complex material systems exhibit local perturbations of their average crystal structure, or short-range lattice disorder, which modifies their functional response1,2. While the average crystal structure is then satisfactory revealed through well-stablished experimental techniques, the underlying lattice disorder is likely to remain unnoted and may be exposed only on the basis of theoretical analyses and computations. Along these lines, the development of lattice disorder emerges as an elusive element to classic ferroelectricity in ATiO3 perovskites.
As opposed to the ‘displacive’ model where the Ti atoms lie at the lattice center or along the quasi-cubic <111>, <110>, and <100> directions of the perovskite phases, experimental and theoretical analyses led to the notion that these cations are shifted in specific <111> directions3,4,5,6,7,8,9,10,11,12. As illustrated in Fig. 1a, the combination of the two <111> Ti shifts arising in the orthorhombic phase produces the net or perceived <110> overall polarization direction. With increases in temperature, the incepted tetragonal isomorph exhibits the four <111> Ti shifts that produce the distinctive <100> stretching and polarization direction (Fig. 1a). Finally, the higher temperature paraelectric cubic isomorph contains the eight possible <111> Ti shifts, so that, in average, the Ti atoms remain at the center of the perovskite structure -without any preferred vibrational mode. A central feature to this lattice disorder or eight-site model is the conception that the Ti shifts are ruled by zig-zagged O-Ti-O chains that extend along the <100> directions of the perovskite phases5,6,13.

Panel a provides schematic representations of the <111> Ti shifts as proposed in the static eight-site model (small black arrows), the net polarization in the supercell (larger orange arrows pointing towards the [111], [110] and [100]-type directions of the rhombohedral, orthorhombic and tetragonal phases), the governing O-Ti-O chains (marked with thicker lines), and the mean lattice parameters from our MD simulations. The stereographic projections in panel b indicate the dynamical distributions of the Ti shifts assessed though the local lattice polarization vector \(P=({P}_{x}{,P}_{y},{P}_{z})\). The scattering of the Ti shifts is indicative of the breakdown of the modeled supercells in a plethora of disordered BTO lattices, each with its own \(P\) vector. Panel c provides the distributions of configurational energy, \(E\), of the disordered lattices as a function of the \(P\) vector. The configurational energy distributions in three-dimensional space are schematically illustrated at the top of panel c along with the active <100> pathways for the Ti oscillations. The actual distributions of configurational energy from the MD simulations, projected in the \({P}_{x}-{P}_{z}\) and Pz – Py horizontal planes, are then provided for each phase at the bottom of the panel. These projections resemble those of the Ti shifts distributions in b. Complementary panel d finally shows that while the Ti atoms remain closer to the average lattice center in the ordered, RT tetragonal PTO phase, a drastic enhancement of the Ti fluctuations is instated as the cubic PTO phase becomes stable at elevated temperature.
The investigation of the lattice disorder has been framed in the context of the potential energy landscape that sets the locations of the Ti atoms throughout the perovskite structures14,15. The shifting of the Ti atoms in the essentially disordered tetragonal and cubic phases of the rhombohedral ground-state BaTiO3 (BTO) perovskite is then dictated by minimum potential energy loci located along <111> directions, a feature that is absent in the ordered tetragonal ground-state PbTiO3 (PTO) perovskite. While experimental results from Raman spectroscopy and X-ray Absorption Fine Structure (XAFS) techniques have however provided apparently contradictory findings in support of both the displacive and the lattice disorder models, it was readily recognized that these results could be reconciled under a dynamical paradigm characterized by the hopping of the Ti cations across the minimum energy <111> sites7,12. Since the observation of these atomic hopping or tunneling processes is precluded under the relatively large observation timeframes of the Raman spectroscopy measurements, this technique could only render ‘mean atomic positions’, which inevitably support the displacive model for ordered structures. On the other hand, the much smaller observation timeframe in the XAFS measurements furnishes tentative access to the instantaneous positions of the central cations, which helps differentiate between the onset of ordered and disordered structures.
A distinctive feature in the phase transitions of perovskites involves the emission of soft phonon modes, which transcends the static eight-site model16,17,18,19,20,21,22,23. Although these phonon modes have been associated with the onset of disordered (higher temperature) phases in which the central cations display low-frequency hops across <111> sites, this interpretation is still uncertain since, to the present, the soft modes have been correlated only with the bond softening phenomenon that occurs along specific directions in ‘quasi-static’ density functional theory (DFT) simulations of ordered structures -where the atoms stay at their equilibrium points24,25,26,27. Along these lines, effective Hamiltonian and molecular dynamics (MD) approaches have provided a theoretical foundation to the hopping processes of the central cations and to the associated ferroelectric properties at the transition to the higher temperature cubic phase, further showcasing the possibly mixed order-disorder character of this transition22,23,24,26,28,29,30,31,32.
Our MD simulation results with large computational systems highlight the pervasive dynamical nature of the lattice disorder in classic ferroelectricity at different length scales. It is demonstrated that the lattice disorder is ubiquitous to the breakdown of all perovskite phases into an ensemble of dynamically distorted, minimum energy rhombohedral configurations or motifs, produced by the excursions of the central cations within the non-stationary A and O cages of the perovskite. These atomic excursions, or tunneling effects, lie at the origin of the scattered soft phonon modes observed across the phase transitions in all disordered perovskite phases, unequivocally indicating the onset of lattice disorder according to our dynamical analyses of the finite-temperature phonon dispersion curves. We also show that the long-range coupling of polarization produces ultrafast, dynamically evolving nanocluster morphologies in the disordered orthorhombic, tetragonal and cubic phases, which serve as the effective carriers of electric polarization across the material. Our findings shed light into the pivotal role of the lattice disorder on the domain wall (DW) structures, fundamentally advancing our knowledge that is currently based on quasi-static, vanishing temperature simulations into more realistic, finite-temperature dynamical conditions. Implications of the discovered DW structures to their light-assisted mobilization are also proposed. Finally, our massive MD simulations illustrate the occurrence of pressure-assisted tetragonal-to-monoclinic phase transitions within preexisting polarization domains. This provides insights to the hierarchical nanostructures which characterize the morphotropic phase boundaries in disordered perovskites. We further show that the lattice disorder can be potentially detected and manipulated by means of deliberately applied shear strains.
Results
Configurational energy and dynamic disorder across the phase fields
The development of ordered and disordered phases in our MD simulations is governed by the bonding energy between the A and O atoms in the perovskite, as described through the core-shell atomic potential model (see Methods). Milder bonds thus result in the disordered BTO phases along with the enhanced mobility of the central Ti cations, whereas the hybridization between O and Pb leads to stronger bonds and to the onset of the ordered, tetragonal low temperature phase in PTO.
Figure 1a, b show the trajectories of the Ti atoms (or Ti shifts) within the enclosing A and O cages of BTO, as traced by the local polarization vector P of the unit lattices which constitute the modeled supercells. As shown in Supplementary Information, the P vector provides an essential measure to the relative Ti shifts as well as to the local lattice distortions, where the 42 independent degrees of freedom produced by the vibrations of the 14 nearest neighbors to each Ti atom along the 3 symmetry axes are conveniently reduced to the three magnitudes Px, Py, and Pz. Figure 1 is also illustrative of the similarity between the distributions of the dynamically distorted BTO lattices, given as a function of the P vector in Fig. 1b, and those of the configurational energy of these lattices, given in Fig. 1c. It is emphasized that while the most likely lattice configurations exhibit minimum configurational energy (cf. panels (b) and (c)), this correlation is lost when the energy is computed so as to account only for the Ti interactions while neglecting those of the O and Ba ions with their nearest neighbors (see Methods). It thus follows that the onset of the distorted lattice configurations is dictated by their overall configurational energy rather than by the potential energy landscape for the Ti displacements throughout an otherwise steady arrangement of O and Ba neighbors, as assumed in the absence of dynamical disorder6,7. A limitation in our analytical framework concerns the fact that different lattice configurations with distinct configurational energy may exhibit nearly identical polarization vectors. To account for this, each data point in the projected energy distributions of panel (c) represents the mean (average) energy level for all lattice configurations within a small window of P vectors. Our MD simulation results are further indicative of the ergodic nature of the lattice disorder where the same distributions of Ti shifts in panel (b) emerge either when the instantaneous lattice polarization vectors of all constituent BTO lattices in the supercell are examined or when the polarization vector of a single lattice is evaluated over an extended timeframe. The P vector distributions for all instantaneous lattice configurations in the supercell (panel (b)) are then equally representative of the Ti excursions occurring within a single lattice over time.
The following observations from our MD simulations concern the disordered nature of the BTO phases. First, the rhombohedral ground-state is found to spontaneously arise in the modeled supercell below T ≈ 150 K, where the collective shifting of the Ti atoms towards the [111] direction marked in Fig. 1b produces the overall polarization of ≈0.26 C m−2. The orthorhombic phase spontaneously sets-in as the temperature is raised above 150 K. The dynamical distortion of the BTO lattices then produces a rather homogeneous Ti shift, or P vector, distribution of minimum configurational energy that extends throughout the [001] direction traversing across the marked [\(11\bar{1}\)] and [111] poles (cf. panels (b) and (c)). This [001] direction thus becomes an effective pathway for the excursions of the Ti atoms within the distorted lattice configurations. In view of these observations, the Ti shifts are taken to occur via gradual atomic oscillations, which produce the observed homogeneous distribution of Ti shifts rather than through a conceptual hopping process where these atoms should tentatively stay at the vicinity of the <111> poles during greater time periods. Attention is drawn to the fact that since the internal electric field in the modeled supercell redirects the Ti atoms towards the [110] pole, the onset of P vectors that point precisely at the [\(11\bar{1}\)] and [111] directions is clearly suppressed from the distribution in panel (b) for the orthorhombic phase.
At temperatures above 250 K, the tetragonal BTO isomorph spontaneously arises in the modeled supercell. This leads to a reduction in overall polarization to ≈0.18 C m−2. The four-fold symmetrical Ti shift or P vector trajectory of minimum configurational energy in Fig. 1b, c is then found to emerge, which is noticeably displaced away from the four <111> poles due to the force exerted by the internal electric field in the supercell. As the temperature continues to rise, the transition towards the paraelectric cubic BTO phase occurs at 350 K. In this phase, the trajectories of Ti shifts along the <100> pathways finally trace the eight <111> poles of minimum potential energy. It is worth noting that as the internal electric field is suppressed in the cubic phase, the Ti shift distribution precisely incorporates the {111} poles, which contrasts with the observations made for the tetragonal phase under non-vanishing internal electric field where these poles are excluded. It is finally argued that the heterogeneous Ti shift distributions observed in panel (b) for the tetragonal and cubic isomorphs support a conceptual atomic hopping process. As the Ti atoms thus traverse more rapidly across the <100> pathways while staying at the <111> poles for a longer time, a higher density of lattice configurations results at the vicinity of the poles.
The MD simulations of the PTO perovskite show the development of the ordered tetragonal ground-state and its spontaneous transition to the cubic phase at elevated temperature. Therefore, for temperatures close to RT, the Ti cations remain at the equilibrium points of the tetragonal structure whereas greater thermal vibrations are induced in the elevated temperature cubic phase (see Fig. 1d).
Spaciotemporal evolutions of lattice disorder: polarization nanoclusters
Figure 2 shows the dynamical evolution of the lattice disorder characterized by the spatial and temporal modulations of the distorted BTO lattices. The figure highlights the development of polarization nanocluster arrangements in all BTO phases, as evidenced through the autocorrelation analyses in the Supplementary Information. While the stronger coupling of polarization is found to occur along the primary <100> pathways of each isomorph, milder polarization coupling is also observed normal to these pathways, which facilitates the development of irregular nanocluster shapes.

a–c Spatial polarization modulation across the main perovskite \(\xi\) and \(\eta\) axes produces nanocluster arrangements [Individual nanocluster units are identified within \(\approx \pm\)10° variability in polarization direction]. Polarization nanovortices are identified with white arrows. d–f Ultrafast nanocluster propagation results in the transmission of electric polarization packets (dashed rectangles in d) along the active O-Ti-O chains, as prescribed by arrows with angle \(\theta =\Delta t/\Delta \xi\). The period of the polarization fluctuations of individual BTO lattices is highlighted with solid rectangles of 0.2 to 0.5 ps. g Detail of the temporal fluctuations of the Ti shifts as measured through the polarization vector \(P\) in four neighboring rows of the crystal lattices in the orthorhombic isomorph. While the governing O-Ti-O chains lie along the \(\xi\) axis (see the inset to part d), mild polarization coupling is observed along the \(\eta\) axis, as marked with black rectangles. The aforementioned, characteristic fluctuation period of \(\approx\)0.2 to \(\approx\)0.5 ps can be also observed in g.
Since the nanoclusters breakdown into smaller units depending on local polarization fluctuations, an objective assessment of the nanocluster size distribution is hindered. Regardless of this observation, it becomes evident that the greater oscillations and increased number of intercepting <100> pathways in the higher temperature phases inevitably lead to smaller nanocluster sizes (cf. Fig. 2a–c). Along these lines, polarization nanovortices are also frequently observed at elevated temperature, which suggests interaction between multiple O-Ti-O chains. Unsuccessful Ti hops are further detected where the oscillation direction is reversed halfway across the <100> pathways.
Figure 2d–f show the temporal evolution of the polarization nanoclusters in the BTO phases, where rapid nanocluster propagation occurs along the <100> directions of the governing O-Ti-O chains. Since this propagation phenomenon occurs for a fixed angle θ in the space-time representation of Fig. 2d, it is arguably attained at constant velocity. A resemblance is further noted between nanocluster propagation and the traveling of solitons, or solitary waves, whose envelope can be envisioned to carry polarization packets along the prime <100> directions. Since ultrafast variations of lattice polarization are therefore attained, the overall or net polarization in the BTO phases is given by the time-averaged P vector in any constituent, distorted lattice.
The time-averaged polarization nanopatterns developing in the cubic BTO phase are shown in Fig. 3. The results indicate that when the lattice polarization is averaged over small bins of 15 ps, the intricate nanocluster arrangements in Fig. 2c can still be discerned. Nevertheless, as a vast number of nanoclusters populate the modeled supercells when the polarization is averaged over larger 150 ps bins, the instantaneous nanocluster structure becomes concealed (cf. Figure 3a with b). While the onset of the currently observed polarization nanostructures thus challenges experimental assertion, complementary MD simulations performed with a realistic concentration of randomly distributed electric charge defects show the prevalence of coarser, extrinsic polarization arrangements around the incepted defects (cf. Fig. 3c, d). This indicates that irrespective of the ultrafast nanocluster propagation phenomenon in the defect-free regions, the stable polarization arrangements that develop around the charge defects would become potentially amenable to experimental observation (see the Discussion section).

a and b Polarization configurations arising in a small defect-free material region where the polarization is averaged over bins of 15 ps and 150 ps (color code for the \({\boldsymbol{P}}\) vectors is given in b). Since ultrafast nanocluster propagation produces stochastic variations of the lattice polarization, the instantaneous nanocluster arrangements are concealed when binning over 150 ps. This leads to the vanishing local polarization in the cubic paraelectric phase (see b). c Extrinsic polarization nanoclusters become stabilized around randomly distributed electric charge defects (marked with back squares). Notice that this extrinsic nanocluster arrangement is maintained even when the polarization is averaged over large bins of 250 ps. d As the polarization is averaged over smaller bins of 15 ps, the ultrafast intrinsic nanoclusters already become visible in defect-free regions, as marked with colored rectangles. Polarization nanovortices are highlighted with circular black arrows.
Finite-temperature analyses of phonon dispersion
Figure 4 provides the phonon dispersion curves from the MD simulations of the disordered BTO and ordered PTO perovskites, as well as those from the DFT simulations. These curves include the 3 lower frequency acoustic modes (\(\omega \lesssim\) 200 cm−1) and the 12 higher frequency optical modes (\(\omega \gtrsim\) 200 cm−1) that arise in ATiO3 compounds. Although well-defined phonon modes develop in the lower temperature rhombohedral phase, where the atoms exhibit small-amplitude harmonic vibrations, increasing phonon mode scattering or delocalization is observed as the temperature increases across the phase transitions (cf. the partial phonon dispersion curves in panel (a)). Our results show that the phonon delocalization is accompanied by specific phonon softening, phonon mode interference, and phonon branch broadening phenomena, as marked in Fig. 4.

The partial phonon dispersions for all atoms and for the individual Ti atoms along the x, y, and z axes of the disordered BTO phases are shown in panels a and b, respectively. Notice the softening and highly scattered nature of the low-frequency transverse optical mode (sTO, highlighted in orange), spreading throughout the marked 100 cm−1, as well as the softening of the associated longitudinal optical mode (sLO). Since these softening phenomena are observed across the phase transitions in both panels a and b (labelled as 1), the onset of the sTO and sLO modes is associated with the Ti oscillations throughout the <100> pathways that increasingly activate with temperature (see main text). The experimental results in [8] (squares) and [10] (circles) concerning the sTO phenomenon are included at the bottom of panel a. The phonon dispersion curves are further illustrative of the interaction between the soft transverse optical and acoustic modes, labeled as (2), as well as of the broadening ((3), (4), and (5)) and merging ((6), (7), and (8)) of the phonon modes across the phase transitions (marked with circles in panel a). Partial phonon dispersion curves for all atoms in the ordered tetragonal PTO phase are given in panel c, where the sTO modes clearly become less scattered or delocalized. Finally, panel d provides the full phonon dispersions from the DFT simulations of the BTO phases, where the onset of the sTO and sLO modes is also marked.
Following Fig. 4a, the transition from the rhombohedral to the orthorhombic phases is characterized by the highly delocalized softening, or overdamping, of the low-frequency transverse optical (TO) mode along the [100] direction, or z-axis, for wavevectors along the Γ-X-S path of the Brillouin zone. As the temperature increases, additional soft TO modes are emitted along the normal [010] direction, or y-axis, of the tetragonal isomorph, and along the horizontal [100] direction (x-axis) in the cubic isomorph. Since these softening phenomena are replicated in the individual phonon dispersions of the Ti atoms in Fig. 4b, it is argued that the soft phonon modes originate from the Ti vibrations along the active <100> pathways, which produce transverse wave propagation throughout the main crystal axes. This observation is supported by the coincidence between the period of the Ti excursions in the individual BTO lattices, marked in Fig. 2d–f, which lies within 0.2 and 0.5 ps for a frequency range ω from 165 to 65 cm−1, and the scattering of ≈100 cm−1 observed in the soft TO modes along the governing <100> pathways (or X directions of the Brillouin zones). As the temperature increases and the cubic phase is stabilized, the coincidence between the lattice fluctuations and the soft phonon modes occurs along Z direction of the Brillouin zone, which is now aligned with the newly incepted <100> pathway of the cubic phase. This is observed in the x-partial phonon dispersion of panel (a). It is also noted that the emission of the soft TO modes is accompanied by the softening of the longitudinal optical mode near Γ, as marked in panel (a), which is further replicated in panel (b) for the Ti atoms.
A key distinction between the MD simulations of the two perovskites concerns the delocalized nature of the soft modes observed in the disordered BTO phases as compared to more localized soft modes that result in the ordered PTO (cf. panels (a) and (c) in Fig. 4 for the tetragonal phase). Under similar anharmonic effects at 300 K, the highly marked scattering of the soft TO modes in the disordered phases thus becomes a signature of the wider Ti vibrations across <100> pathways. On the other hand, the localized soft TO modes occurring in the ordered PTO perovskite involve narrower Ti vibrations, which develop at the proximity of the equilibrium points in the tetragonal structure.
In contrast to the full-fledged lattice dynamics modeled through the MD simulations, the DFT simulations provide accurate reproductions to the atomic fluctuations occurring under the harmonic approximation, which is strictly valid at vanishing temperature. Although the disordered BTO phases naturally arise in the MD simulations, the onset of ordered BTO phases is assumed to occur in the DFT simulations, where the Ti atoms remain at the vicinity of the equilibrium points of the crystal. In the overall, a relatively good qualitative agreement is reached between the phonon dispersion curves from the MD and DFT simulations concerning the low-frequency acoustic modes and the highest frequency optical modes, while the shapes of the intermediate phonon branches along the Brillouin zone increasingly depart in the higher temperature phases (Supplementary Information). It is finally noted that while the soft TO phonon modes in the DFT simulations undergo a rapid decay towards the negative (imaginary) frequency region, which suggests onset of negative stiffness values (see the X direction for the tetragonal and cubic phases in Fig. 4d), this is an artifact of the computational scheme that is not obviously reproduced in the MD simulations where the atomic vibrations occur at effective (real) frequencies (see Methods).
Domain walls and lattice disorder
The disordered, time-averaged lattice configurations that emerge across the domain walls (DWs) of the tetragonal BTO phase in our massive room temperature MD simulations are shown in Fig. 5. In the stable 180o head-to-tail DW structure of panel (a), each of the impinging tetragonal domains are comprised of the four-fold symmetrical polarization distributions discussed in section “Configurational energy and dynamic disorder across the phase fields”. Since the four prevalent polarization vectors are abruptly reversed across the DW, in accordance with the Ising model, the wall thickness essentially vanishes. The simulation results for the stable, Nèel-type 90o head-to-tail configuration are shown in panel (b), where the local polarization rotates gradually33 through successive activation and deactivation processes of <100> pathways. This is schematically illustrated through the cubic lattices at the top of panel (b) in conjunction with the stereographic projections of the Ti shift distribution across the DW. Notice that the hopping of the Ti atoms across the equal probability <100> pathway incepted at the DW center results in the observed net [\(1\bar{1}0\)] local polarization. The thickness of this 90o head-to-tail DW lies in the ≈1.5 nm range.

a Stable 180° head-to-tail DW characterized by the abrupt reversal of the four-fold symmetrical Ti shift distribution. b Stable 90° head-to-tail DW structure distinguished by the gradual clockwise rotation of the polarization produced by the selective activation and deactivation of <100> pathways. c and d Morphologies of the thicker, unstable 90° and 180° head-to-head DW structures characterized by the confluence of polarization vectors and by positive electric change accumulation. Schematic representations of the polarization rotations across the DWs are provided at the figure top for the different examined DW configurations. The main crystal axes are marked with white arrows in the domains at the center of the figure, where the employed color code for the lattice polarization vector \({\boldsymbol{P}}\) is provided at the bottom right (notice that the main crystal axes are different to those of the \({\boldsymbol{P}}\) vector depicted in the color code representation). The associated Ti shift distributions halfway through the DWs are shown at the bottom of the figure using the stereographic projections of the \({\boldsymbol{P}}\) vectors (cf. Figure 1b for the tetragonal phase).
The 90o and 180o head-to-head DW structures in tetragonal BTO are shown in Fig. 5c, d, respectively. These structures become clearly unstable as the impinging domains retract upon the removal of the externally applied electric fields or boundary conditions. The Nèel-type 90o head-to-head structure in panel (c) arises from reversing of the polarization rotation on both sides of the wall. The unstable 180o head-to-head structure in panel (d) is instated from an abrupt 45o head-to-tail polarization rotation, followed by a gradual 90o head-to-head polarization rotation. These polarization rotations entail selective activation and deactivation of the <100> pathways, as shown by the cubic lattices at the top of panels (c) and (d), along with the evolution of the Ti shift distributions at the bottom of panel (c). These high-energy unstable DW configurations are also characterized by the onset of locally orthorhombic lattices -where only one <100> pathway remains active at the DW center- and by their relatively large thicknesses of ≈4 nm. It is further noted that the DW structure in panel (d) exhibits an overall zig-zagged or stepped morphology where the left-to-right lattice polarization sequence depicted in the cubic lattices at the top is periodically reversed throughout the DW length.
The role of mechanical stress in the phase transitions of disordered perovskites
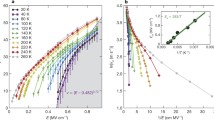
Figure 6a demonstrates the impact of externally applied shear strain, γ, on the Ti shift redistribution in the RT tetragonal BTO and cubic PTO phases. Both phases exhibit a monoclinic lattice configuration (a ≠ b ≠ c) when subjected to γ = tg(θ), where the electric polarization rotation measured by the deviation angle δ is a linear function of γ. Lattice disorder has a drastic effect on this relationship, resulting in larger δ values induced by applied γ in tetragonal BTO, where polarization rotation can be easily accommodated by redistributing the Ti shifts in two of the four regions with higher Ti occupancy (panel (a)). Conversely, the ordered PTO isomorph has hindered Ti shifts, and the rotation of polarization is relatively stiffer, leading to smaller δ values under the applied γ.

a Externally applied shear strain γ results in the emergence of monoclinic structures characterized by the rotation of the main c-axis in the RT tetragonal BTO and cubic PTO structures, measured through the angle \(\theta\), which produces polarization rotations measured through the angle\(\,\delta\). The evolution of the Ti shift distributions under the imposed shear are further illustrated through the stereographic projections of the lattice polarization vector \({\boldsymbol{P}}\). Polarization rotation is facilitated in BTO due to the disordered character of its crystal structure (as discussed in the main text). b Monoclinic subdomain structure incepted within a preexisting 90° head-to-tail DW structure in tetragonal BTO under the applied biaxial pressure (black arrows at the top part of the panel denote stress application whereas the smaller grey arrows indicate the polarization directions in the preexisting head-to-tail structure). Note the spontaneous, sudden development of a self-assembled subdomain structure showcased at the center of panel b where the incepted monoclinic subdomains are highlighted in pink, yellow, blue and black in accordance with the provided polarization color plot. The associated fluctuation trajectories for the Ti atoms within each subdomain are represented at the bottom of the panel, where the local polarization is colored according to the provided color code.
In Fig. 6b, our massive MD simulations reveal the instability of the 90o head-to-tail DW structure in tetragonal BTO when subjected to externally applied pressure under displacement controlled loading conditions. Equibiaxial compressive stresses are applied to the two orthogonal <100> directions of the stable 90o head-to-tail DW structure, while the third orthogonal direction is allowed to relax, as shown at the top of panel (b). As the applied pressure reaches ≈300 MPa (yielding normal stresses σxx = σyy = 450 MPa and σzz = 0), a self-assembled monoclinic substructure suddenly emerges across the compressed parent tetragonal stripes at the center of panel (b). The monoclinic phase in the incepted subdomains clearly differs from that produced through shear strain application in panel (a) since, for the measured value of θ ≈ 0.18o in the subdomain structure, the polarization deviation angle δ already approaches the characteristic 45o level of the stable orthorhombic phase, as depicted through the [110]-oriented orange arrow in panel (a) of Fig. 1. The Ti atoms in the monoclinic subdomains are then found to oscillate across the same minimum energy <100> pathway of the orthorhombic phase, as illustrated by the equal probability Ti shift distribution at the bottom of Fig 6b. It is further observed that the overall polarization within any given pressure-induced subdomain arrangement matches that in the parent tetragonal stripe (see Fig. 6b). Since 90o rotations of the polarization direction occur across the adjacent, zig-zagged monoclinic subregions, fully coherent twin boundaries develop, as detected in the low energy head-to-tail DW configuration described in section “Domain walls and lattice disorder”. This is necessarily attendant on the development of alternating local shear strains in the out-of-plane [001] and [00\(\bar{1}\)] directions of panel (b), which triggers the complete unloading of the modeled system under the imposed (fixed) displacements ux and uz.
Discussion
This investigation reveals the intricate connection between classic ferroelectricity and the finite-temperature dynamics of lattice disorder in model ATiO3 perovskites. Our results systematically show that the disordered perovskite phases consist of large assemblages of dynamically distorted rhombohedral lattices, or structural motifs, with minimum configurational or potential energy. By mapping the distributions of potential energy in terms of the polarization vectors of the dynamically distorted rhombohedral lattices in the orthorhombic, tetragonal and cubic phase fields, we have identified one, two and three <100> pathways of minimum configurational energy, respectively, which traverse across the vicinity of the most likely <111> Ti shifts. These minimum energy pathways coincide with the hopping trajectories and fluctuations of the Ti atoms in the disordered perovskites.
Our findings extend recent DFT simulations which revealed that certain disordered structural motifs possess lower energy levels than the hypothetical ordered cubic phase27,34,35. Our large-scale MD simulations of the orthorhombic, tetragonal and cubic perovskite phases demonstrate that, irrespective of any reference ordered cubic structure, the diverse distributions of disordered motifs within which the Ti excursions occur over time indeed have minimum potential energy. Our analysis pioneers the use of the local lattice polarization vector P as a direct and robust indicator of the lattice distortion which, in addition to accounting for the Ti excursions, inherently evaluates the geometrical irregularities of the enclosing A and O perovskite cages. This parameter is thus central in establishing the observed correspondence between the distributions of distorted lattices and potential energy (cf. Fig. 1b, c).
The results of our investigation indicate a clear connection between the onset of the soft phonon modes observed throughout the phase transition temperatures and the transverse waves produced by fluctuations of the Ti atoms across the active <100> pathways of disordered perovskites. This connection is derived from high-resolution phonon dispersion computations, which account for finite-temperature anharmonic effects in multiple MD realizations (see Methods). Although similar soft phonon modes are produced in the ordered perovskite structures due to reductions in interatomic force constants, as shown through the DFT simulations where the Ti atoms stay at the equilibrium points, our study demonstrated that the markedly delocalized (or scattered) nature of these modes becomes a fingerprint of the disordered phases.
The long-range modulation of the lattice disorder is found to produce rich spatial correlations of polarization (or polarization nanoclusters) in the dynamically disordered lattice configurations of the perovskite, as anticipated in early investigations3,6,30. The results of our large-scale MD simulations show that the spatial polarization correlations observed in a disordered perovskite phase at thermal equilibrium are not exclusive to the previously assumed transition from an ordered tetragonal precursor phase to a disordered paraelectric cubic structure30,35,36. Instead, these polarization correlations are attendant to the equilibrium states in all disordered perovskite phases. In accordance with the microstructural models for relaxor materials2, the nanocluster structures are found to populate the perovskite phases in the absence of any static, nanocluster-free background or matrix. Our simulations also revealed other notable features, such as the ultrafast, soliton-like propagation of the nanoclusters along the active <100> pathways in all phases, and their multiple interactions resulting from the higher number of active O-Ti-O chains with temperature increases across the BTO phases. The driving force to the emergence of the polarization nanoclusters is the conceivable reduction of the system’s configurational entropy through the development of ‘partially ordered’ microstates or nanotopologies. While the attainment of large nanocluster sizes is thus energetically favorable, we argue that the depolarization field produced by neighboring nanoclusters naturally limits their collective growth to less than, say, 10 consecutive lattices, which varies as a function of thermal fluctuations and the number of intercepting O-Ti-O chains in the structure. While the polarization direction of a nanocluster unit can then be transferred to the adjacent disordered lattices in one sense of the prevailing <100> propagation direction, the lattices at the opposite side of the nanocluster would lose their prevalent polarization so as to preserve the mean nanocluster size.
In spite of the elusive character of the observed spatial polarization correlations, intriguing apparently stable polarization nanoclusters have been recently observed in cubic BTO -a feature that is attributed to the stabilizing role of preexisting charge defects in the material37. Although our simulation results provide clear evidence in that coarser nanoclusters can become effectively stabilized by the charge defects, these extrinsic stable polarization arrangements shall not be mistaken with the rich, dynamically evolving smaller nanoclusters that populate the defect-free regions of the perovskite. Similar considerations apply concerning the potential role of charged free surfaces with O−2, Ti+4, or Ba+2 terminations on the stabilization of the electric polarization at the nanoscale, tentatively allowing for the experimental observation of extrinsic nanoclusters in thin films36,38.
Our MD simulations provided clear insights into the disordered nature of the room temperature domain wall (DW) structures that build on previous knowledge gathered from quasi-static, vanishing temperature DFT simulations39. We demonstrated that the polarization variations in the Néel and Ising models are accommodated by suitable rearrangements of the lattice disorder across the DWs. Specifically, our simulations reveal the onset of local orthorhombic structures whose <100> pathways are selectively activated and deactivated. Charged domain walls40,41 are then incepted at the centers of the thicker, unstable DW arrangements due to the confluence of polarization vectors in neighboring orthorhombic lattices that share the same active <100> pathway. The observed disordered lattice arrangements at the DWs allow us to envision a possible mechanism to the recent discovery of the light-induced DW mobilization reported in tetragonal BTO42,43, which is associated with the onset of a photoinduced electric field that drives head-to-tail or head-to-head configurations44,45. While this light-induced DW mobilization phenomenon is intrinsically linked with the atomic-scale properties, little is presently known on the extrinsic role of the DW structure itself, where the presently studied larger length scales come into play. Our proposal is that the application of a coherent light source with oscillatory electric field along the [010] and [\(0\bar{1}0\)] directions of the 90° head-to-tail DW depicted in Fig. 5b favors the spreading of the dual [010] and [001] Ti pathways of the left-hand side towards the domain wall center, effectively mobilizing the DW from left to right. On the other hand, the application of a similar oscillatory electric field along the [100] and [\(\bar{1}00\)] directions would favor the spreading of the dual [001] and [100] Ti pathways of the right-hand side towards the domain wall center, effectively mobilizing the DW from right to left. Similar processes, however, cannot be conceived when the electric field carried by the polarized light source is applied onto the 180° head-to-tail DW in Fig. 5a, regardless of its oscillation direction.
Our large-scale MD simulations also demonstrated the sudden shifting of the phase equilibrium that occurs in a preexisting head-to-tail tetragonal domain structure when exposed to a combination of biaxial pressure and strain relaxation. The simulation results show that as the applied pressure exceeded approximately 300 MPa, the system’s energy was effectively reduced due to the expansion of each parent tetragonal domain along the relaxed, out-of-plane direction. This was facilitated by the emergence of zig-zagged, twinnined monoclinic subregions with orthogonal polarization directions, which locally accommodated alternating out-of-plane shear deformations at the atomic level. The distorted BTO lattices in these monoclinic subregions were found to exhibit a Ti shift modulation similar to that seen in the lower temperature orthorhombic structure with a single <100> pathway or active O-Ti-O chain.
The aforementioned MD simulations on the emergence of polarization subdomains have clear implications to our understanding of the morphotropic phase boundaries (MPBs), which yield exceptional piezoelectric properties in perovskite materials used in new generations of sensors and actuators. While the MPBs are incepted via electric tuning of tetragonal-to-monoclinic phase transitions in alloyed perovskite systems, our results provide a proof-of-concept in that these phase transitions can be easily triggered under the mechanical solicitations found at the vicinity of pure BTO surfaces, which undergo strain relaxation. The similarity between the experimentally observed MPBs and those modeled here is evident from the resulting hierarchical domain structures, which feature local polarization rotations46,47,48,49,50,51,52,53,54. It should be emphasized that earlier experimental findings hinted at the possibility of the monoclinic MPBs being constituted by nanoscopic ensembles of tetragonal regions51,55,56. Our simulations, however, suggest that these nanoregions readily convert into locally monoclinic nanostructures that effectively carry the characteristic zig-zagged polarization rotations from which the piezoelectric properties of MPBs originate. Since the transformation to this monoclinic nanostructure is triggered under a moderate biaxial pressure of ~300 MPa, the material processing route could play a central role in tailoring MPB formation. This opens up prospects for thin-film processing57,58,59,60,61, where the stress relaxation at the vicinity of the surface would help in the onset of the local polarization rotations and accompanying out-of-plane shear strains. Our MD simulations offer clear evidence to this argument through the attainment of alternating surface uplift patterns produced by the rotation of <100> Ti pathways.
It is worth noting that previous studies have shown that the pressure-induced tetragonal to monoclinic transformation can arise in pure BTO and PTO in the absence of any compositional destabilization48,62,63. While these investigations have focused on bulk crystals subjected to extreme mechanical solicitations of several GPa and applied strains exceeding 1%, our simulations are indicative in that these large pressure levels can be drastically reduced through surface strain relaxation. Our results further illustrate on the potential of large-scale MD simulations with ab initio potentials in the investigation of the MBP nanostructures. These modeling strategies offer notable advantages over the previously employed phenomenological or mean-field methods, involving phase-field and Ginzburg-Landau theories64,65,66. In contrast to MD simulations, these other methods may not furnish precise quantitative insights into the critical transformation pressure levels in small-scale piezoelectric applications subjected to three-dimensional stress fields, or when significant surface relaxation is present.
Finally, our investigation suggests avenues for distinguishing between the onset of ordered and disordered ferroelectrics. We discovered that the <100> pathways in disordered structures are significantly altered by small amounts of the externally applied shear strains, facilitating the rotation of electric polarization compared to ordered phases where the Ti shifts are absent. In contrast to the pressure-induced tetragonal-to-monoclinic phase instabilities, exploring lattice disorder by mechanical means involves gradually inducing a monoclinic structure in a ferroelectric crystal through externally applied shear solicitations. In situ measurements of the relationship between the applied shear strain (γ) and the resulting polarization angle (θ) then provide insights into the emergence of lattice disorder.
In summary, our research offers comprehensive views into the interplay between lattice disorder, finite-temperature DW arrangements, and MPB transitions. This understanding is essential for elucidating the correlation between the structural complexities arising at different length scales and the emergence of ferroelectric responses. Our results are based upon massive MD simulations, which reveal the dynamical intricacies of lattice disorder and its spatiotemporal evolution, as governed by the presently discovered minimum configurational (or potential) energy distributions. Our high-resolution, anisotropic phonon dispersion analyses, conducted on multiple MD realizations, further provided a clear correlation between the emission of soft phonon modes and the spreading of the <100> pathways across the disordered phases.
Methods
Molecular dynamics simulations
Our large-scale MD simulations were carried-out with an isotropic anharmonic core-shell potential model that was fitted to first-principle density functional theory calculations under the Perdew-Burke-Ernzerhof generalized gradient approximation67,68. A net atomic polarization is allowed to develop in this core-shell model where each atom is represented by two particles: a positively charged core particle with finite mass that takes the role of the atomic core, and a negatively charged shell particle with almost vanishing 2 a.u. mass. A short-range interaction is then introduced between the shells of the neighboring atoms along with an electrostatic pair interaction calculated through the computationally efficient Wolf method with a cutoff radius and damping parameter of 11 Å and 0.25 Å−1, respectively69. While the MD simulations for the BTO perovskite reported in the main text were carried-out with the core-shell potential parameters fitted by Vielma et al., a full simulation set was also conducted with the parameters proposed in the early work by Sepliarsky et al.67,70. The complementary MD simulations of the PTO perovskite were then carried-out with the parameters proposed by Wu and Cohen71. It is emphasized that while the parameters of the MD potentials are optimized on the basis of vanishing temperature ab initio computations, they become sufficiently robust so as to reproduce the dynamically disordered character of the BTO phases as well as the ordered tetragonal PTO phase when subjected to finite-temperature. This is indicative in that the potential parameters capture the stronger hybridization between the Pb and O atoms which occurs in PTO, as well as the relatively weaker bonding between the Ba and O atoms in BTO15.
The current adiabatic molecular dynamics simulations were conducted with the LAMMPS code, where the short-range atomic interactions of the core-shell model are implemented using the Born-Mayer-Huggins potential with cutoff radius of 9 Å. The assessments of the lattice disorder and the onset of polarization nanoclusters as well as the phonon spectra calculations all relied on MD simulations performed with exceptionally large triclinic supercells containing 60 × 60 × 60 BTO unit lattices (≈1 million atoms) subjected to periodic boundary conditions. The supercells were first poled under an externally applied electric field of 20 MV m−1 so as to ensure initial polarization homogeneity across the computational domain. This was followed by a 20 ps relaxation step under vanishing pressure using the isothermal isobaric ensemble (NPT). At the temperature of this relaxation step, the stable isomorph spontaneously set-in throughout the entire supercell. A 0.4 fs time step was employed in all simulations.
The microcanonical NVE ensemble was invoked in the MD simulations that served as input to the phonon spectra computations, as the assumption of barostats and thermostats may potentially affect on the Hamiltonian character of the simulations. A set of 6 MD realizations (each representing 20 ps of actual atomic vibrations) were employed to improve the robustness of the statistical representation. In the investigations of the nanodomain substructures, an idealized point defect model was used where 0.025% of the barium atoms in the BTO supercell were randomly charged with +1 qe while an equal number of barium atoms were charged with −1 qe. A BTO structure containing 0.01 at% charge defects was thus produced. Finally, the DW simulations were performed at 300 K under vanishing pressure using the isothermal isobaric ensemble. The stable 180° and 90° head-to-tail DWs were incepted in a supercell containing 100 × 100 × 20 BTO unit cells. The polarized domains were produced under an externally applied electric field of 20 MV m−1 over 1 ps. The imposed polarization was then removed throughout a 4 ps step, and the resulting computational cell was allowed to relax for an additional 40 ps. This helped in the stability of the resulting DWs. The unstable 90° and 180° head-to-head DWs were simulated with supercells containing 150 × 100 × 20 BTO lattices. These DWs were maintained under a sustained electric field of 20 MV m−1, applied at both sides of the walls. The role of mechanical stresses on the development of a monoclinic subdomain structure was investigated by constraining the 90° DW configuration under equibiaxial strains of εx = εy = 0.2% (produced by the imposed displacements ux and uy), while the computational cell was free to relax (or expand) in the “z” axis (see Fig. 6).
A typical large-scale MD simulation involved ≈100 h of computational time using 128 CPU cores at 2.4 GHz interconnected through a QDR Infiniband switch. A six-fold increase in computational time was required to produce the MD realizations used in one phonon dispersion analysis.
Density functional theory simulations
Density functional theory (DFT) calculations for the rhombohedral, orthorhombic, tetragonal and cubic BTO phases were performed with the Vienna Ab initio Simulation Package (VASP)72 within the projector augmented-wave approach73 of the generalized gradient approximation with the Perdew-Burke- Ernzerhof functional (PBE)74. The plane-wave energy cutoff was set at 530.00 eV, and the electronic energy convergence was 10−8 eV. During the structural relaxations, the force convergence for the ions was set to 10−7 eV Å−1. The phonon dispersions were then calculated through the frozen phonon method for a 2 × 2 × 2 supercell, and subsequently postprocessed using the Phononpy package75.
It is noted that since the ratio between the stiffness matrix and the atomic mass prescribe the vibration frequency ω in the DFT simulations of the phonon dispersion curves, i.e., \(\omega =\sqrt{k/m}\) where k is the stiffness and m is the mass, negative k values result in complex phonon frequencies. These complex magnitudes are represented in the negative side of the frequency axis of Fig. 4 from the main text. It is emphasized that such complex frequencies cannot possibly arise when the phonon branches are computed through the MD simulation results, as these simulations give access to the effective anharmonic nature of the finite-temperature atomic vibrations in the supercell as well as to necessarily positive ω values.
Calculation of the lattice polarization
An accurate description of the local lattice polarization P is obtained through the following relation that accounts for the instantaneous positions of the Ti atom and its 14 nearest neighbors (i.e., 8 barium atoms and 6 oxygen atoms):
where the summation index \({a}\) refers to each of the 15 atoms in the lattice, \({r}_{j}\) indicates the position coordinates of each “core” and “shell” particles while \(q\) prescribes their electric charges, \(j=x,{y},{z}\), and \(V\) is the unit cell volume estimated through an irregular hexahedron, which contains the 8 barium atoms at the lattice vertices. The charges \({q}_{(\text{core})}^{a}\) and \({q}_{(\text{shell})}^{a}\) are then given in the core-shell parameter set for BTO67 and for PTO71. The weight factor \({w}\) finally accounts for the actual stoichiometry of the modeled perovskite. [Since the summation in Eq. (1) is performed for 1 Ti, 6 O and 8 Ba atoms in the Ba1Ti1O3 stochiometric relation, it follows that \(w=\) 1, 1/2 and 1/8 for titanium, oxygen and barium, respectively.]
Configurational energy calculations
While the potential energy of the Ti atoms in each lattice configuration is simply given by the sum of its core and shell particles, \({U}_{({\rm{core}})}+{U}_{({\rm{shell}})}\), the full lattice configurational energy necessarily accounts for the potential energy of each of the constituent anions and cations. As the thermally assisted atomic vibrations naturally lead to local distortions of the crystal lattices in the modeled supercells, the configurational energy \(U\) was estimated through the following weighted sum of the potential energy of the central titanium atom and of its neighboring six oxygen and eight barium atoms:
It is noted that since the potential energy \({U}_{({\rm{core}})}\) and \({U}_{({\rm{shell}})}\) magnitudes are indeed affected by the surrounding lattice configurations, the configurational energy \(U\) becomes a non-local quantity.
Phonon spectra analyses
The phonon dispersion curves from the MD simulations were computed using the atomic trajectories and velocities76. This calculation essentially involves the core particles, which carry the atomic masses, as the shell particles have vanishing mass and can be viewed only as mediators to the interatomic interactions. First, the power spectral density is estimated through the spectral decomposition of the autocorrelation k-space velocity function77. The phonon spectra are then constructed through the computed power spectral density using a selected point sequence in the reciprocal space. This procedure is performed recurrently for each individual atom in the unit cell and its Cartesian space components, so as to account for all degrees of freedom. The power spectral density results are subsequently merged to obtain the total phonon spectra, or analyzed separately to obtain the partial phonon spectra (see Fig. 4 from the main text).
The velocity distribution in reciprocal space for the \(a\)-th atom in the unit cell at fixed wavevector \({\bf{k}}\) is given by
where the summation index \(p\) accounts for all unit cells, \({v}_{j}\) are the atomic velocity components, \(t\) is the time, and \({\bf{k}}\,\cdot \,{\bf{r}}\) is the dot product between the wavevector and the atomic position vector. Since the MD simulations provide the atomic trajectories in terms of a finite (truncated) time sequence, \({t}_{m}\), the following velocity autocorrelation estimator \(s\) was used in our computations:
where \(N\) is the number of data samples, \(m\) is the positive time index for the entire data sample sequence, which runs from 1 to \(N\), and the asterisk refers to the complex conjugate. Finally, the power spectral density is calculated through Fast Fourier Transform of each autocorrelation sequence.
Along these lines, the results in section “Finite-temperature analyses of phonon dispersion” illustrate the potential of our large-scale simulations with multiatomic lattices and computationally intensive MD potentials in rendering high resolution, rather continuum, phonon dispersion curves with a large number of points in k space. Moreover, while the conventional approaches using the dynamical matrix furnish the relationship between the phonon frequency \(\omega\) and the wavevector k, our phonon dispersions obtained through the actual thermally enhanced atomic fluctuations from the MD simulations reveal important lattice dynamics phenomena such as the fundamental anisotropy of the mode softening. Although this information is concealed in the overall spectra, it can be clearly discerned when the spectra are decomposed in Cartesian directions. A detailed comparison between the phonon dispersions from the MD simulations and the DFT simulations under the harmonic approximation is provided in the Supplementary Information (Supplementary Fig. 5).
Data availability
Data are available from the authors upon reasonable request.
Code availability
All molecular dynamics simulations were performed with the open source LAMMPS code (https://github.com/lammps/lammps). The electronic structure code Vienna Ab initio Simulation Package (VASP) can be downloaded upon completion of the license agreement in the webpage of the package. The phonon postprocessing from the DFT calculations was performed with the open source code Phonopy, available at https://github.com/phonopy/phonopy. The MATLAB codes used in the data analyses from the MD simulations are available upon request to the authors.
References
Eremenko, M. et al. Local atomic order and hierarchical polar nanoregions in a classical relaxor ferroelectric. Nat. Commun. 10, 2728 (2019).
Pramanick, A. & Nayak, S. Perspective on emerging views on microscopic origin of relaxor behavior. J. Mater. Res. 36, 1015–1036 (2021).
Chaves, A. S., Barreto, F. C. S., Nogueira, R. A. & Zẽks, B. Thermodynamics of an eight-site order-disorder model for ferroelectrics. Phys. Rev. B 13, 207–212 (1976).
Bersuker, I. B. On the origin of ferroelectricity in perovskite-type crystals. Phys. Lett. 20, 589–590 (1966).
Cochran, W. Crystal stability and the theory of ferroelectricity. Adv. Phys. 9, 387–423 (1960).
Comes, R., Lambert, M. & Guinier, A. The chain structure of BaTiO3 and KNbO3. Solid State Commun. 6, 715–719 (1968).
Ravel, B., Stern, E. A., Vedrinskii, R. I. & Kraizman, V. Local structure and the phase transitions of BaTiO3. Ferroelectrics 206–207, 407–430 (1998).
Stern, E. A. Character of order-disorder and displacive components in barium titanate. Phys. Rev. Lett. 93, 037601 (2004).
Ravy, S., Itié, J.-P., Polian, A. & Hanfland, M. High-pressure study of X-ray diffuse scattering in ferroelectric perovskites. Phys. Rev. Lett. 99, 117601 (2007).
Kwei, G. H., Billinge, S. J. L., Cheong, S.-W. & Saxton, J. G. Pair-distribution functions of ferroelectric perovskites: direct observation of structural ground states. Ferroelectrics 164, 57–73 (1995).
Cohen, R. E. Origin of ferroelectricity in perovskites: the principal problems from a theoretical perspective. Ferroelectrics 150, 1–12 (1993).
Blinc, R. Order and disorder in ferroelectrics. Ferroelectrics 301, 3–8 (2004).
Chapman, B. D. et al. Diffuse x-ray scattering in perovskite ferroelectrics. Phys. Rev. B - Condens. Matter Mater. Phys. 71, 2–5 (2005).
Cohen, R. E. Origin of ferroelectricity in perovskite oxides. Nature 358, 136–138 (1992).
Cohen, R. E. & Krakauer, H. Electronic structure studies of the differences in ferroelectric behavior of BaTiO3 and PbTiO3. Ferroelectrics 136, 65–83 (1992).
Shirane, G., Frazer, B. C., Minkiewicz, V. J., Leake, J. A. & Linz, A. Soft optic modes in barium titanate. Phys. Rev. Lett. 19, 234–235 (1967).
Comès, R. & Shirane, G. Neutron-scattering analysis of the linear-displacement correlations in KTaO3. Phys. Rev. B 5, 1886–1891 (1972).
Harada, J., Axe, J. D. & Shirane, G. Neutron-scattering study of soft modes in cubic BaTiO3. Phys. Rev. B 4, 155–162 (1971).
Lima, C. A. S., Scalabrin, A., Miranda, L. C. M., Vargas, H. & Porto, S. P. S. Temperature behaviour of the dielectric constant of tetragonal BaTiO3. Phys. Status Solidi 86, 373–378 (1978).
Luspin, Y., Servoin, J. L. & Gervais, F. Soft mode spectroscopy in barium titanate. J. Phys. C. Solid State Phys. 13, 3761–3773 (1980).
Vogt, H., Sanjurjo, J. A. & Rossbroich, G. Soft-mode spectroscopy in cubic BaTiO3 by hyper-Raman scattering. Phys. Rev. B 26, 5904–5910 (1982).
Girshberg, Y. & Yacoby, Y. Ferroelectric phase transitions in perovskites with off-center ion displacements. Solid State Commun. 103, 425–430 (1997).
Sepliarsky, M., Migoni, R. L. & Stachiotti, M. G. Ab initio supported model simulations of ferroelectric perovskites. Comput. Mater. Sci. 10, 51–56 (1998).
Ghosez, P., Gonze, X. & Michenaud, J. P. Ab initio phonon dispersion curves and interatomic force constants of barium titanate. Ferroelectrics 206–207, 205–217 (1998).
Ghosez, P., Cockayne, E., Waghmare, U. V. & Rabe, K. M. Lattice dynamics of BaTiO3, PbTiO3, and PbZrO3: a comparative first-principles study. Phys. Rev. B 60, 836–843 (1999).
Yu, R. & Krakauer, H. First-principles determination of chain-structure instability in KNbO3. Phys. Rev. Lett. 74, 4067–4070 (1995).
Kotiuga, M. et al. Microscopic picture of paraelectric perovskites from structural prototypes. Phys. Rev. Res. 4, 1–7 (2022).
Edwardson, P. J. Corridors-between-adjacent-sites model of the four phases of KNbO3. Phys. Rev. Lett. 63, 55–58 (1989).
Gigli, L. et al. Thermodynamics and dielectric response of BaTiO3 by data-driven modeling. npj Comput Mater 8, 209 (2021).
Tinte, S., Stachiotti, M. G., Sepliarsky, M., Migoni, R. L. & Rodriguez, C. O. Order-disorder, local structure and precursor effects in BaTio3. Ferroelectrics 237, 41–48 (2000).
Zhong, W., Vanderbilt, D. & Rabe, K. M. Phase Transitions in BaTiO3 from First Principles. Phys. Rev. Lett. 73, 1861–1864 (1994).
Pirc, R. & Blinc, R. Off-center Ti model of barium titanate. Phys. Rev. B - Condens. Matter Mater. Phys. 70, 1–8 (2004).
Zhang, Q. & Goddard, W. A. Charge and polarization distributions at the 90° domain wall in barium titanate ferroelectric. Appl. Phys. Lett. 89, 1–4 (2006).
Zhao, X. G., Malyi, O. I., Billinge, S. J. L. & Zunger, A. Intrinsic local symmetry breaking in nominally cubic paraelectric BaTi O3. Phys. Rev. B 105, 224108 (2022).
Paściak, M., Boulfelfel, S. E. & Leoni, S. Polarized cluster dynamics at the paraelectric to ferroelectric phase transition in BaTiO3. J. Phys. Chem. B 114, 16465–16470 (2010).
Tsuda, K. & Tanaka, M. Direct observation of the symmetry breaking of the nanometer-scale local structure in the paraelectric cubic phase of BaTiO3 using convergent-beam electron diffraction. Appl. Phys. Express 9, 1–5 (2016).
Bencan, A. et al. Atomic scale symmetry and polar nanoclusters in the paraelectric phase of ferroelectric materials. Nat. Commun. 12, 3509 (2021).
Tsuda, K., Sano, R. & Tanaka, M. Nanoscale local structures of rhombohedral symmetry in the orthorhombic and tetragonal phases of BaTiO3 studied by convergent-beam electron diffraction. Phys. Rev. B - Condens. Matter Mater. Phys. 86, 2–6 (2012).
Lee, D. et al. Mixed Bloch-Néel-Ising character of 180° ferroelectric domain walls. Phys. Rev. B - Condens. Matter Mater. Phys. 80, 060102 (2009).
Sluka, T., Tagantsev, A. K., Bednyakov, P. & Setter, N. Free-electron gas at charged domain walls in insulating BaTiO3. Nat. Commun. 4, 1808 (2013).
Bednyakov, P. S., Sturman, B. I., Sluka, T., Tagantsev, A. K. & Yudin, P. V. Physics and applications of charged domain walls. npj Comput Mater 4, 65 (2018).
Rubio-Marcos, F., Campo, A. Del, Marchet, P. & Fernández, J. F. Ferroelectric domain wall motion induced by polarized light. Nat. Commun. 6, 6594 (2015).
Rubio-Marcos, F. et al. Experimental evidence of charged domain walls in lead-free ferroelectric ceramics: light-driven nanodomain switching. Nanoscale 10, 705–715 (2018).
Ordoñez-Pimentel, J. et al. Light-driven motion of charged domain walls in isolated ferroelectrics. Phys. Rev. B 106, 224110 (2022).
Rubio-Marcos, F. et al. Reversible optical control of macroscopic polarization in ferroelectrics. Nat. Photon. 12, 29–32 (2018).
Bai, F., Li, J. & Viehland, D. Domain hierarchy in annealed (001)-oriented Pb(Mg 1/3Nb 2/3)O 3-x%PbTiO 3 single crystals. Appl. Phys. Lett. 85, 2313–2315 (2004).
Jin, Y. M., Wang, Y. U., Khachaturyan, A. G., Li, J. F. & Viehland, D. Adaptive ferroelectric states in systems with low domain wall energy: tetragonal microdomains. J. Appl. Phys. 94, 3629–3640 (2003).
Fu, H. & Cohen, R. E. Polarization rotation mechanism for ultrahigh electromechanical response in single-crystal piezoelectrics. Nature 403, 281–283 (2000).
Bian, J. et al. Fingerprints of relaxor ferroelectrics: Characteristic hierarchical domain configurations and quantitative performances. Appl. Mater. Today 21, 100789 (2020).
Roleder, K. et al. Monoclinic domain populations and enhancement of piezoelectric properties in a PZT single crystal at the morphotropic phase boundary. Phys. Rev. B 105, 144104 (2022).
Wang, H. et al. Hierarchical micro-/nanoscale domain structure in MC phase of (1-x)Pb(Mg1/3Nb2/3)O3-xPbTiO3 single crystal. Appl. Phys. Lett. 89, 87–90 (2006).
Hinterstein, M. et al. Interplay of strain mechanisms in morphotropic piezoceramics. Acta Mater. 94, 319–327 (2015).
Lummen, T. T. A. et al. Thermotropic phase boundaries in classic ferroelectrics. Nat. Commun. 5, 3172 (2014).
Fu, J., Zuo, R. & Xu, Z. High piezoelectric activity in (Na,K)NbO 3 based lead-free piezoelectric ceramics: Contribution of nanodomains. Appl. Phys. Lett. 99, 3–6 (2011).
Wang, Y. U. Three intrinsic relationships of lattice parameters between intermediate monoclinic MC and tetragonal phases in ferroelectric Pb [(Mg1/3 Nb2/3) 1-x Tix] O3 and Pb [(Zn1/3 Nb2/3) 1-x Tix] O3 near morphotropic phase boundaries. Phys. Rev. B Condens. Matter Mater. Phys. 73, 1–13 (2006).
Jin, Y. M., Wang, Y. U., Khachaturyan, A. G., Li, J. F. & Viehland, D. Conformal miniaturization of domains with low domain-wall energy: monoclinic ferroelectric states near the morphotropic phase boundaries. Phys. Rev. Lett. 91, 1–4 (2003).
Nakajima, M. et al. Large Electromechanical responses driven by electrically induced dense ferroelastic domains: beyond morphotropic phase boundaries. ACS Appl. Electron. Mater. 2, 1908–1916 (2020).
Diéguez, O. et al. Ab initio study of the phase diagram of epitaxial BaTiO3. Phys. Rev. B - Condens. Matter Mater. Phys. 69, 1–4 (2004).
Choi, K. J. et al. Enhancement of ferroelectricity in strained BaTiO3 thin films. Science 306, 1005–1009 (2004).
Pramanick, A., Prewitt, A. D., Forrester, J. S. & Jones, J. L. Domains, domain walls and defects in perovskite ferroelectric oxides: a review of present understanding and recent contributions. Crit. Rev. Solid State Mater. Sci. 37, 243–275 (2012).
Catalan, G., Seidel, J., Ramesh, R. & Scott, J. F. Domain wall nanoelectronics. Rev. Mod. Phys. 84, 119–156 (2012).
Ganesh, P. & Cohen, R. E. Pressure induced phase transitions in PbTiO3. J. Phys. Condens. Matter 21, 064225 (2009).
Ahart, M. et al. Origin of morphotropic phase boundaries in ferroelectrics. Nature 451, 545–548 (2008).
Kumar, A. & Waghmare, U. V. First-principles free energies and Ginzburg-Landau theory of domains and ferroelectric phase transitions in BaTiO3. Phys. Rev. B - Condens. Matter Mater. Phys. 82, 1–10 (2010).
Zhang, L. et al. A new strategy for large dynamic piezoelectric responses in lead-free ferroelectrics: the relaxor/morphotropic phase boundary crossover. Adv. Funct. Mater 30, 2004641 (2020).
Ke, X. Q., Wang, D., Ren, X. & Wang, Y. Formation of monoclinic nanodomains at the morphotropic phase boundary of ferroelectric systems. Phys. Rev. B Condens. Matter Mater. Phys. 88, 1–6 (2013).
Vielma, J. M. & Schneider, G. Shell model of BaTiO3 derived from ab-initio total energy calculations. J. Appl. Phys. 114, 20–24 (2013).
Tinte, S., Stachiotti, M. G., Sepliarsky, M., Migoni, R. L. & Rodriguez, C. O. Atomistic modelling of BaTiO3 based on first-principles calculations. J. Phys. Condens. Matter 11, 9679–9690 (1999).
Boddu, V., Endres, F. & Steinmann, P. Molecular dynamics study of ferroelectric domain nucleation and domain switching dynamics. Sci. Rep. 7, 806 (2017).
Sepliarsky, M., Asthagiri, A., Phillpot, S. R., Stachiotti, M. G. & Migoni, R. L. Atomic-level simulation of ferroelectricity in oxide materials. Curr. Opin. Solid State Mater. Sci 9, 107–113 (2005).
Wu, H. H. & Cohen, R. E. Electric-field-induced phase transition and electrocaloric effect in PMN-PT. Phys. Rev. B 96, 054116 (2017).
Kresse, G. & Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169–11186 (1996).
Blöchl, P. E. Projector augmented-wave method. Phys. Rev. B 50, 17953–17979 (1994).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1996).
Togo, A. & Tanaka, I. First principles phonon calculations in materials science. Scr. Mater. 108, 1–5 (2015).
Dickey, J. M. & Paskin, A. Computer simulation of the lattice dynamics of solids. Phys. Rev. 188, 1407–1418 (1969).
Marple, S. L. Digital Spectral Analysis: with Applications (Prentice-Hall International, 1987).
Acknowledgements
J.A. acknowledges financial support by Ministerio de Ciencia e Innovación (Grant PID2019-106744GB-I00 to the UPC). The work by J.O. and J.M. was sponsored by The Ministry of Education, Youth and Sports under grant CEDAMNF CZ.02.1.01/0.0/0.0/15_003/0000358 (Czech Republic).
Author information
Authors and Affiliations
Contributions
J.O and J.A. designed the research. J.O. and J.M. performed the MD and DFT simulations, respectively. J.O. analyzed the results. J.A wrote the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Očenášek, J., Minár, J. & Alcalá, J. Dynamics of lattice disorder in perovskite materials, polarization nanoclusters and ferroelectric domain wall structures. npj Comput Mater 9, 118 (2023). https://doi.org/10.1038/s41524-023-01069-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41524-023-01069-6
- Springer Nature Limited