

Abstract
Germline and somatic TP53 variants play a crucial role during tumorigenesis. However, genetic variations that solely affect the alternatively spliced p53 isoforms, p53β and p53γ, are not fully considered in the molecular diagnosis of Li-Fraumeni syndrome and cancer. In our search for additional cancer predisposing variants, we identify a heterozygous stop-lost variant affecting the p53β isoforms (p.*342Serext*17) in four families suspected of an autosomal dominant cancer syndrome with colorectal, breast and papillary thyroid cancers. The stop-lost variant leads to the 17 amino-acid extension of the p53β isoforms, which increases oligomerization to canonical p53α and dysregulates the expression of p53’s transcriptional targets. Our study reveals the capacity of p53β mutants to influence p53 signalling and contribute to the susceptibility of different cancer types. These findings underscore the significance of p53 isoforms and the necessity of comprehensive investigation into the entire TP53 gene in understanding cancer predisposition.
Similar content being viewed by others


Introduction
A substantial proportion of cancer patients have a familial history of disease, although the extent of this phenomenon is dependent on the type of cancer1. For instance, disease clustering within families can be observed in 20–30% of colorectal cancer patients, 15–20% of breast cancer patients, and ~5% of papillary thyroid cancer patients2,3,4,5. However, the genetic cause of familial cancer predisposition is only known in approximately half the familial colorectal and breast cancer cases and remains largely elusive for most of the familial papillary thyroid cancers. In contrast to most inherited cancer syndromes, that predominantly affect specific organs, Li-Fraumeni syndrome (OMIM#151623) predisposes to a variety of early-onset cancers, including breast cancer, bone and soft tissue sarcomas, brain cancer, and to a lesser extent, colorectal cancer and thyroid cancer (2015 revised Chompret criteria)6. Pathogenic germline variants in the TP53 gene are responsible for most cases of Li-Fraumeni syndrome, however different pathogenic variants confer varying cancer risks and are associated with different types of tumors7,8,9,10,11.
The TP53 gene encodes at least 12 different p53 protein isoforms. This results from the combination of four translation initiation sites (full-length, Δ40, Δ133, and Δ160) and three distinct C-terminal protein isoforms (α, β, and γ). The C-terminus of the p53β and p53γ isoforms are encoded by two cryptic exons (9β and 9γ, respectively), located in intron-9, that contain premature stop codons12. In suspected Li-Fraumeni syndrome patients only the canonical coding exons of the TP53 gene (encoding full-length p53α) are sequenced, as the pathogenicity of genetic variants located in the 9β and 9γ exons has not yet been established13. Advances in genomic technologies have facilitated the identification of additional germline susceptibility loci.
In this work, we perform whole-exome sequencing on the germline DNA of individuals that are considered likely of carrying unidentified cancer-predisposing genetic variants. We identify a candidate cancer-predisposing variant that specifically affects p53β isoforms, but not full-length p53α. Functional characterization of this variant supports the oncogenic effect by increasing oligomerization to canonical p53α and dysregulating the expression of p53’s transcriptional targets.
Results
Identification of the cancer susceptibility variant
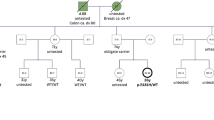
We performed germline whole-exome sequencing analysis of 94 individuals suspected of being genetically predisposed to colorectal cancer (Online Methods). These individuals were either members of families with high colorectal cancer burden, early-onset colorectal cancer patients, and/or patients presenting with multiple colonic adenomas or hyperplastic polyps. Known Mendelian syndromes had previously been excluded by multigene screening approaches in a diagnostic context. We identified one recurrent heterozygous variant in two unrelated families (Fig. 1a, b), which corresponds to a stop-lost variant in the cryptic exon 9β of the TP53 gene ((hg38): chr17:7673308T > G, (NM_001126114.2): c.1025A > C, (NP_001119586.1): p.(*342Serext*17)). This exon encodes the 10-amino acid C-terminus of the p53β isoforms (DQTSFQKENC*), of which the exact function remains largely elusive. The stop-lost variant is predicted to elongate all p53β isoforms by 17 amino acids (SREHENGSMTLPDTDAT*; Fig. 2 and Supplementary Fig. 1). This genetic variant and its protein isoforms will, hereafter, be referred to as the TP53β-stop-lost variant and the p53β-stop-lost protein isoforms.

The pedigree of (a) family 1, (b) family 2, (c) family 3, and (d) family 4 are shown. Genotypes of all tested individuals are indicated: Mut, mutation carrier; Mut*, obligate carrier; WT, wild-type. Symbols: filled quarters represent cancer patients; filled top left quarter, colorectal cancer; filled top right quarter, breast cancer; filled bottom right quarter, thyroid cancer; filled bottom left quarter, other cancers; question mark, unknown phenotype. Numbers in brackets indicate relatives merged in the pedigree for clarity. The abbreviations under each symbol indicate the diagnosis of a malignancy or carcinoma in situ (asterisk), followed by the age at diagnosis or age at death (d) reported in 5-year aggregates: B breast, Bl bladder, Br brain, C colorectum, Cx cervix, Ga gallbladder, Ki kidney, Lu lung, Me melanoma, NHL non-Hodgkin lymphoma, O ovary, SL serrated colonic lesions, St stomach, Thr throat, Thy thyroid. The abbreviations above each symbol indicate E, whole-exome sequencing analysis of leukocyte DNA; T, somatic hotspot analysis of neoplasia; B, patient-derived B-lymphocyte cell experiments. Clinicopathological characteristics of malignant and benign tumors are provided in Supplementary Table 2.

a The TP53 gene locus is composed of 11 exons and two cryptic exons (9β and 9γ). Its two promoters (P1 and P2) result in four different translation initiation sites (full-length, Δ40, Δ133 and Δ160). b The TP53 gene encodes for at least 12 different p53 protein isoforms, where alternative splicing of intron-9 leads to three distinct C-terminal domains (α, β, and γ). p53α is composed of the transactivation domain, the DNA-binding domain, the hinge domain, the oligomerization domain, and the p53α regulatory domain. The p53β and p53γ isoforms only contain the first 7 amino acids of the oligomerization domain. The TP53β-stop-lost variant is predicted to elongate the p53β-domain (cyan) by 17 amino acids (red) for all p53β protein isoforms.
We further investigated the presence of the TP53β-stop-lost variant in a whole-exome sequencing dataset composed of 18 unexplained, cancer-prone families (26 individuals), and identified this variant in a family affected by, amongst others, breast and papillary thyroid cancer (Fig. 1c). Strikingly, whole-exome sequencing analysis performed in a diagnostic context found a fourth family containing multiple papillary thyroid cancer patients (Fig. 1d). Also, in these two families no known pathogenic variants were identified in other cancer-associated genes. The TP53β-stop-lost variant was not present in a local cohort of consecutive colorectal and breast cancer patients (0/1960; Online Methods, Supplementary Table 1). We analyzed public sequencing databases to determine the frequency of this variant (annotated as rs764562217) in the general population (Supplementary Table 1). In the Genomes of the Netherlands (GoNL) database no carriers were found (0/498). In the Genome Aggregation Database14 (gnomAD v2.1.1), which largely contains non-cancer individuals, only seven heterozygous carriers were reported (7/124,924 individuals), with the highest frequency amongst North-Western Europeans (5/23,366). This corresponds to a higher reported prevalence than other known pathogenic TP53 variants, such as the variants encoding p.Arg273His (4/125,527) and p.Arg248Gln (3/125,737). In addition, we searched the Catalog of Somatic Mutations in Cancer (COSMIC v96) database for somatic mutations. This variant was reported only once (1/149,537), in a patient diagnosed with a chronic hematological malignancy (myelodysplatic syndrome with single lineage dysplasia)15. A germline origin of this variant cannot be excluded as no germline DNA analysis was performed. Furthermore, no additional genetic variants (i.e., single nucleotide variants or frameshift mutations) have been described that result into a stop-lost mutation in the TP53 9β exon.
Enhanced cancer susceptibility in families with the TP53β-stop-lost variant
The TP53β-stop-lost variant was identified in four families, all of whom exhibited a spectrum of malignancies and demonstrated inheritance patterns consistent with an autosomal dominant cancer syndrome (as shown in Fig. 1). Subsequent genotyping of 36 DNA samples from individuals belonging to family 1, 3 and 4 revealed an additional 22 mutation carriers (details in Online methods). Altogether, we identified a total of 27 carriers, comprising 14 individuals with cancer, 11 unaffected individuals at the end of the follow-up period, and 2 relatives of whom clinical data was unavailable. Furthermore, 7 out of these 14 cancer patients were diagnosed with multiple primary cancers. In total, 26 cancers were diagnosed in carriers, most frequently (mismatch-repair proficient) colorectal cancer (n = 8), different subtypes of breast cancer (n = 7), and papillary thyroid cancer (n = 6). All genotypes and clinicopathological characteristics are listed in Supplementary Table 2. For all carriers and cancers combined, based on these four families severely affected by cancer, the penetrance at the ages of 30 years, 50 years, 60 years, and 70 years was estimated at, respectively, 0%, 31%, 62%, and 69% (Kaplan-Meier curve shown in Supplementary Fig. 2a). Of note, also in non-carriers a relatively high cancer risk was observed, suggesting that the presence of additional risk factors might be at play in these families.
We investigated recent shared ancestry between these four families by performing identity-by-descent (IBD) analysis in PLINK16 using genotypes extracted at 2364 independent locations from 54 whole-exome sequencing dataset (Online Methods). Based on individuals with documented family relationships, including two individuals from family 1 and 3, a discrimination was possible for first, second and third-degree relatives and unrelated individuals (Supplementary Fig. 2b). The proportion of shared alleles between the four TP53β-stop-lost families was 0% for all pairs, indicating that a shared recent ancestor is unlikely. Nevertheless, the TP53β-stop-lost variant is likely located on the locus containing the TP53 c.215 C > G, p.Arg72 allele (annotated as rs1042522), since all TP53β-stop-lost carriers were either heterozygous or homozygous for this variant. This is in accordance with the predominance of the p.Arg72 allele in North-Western Europeans (allele frequency = 0.74 in gnomAD).
To investigate the four pedigrees for segregation of the TP53β-stop-lost variant with the cancer phenotype, we performed co-segregation analysis based on the full-likelihood Bayes (FLB) factor17 (Online Methods). We considered as phenotype of interest any cancer, excluding lung cancer, diagnosis before 60 years of age, due to restricted follow-up data past this age. Cumulative risk of non-carriers was assumed to be equal to the general population. Using a dominant disease model with 50% penetrance resulted in FLB scores ranging 1.00–3.44 (Supplementary Fig. 2c). A combined FLB score of the four families of 6.5 provided supportive evidence for pathogenicity (FLB > 4), according to the thresholds suggested by Jarvik and Browning18.
Retained TP53β-stop-lost variant in neoplasia
Somatic TP53 mutations with loss of heterozygosity (LOH) of the wild-type allele, are frequent in many tumor types19. Recently, LOH has also been shown as an early event in tumorigenesis of Li-Fraumeni syndrome tumors20, contrary to previous reports21,22. In 11 neoplasia from 7 TP53β-stop-lost carriers (7 carcinomas, 1 carcinoma in situ, and 3 benign lesions, indicated in Fig. 1), we examined somatic mutations and LOH in the TP53 gene and other cancer genes by comparing tumor DNA to that of adjacent normal tissues or leukocytes (Online Methods). Heterozygosity at the TP53 locus was retained in 82% of the tissues, with only one sample showing loss of the TP53β-stop-lost variant and one sample showing loss of the wild-type allele (Supplementary Table 3). The colorectal carcinoma, that had lost the TP53β-stop-lost variant, harbored a pathogenic somatic TP53 mutation (encoding p.Gly262Val) at the other allele (Supplementary Table 4)23. In another colorectal cancer, we observed two pathogenic somatic TP53 mutations (encoding p.Arg248Trp and p.Arg283Pro), however, we were unable to determine on which allele these mutations were located, due to genetic spacing. Hence, these tumors commonly retained heterozygosity of the stop-lost allele and did not show other distinctive genetic features.
Detection of TP53β isoforms in B cell lines
To investigate the expression of the different TP53 isoforms at the transcriptional and protein level, we analyzed Epstein-Barr virus (EBV)-immortalized B-lymphocyte cells established from a TP53β-stop-lost carrier (NA50, indicated in Fig. 2) and eight unrelated p53 wild-type colorectal cancer patients. We performed targeted transcriptome sequencing of each TP53 exon in mutant NA50 cells compared to one p53 wild-type cell line (Online Methods). In the mutant NA50 cells, the expression of the 9β exon was specifically increased (Fig. 3a). Quantification of TP53β transcripts, by isoform-specific qPCR assay, showed 4-to-7-fold higher expression (p < 0.001) of TP53β in mutant NA50 cells when compared to two wild-type samples NA16 and NA68 (Fig. 3b). Quantification of allele-specific expression, by KASPar assay, showed a 5-fold (mean ± standard deviation: 4.7 ± 1.9; p = 0.01) higher expression of the mutant allele versus the wild-type TP53β allele, indicating dominant mRNA expression from the TP53β-stop-lost allele (Supplementary Fig. 3a), which was consistent with the results from transcriptome reads (Supplementary Fig. 3b). We investigated whether the increased expression of the TP53β transcripts and the allelic imbalance was a consequence of the TP53β-stop-lost variant gaining resistance to non-sense mediated decay, which is reported to degrade TP53β transcripts, but not TP53α transcripts, due to the premature stop codon24. After inhibition of non-sense mediated decay in B-cells (Online Methods), we observed no changes in TP53α levels and a 10-fold increase in TP53β expression, irrespective of mutation status, indicating that the allelic imbalance is not the result of resistance to non-sense mediated decay (Supplementary Fig. 3a–c). Taken together, we showed high transcriptional levels of TP53β isoforms, mostly derived from the TP53β-stop-lost allele, in carrier-derived B-cells.

a Targeted transcriptome sequencing of the whole TP53 gene showed a relative increase in 9β exon expression in TP53β-stop-lost carrier-derived (NA50) compared to non-carrier-derived (NA68) B-cells. b Increased TP53β expression was confirmed in NA50 cells compared to p53 wild-type B-cells (NA68, NA16). Difference in TP53α expression were <2-fold between the samples. Expression was measured with isoform-specific qPCR primers. Mean and standard deviations are plotted of biological replicates (n = 3). P-values were calculated with an unpaired two-sided t test (NA16:NA68: TP53α p < 0.001, TP53β p = 0.12; NA50:NA68: TP53α p < 0.001, TP53β p = 0.01). c Immunoblotting using the p53β-specific KJC8 antibody and full-length p53-specific antibody DO-7 was performed on nine B-cell samples. DO-7 preferentially detects p53α. Wild-type p53β and p53β-stop-lost co-migrate with gel electrophoresis, making it impossible to distinguish them using generic p53 antibodies. All samples showed comparable protein expression of p53 isoforms, including p53α, p53β, Δ133p53β and Δ160p53β. d The p53β-stop-lost-specific (p53β-SL) antibody was generated against synthetic p53β-stop-lost (p53β-SL) peptide (CSREHENGSMTLPDTDAT) and showed specificity for p53β-stop-lost protein isoforms in p53-null cells (H1299) transiently transfected with different p53 isoform expression vectors. Proteins were analyzed by immunoblotting. DO-1 blot was cut. e The p53β-SL and Δ133p53β-SL proteins were detected in mutant B-cells (NA50) but not in non-carrier B-cells (NA16). Both samples showed comparable expression of other p53 isoforms, shown by re-probing with full-length specific DO-1 and pan-p53 antibody SAPU. f The mRNA expression of 21 p53-target genes was measured for biological replicates (n = 3) with a targeted qPCR panel, showing altered regulation in mutant NA50 compared to p53 wild-type (NA16, NA68) B-cells. 13 genes (in bold) are consistently altered in the same direction. Median fold changes and p-values, calculated with an unpaired two-sided t test are shown in Supplementary Table 5. g Protein expression of the p53-target protein p21 (encoded by CDKN1A) was assessed with immunoblotting. Mutant B-cells (NA50) showed higher basal expression of p21 and an altered response to cellular stress, tested by doxorubicin treatment (0.5 μM), at different timepoints. All immunoblotting was performed in two independent experiments. Source data are provided as a Source Data file.
Next, we examined the expression of p53 protein isoforms in mutant NA50 B-cells with immunoblotting, using different p53 isoform-specific antibodies (Online Methods). NA50 and B-cells derived from unrelated colorectal cancer patients showed expression of p53 isoforms, predominantly p53α, p53β and Δ160p53β (Fig. 3c). To specifically detect the expression of p53β-stop-lost protein isoforms, we successfully generated a p53β-stop-lost-specific antibody (p53β-SL) against a 13 amino-acid peptide (SREHENGSMTLPD) only present in p53β-stop-lost proteins (Online methods). The specificity of this antibody was assessed in p53-null H1299 cells (lung cancer cell line) which were transiently transfected with expression vectors encoding different p53 isoforms. The p53β-SL antibody only detected p53β-stop-lost containing proteins, but not p53α, p53β nor other proteins in H1299 cells (Fig. 3d). Subsequently, we performed immunoblotting using the p53β-SL antibody and detected expression of p53β-stop-lost and Δ133p53β-stop-lost proteins in the mutant NA50 cells, but not in wild-type NA16 B-cells (Fig. 3e). This confirms endogenous expression of p53β-stop-lost at the protein level in patient-derived cell lines.
Altered p53-target gene expression in B cell lines
We questioned whether the endogenous expression of p53β-stop-lost isoforms resulted into altered expression of p53-target genes in the patient-derived B cells. p53 transcriptionally regulates over 900 genes encoding proteins involved in e.g., cell cycle arrest, apoptosis, autophagy, senescence, DNA repair, and differentiation25,26. We therefore measured the gene expression level of twenty-one p53-target genes using qPCR in mutant NA50 and two p53 wild-type B-cell models (Online Methods). The variability between the two p53 wild-type samples indicates that the genetic and epigenetic background of the B-cells derived from different patients influences the expression of p53-target genes, as previously established27,28,29,30,31. However, 13 genes were consistently up- or down-regulated in the TP53β-stop-lost carrier-derived NA50 cells on top of this baseline variability, including CDKN1A, PTEN, BAX, MYC and TIGAR (Fig. 3f and Supplementary Table 5). Subsequently, we performed immunoblotting of p53-target protein p21 (encoded by CDKN1A) in B-cells (Online Methods). Contrary to the low mRNA expression level, mutant NA50 cells showed increased basal expression of p21 protein compared to wild-type NA16 cells (Fig. 3g). In addition, we induced cellular stress in NA50 and NA16 cells by doxorubicin treatment, a chemotherapeutic agent which causes DNA damage and histone eviction. NA50 cells showed a distinct p21 protein expression profile compared to NA16 with an earlier induction of p21 expression (Fig. 3g). This suggests that B-cells, which express at least full-length p53β-stop-lost and Δ133p53β-stop-lost in addition to the wild-type p53 isoforms, have an altered expression pattern of some p53-target genes, such as CDKN1A encoding p21 protein.
Modulated p53 signaling in full-length p53-β-stop-lost cell line models
p53 transcriptional activity is highly dependent on the cell context, which includes but is not limited to genetics27,28, chromatin structure, cell type, and cell signals29,30,31. Consistently, gene expression patterns in patient-derived B-cells confirmed that cell context is important when studying p53-target gene expression. We therefore compared p53β-stop-lost and wild-type p53β functions in isogenic, wild-type TP53 cell line models; the colon cancer cell line HCT-116 and the breast cancer cell line MCF-7. We transduced these cells with the full-length p53β-stop-lost, full-length p53β, or empty expression vector as a control (Online Methods; Supplementary Fig. 4). The basal mRNA expression of twenty-one p53-target genes, measured by qPCR (Online Methods), was higher for most genes, such as CDKN1A, in the p53β-stop-lost-overexpressing cells (HCT-116 and MCF-7) compared to the corresponding control or p53β overexpressing cells (Fig. 4a and Supplementary Table 6). Furthermore, immunoblotting revealed an elevated basal expression of the cyclin-dependent kinase inhibitor protein p21, encoded by CDKN1A, in HCT-116 cells overexpressing p53β-stop-lost, which aligns with the immunoblotting results obtained from patient-derived B-cells (Fig. 4b and Supplementary Fig. 5). In contrast, all MCF-7 conditions only exhibited p21 induction 8 h after doxorubicin treatment, irrespective of the expression of p53β-stop-lost or p53β.

a p53 wild-type cells (HCT-116 and MCF-7) were transduced with full-length p53β, p53β-stop-lost (p53β-SL) or empty vector. p53β-stop-lost overexpression resulted in increased basal expression of most p53-target genes measured by qPCR, compared to p53β and vector controls. Minor differences in expression between the two cell-lines can be attributed genetic background and/or cell-type. Gene expression was measured for biological duplicates and normalized to GAPDH expression. Median fold changes and p-values, calculated with an unpaired two-sided t test, are shown in Supplementary Table 6. b Immunoblots of p21 protein expression, encoded by CDKN1A, after induction of cellular stress by doxorubicin (1.0 µM continuous for 2 h, 4 h or 8 h). HCT-116 p53β-stop-lost overexpressing cells showed increased basal expression of p21, which did not increase upon doxorubicin treatment, as was seen in both control and wild-type p53β overexpressing cells. MCF-7 cells showed induction of p21 after 8 h, irrespective of the overexpression of p53β-stop-lost or p53β. Immunoblotting was performed in two independent experiments (see Supplementary Fig. 5). c Cell cycle analysis of transduced cancer cell lines and response to doxorubicin treatment. In the controls, doxorubicin treatment (1.0 µM continuous for 24 h) resulted in the accumulation in G2M in HCT-116 and S-phase cells in MCF-7, while treatment caused an increased replication blockade in S-phase in the p53β-stop-lost overexpressing HCT-116 cells, but its MCF-7 counterpart, where wild-type p53β overexpressing cells showed the most pronounced blockade in S-phase. Cell cycle analysis was assessed in independent experiments (n = 3) with the NucleoCounter (Chemometec). Data points, means, standard deviations and significance (* p < 0.05; ** p < 0.01) are plotted. Cell cycle distribution and p-values, calculated with a paired two-sided t test, are shown in Supplementary Table 7). d Unbiased gene expression analysis of bulk RNA-sequencing of the transduced cell lines. Thirteen genes were consistently downregulated (logFC < −0.6) in both p53β-stop-lost-overexpressing cell lines, while two genes were upregulated (logFC > 0.6).Source data are provided as a Source Data file.
To delve into the impact of full-length p53β-stop-lost protein expression, we performed cell cycle analysis of the transduced cell lines without treatment and 24 h after the induction of cellular stress by doxorubicin. Doxorubicin blocked cell cycle progression in both control cell lines, however at different stages i.e., HCT-116 cells accumulated in G2M, while MCF-7 accumulated in the S-phase, indicating cell line-specific responses to doxorubicin (Fig. 4c and Supplementary Table 7). For HCT-116, distinctions in cell cycle progression for p53β-overexpressing cells were only evident after doxorubicin treatment. p53β-stop-lost-overexpressing cells showed reduced G2M arrest over S-phase accumulation (mean ± standard deviation: S = 51 ± 5%, G2M = 42 ± 6%) compared to control (S: 40 ± 6%, G2M: 55 ± 6%) and p53β-overexpressing (S: 45 ± 10%, G2M: 48 ± 12%) HCT-116 cells. For MCF-7 cells, p53β-stop-lost-overexpression resulted into distinct cell cycle dynamics compared to control MCF-7 and p53β-overexpressing cells, both in the absence and presence doxorubicin treatment. Without treatment, the majority of the p53β-stop-lost-overexpressing cells (48 ± 4%) and p53β-overexpressing cells (41 ± 5%) were in S-phase, whereas control MCF-7 cells predominantly resided in G1-phase (54 ± 2%). Conversely, 24 h after doxorubicin treatment, most of the p53β-overexpressing cells arrested in G1-phase (G1: 60 ± 2%, S: 40 ± 2%), while control MCF-7 cells (G1: 50 ± 10%, S: 50 ± 10%) and p53β-overexpressing cells (G1: 26 ± 5%, S: 74 ± 5%) showed accumulation in S-phase. Nevertheless, immunoblotting of HCT-116 and MCF-7 cells after doxorubicin treatment, did not show large differences in the expression of apoptotic markers (cleaved PARP and cleaved caspase-3) and DNA damage response markers (histone γH2AX levels) between p53β-stop-lost-overexpressing, p53β-overexpressing and control cells (Supplementary Fig. 5).
To provide a comprehensive analysis of the intricate differences between overexpressing wild-type p53β and p53β-stop-lost, as well as to explore the influence of p53β-stop-lost on gene regulation beyond the selected canonical TP53 targets, we compared the transcriptome of p53β-stop-lost-overexpressing HCT-116 and MCF-7 cells to their wild-type p53β-overexpressing counterparts. We investigated genes that were either up- or downregulated (logFC < 0.6 or logFC > −0.6, representing a 4-fold change) in both p53β-stop-lost-overexpressing cell lines. We found thirteen downregulated genes and two upregulated genes (Fig. 4d). Interestingly, gene-set enrichment analysis (GSEA) identified five genes as being involved in hypoxic response (ALDOC, ANGPTL4, BNIP3L, PPFIA4, NDRG1). Of note, both BNIP3 and BNIP3L are important pro-apoptotic molecules and have been suggested as tumor suppressors32. Furthermore, NDRG1 is involved in p53-mediated apoptotic response and maintenance of chromosomal stability33. Finally, IFI6 is another apoptotic molecule involved in cellular innate immune responses34 and extracellular insults35,36. Altogether, it appears that p53β-stop-lost leads to an increased mRNA expression of canonical p53 target genes and, compared to the wild-type isoform, a reduced expression of several pro-apoptotic genes, in particular those associated with the hypoxia pathway.
Taken together, p53β-stop-lost has an intrinsic transcriptional activity, when co-expressed with other p53 isoforms including p53α, which differs from wild-type p53β. Like p53α29, p53β-stop-lost activity is dependent on cell context and genetic background.
Increased oligomerization properties of p53β-stop-lost
To further characterize the molecular mechanisms of p53β-stop-lost, we performed protein modeling to predict its secondary protein structure. The C-terminus of p53α contains an oligomerization domain which is used for dimerization and tetramerization. p53β isoforms lack the oligomerization domain as 25 out of 32 amino acids are missing (Fig. 2b)37. The crystal structure of the p53β isoforms has not yet been determined and its 10-amino acid p53β-specific peptide sequence shows no homology with known protein domains. We used Phyre238 to predict the secondary protein structure of p53β and p53β-stop-lost. The oligomerization domains of a p53α dimer (PDB 1SAE39) each consist of a β-sheet followed by an α-helix of ~5.5 turns, orientated antiparallel (Fig. 5a, left panel)40,41. The C-terminal domain of p53β was predicted to consist of the identical β-sheet and a helix of ~1.5 turns (Fig. 5a, middle panel). The TP53β-stop-lost variant extended the C-terminal helix to ~4.5 turns (Fig. 5a, right panel). We therefore theorized that the elongation of p53β C-terminal structure confers oligomerization properties to the p53β-stop-lost isoforms.

a Crystal structure of the oligomerization domains of two p53α proteins (left panel), orientated anti-parallel for dimer formation (PDB 1SAE). Composite model of one p53α oligomerization domain and the predicted structure of the C-terminal domains of p53β (middle panel) and p53β-stop-lost (p53β-SL; right panel) showing the oligomerization domain sequence (dark gray), the β-specific protein sequence (cyan) and the p53-stop-lost peptide sequence (red). The TP53β-stop-lost variant is predicted to extend the α-helical structure. Visualization was performed in PyMOL. b Co-immunoprecipitation was performed using p53α-domain-specific antibodies (421/BP53.10) or p53β-domain-specific antibody (KJC8) on protein lysates of p53-null cells (H1299) transiently co-transfected with p53α/p53β or p53α/p53β-stop-lost, and compared to IgG control and whole cell lysates (WCL). p53β-stop-lost, but not wild-type p53β, co-immuno-precipitated with p53α, and vice versa. c Immunofluorescence staining using the p53β-stop-lost-specific (p53β-SL; green) antibody and DAPI (blue) of p53β-stop-lost expressing H1299 cells shows nuclear and cytoplasmic localization of this protein. d Immunoblot of expression levels of p53α, p53β and p53β-stop-lost protein in luciferase gene reporter assays using polyclonal p53 antibody (SAPU). H1299 cells were co-transfected with luciferase reporter plasmid driven by p53-target gene promoter, SV-renilla and empty vector or p53α; or p53α/p53β; or p53α/p53β-SL. Protein extracts were normalized according to the Renilla activity. e For the promoter sequences of p53-target genes CDKN1A, BAX and IGFBP3, the effect of p53β-stop-lost on p53α transcriptional activity was promoter specific (inducive at CDNK1A (p = 0.032) and inhibitory at BAX (p = 0.001) and IGFBP3 (p < 0.001) promoters), while p53β co-expression did not affect p53α transcriptional activity, at comparable protein levels. Expression of p53β-stop-lost alone, in the absence of p53α, did not transactivate these promoters. Luciferase activity was normalized to the basal activity in the absence of p53, and data points, means, standard deviations and p-values (* p < 0.05, ** p < 0.01, *** p < 0.001) are plotted for independent experiments; p53β and p53β-stop-lost (n = 3), other CDKN1A and BAX conditions (n = 6) and other IGFBP3 conditions (n = 7). Normalized transduction and p-values, calculated with an unpaired two-sided t test are shown in Supplementary Table 8. Source data are provided as a Source Data file.
To test this hypothesis, we performed co-immunoprecipitations with antibodies specific for the p53α- or p53β-domains on protein lysates of p53-null H1299 cells co-expressing full-length p53α + p53β or p53α + p53β-stop-lost (Online Methods). We showed that p53β-stop-lost co-immunoprecipitates with p53α, and that p53α co-immuno-precipitates with p53β-stop-lost, while p53α and p53β do not co-immunoprecipitate together (Fig. 5b). This confirms that p53β-stop-lost protein can directly oligomerize and form a protein complex with p53α.
Modulated p53α transcriptional activity of p53β-stop-lost on p53-responsive promoters
Given that p53α and p53β-stop-lost can form protein complexes, we investigated whether p53β-stop-lost can directly modulate the transcriptional activity of p53α on p53-responsive promoters. First, immunofluorescence staining of full-length p53β-stop-lost transfected H1299 cells (Online Methods), using the p53β-stop-lost-specific antibody, showed that, like p53β42, p53β-stop-lost can locate to the nucleus and the cytoplasm (Fig. 5c and Supplementary Fig. 6). Next, we performed luciferase reporter gene assays driven by the natural promoters of p53 target genes CDKN1A, BAX or IGFBP3 in p53-null H1299 cells to avoid interference with other p53 isoforms. Cells were transfected with p53α, p53β or p53β-stop-lost expression vectors either as single plasmid or in combination p53α + p53β or p53α + p53β-stop-lost (Online Methods). Comparable expression levels of p53α, p53β and p53β-stop-lost proteins were obtained to compare their intrinsic transcriptional activity (Fig. 5d and Supplementary Fig. 7).
p53α alone was able to transactivate the CDKN1A, BAX and IGFBP3 promoters, but p53β or p53β-stop-lost in the absence of p53α did not (Fig. 5d and Supplementary Table 8). The transcriptional activity on these promoters was not affected by co-expression of p53α + p53β compared to expression of p53α alone. Interestingly, the co-expression of p53α + p53β-stop-lost enhanced the transcriptional activity of the CDKN1A promoter but abolished the transcription of the BAX promoter, and resulted in reduced transcriptional activity of the IGFBP3 promoter compared to expression of p53α alone. Therefore, p53β-stop-lost either enhances, reduces, or abolishes the transcription activity of p53α in a promoter-dependent manner and has a distinct intrinsic activity from p53β.
Discussion
In our study, we have identified a heterozygous germline TP53β-stop-lost variant in four families, which presented with a high incidence of various cancer types, including colorectal, breast, and papillary thyroid cancer. The clinical manifestation in these independent families aligned with patterns typical of autosomal dominant cancer syndromes for which co-segregation analysis provides supportive evidence.
Functional characterization of the TP53β-stop-lost variant, which exclusively affects the p53β isoforms, but not the canonical p53α protein isoform, supports its role in oncogenesis. We revealed that the 17 amino acid extension to p53β, giving rise to the p53β-stop-lost protein, confers a unique and intrinsic activity, which enables protein complex formation with p53α. These hetero-oligomers regulate p53α activity in a manner distinct from p53α homo-oligomers due to differential transcriptional activity and promoter selectivity on various p53-target gene promoters. Notably, co-expression of p53α + p53β did not affect the transcription activity of p53α on the tested promoters. These distinct effects on different p53-target gene promoters suggests a mechanism by which p53β-stop-lost can modulate p53α-mediated cell responses. Promoter selectivity has previously been described for other p53 mutations, such as the p53 p.Ser121Phe mutant43.
The importance of genetic background as well as cell-type specific gene expression patterns, including the co-expression of p53 isoforms, in the regulation of the p53-target genes is illustrated by the high degree of variances between the different models investigated in our study, i.e., patient-derived B-cells, p53 wild-type cells and p53-null cells. Throughout the models, however, p53β-stop-lost seems to induce the expression of p21, a cyclin-dependent kinase inhibitor protein, encoded by CDKN1A.The primary function of p21 is promoting cell cycle arrest, inducing senescence, and modulating or inhibiting apoptosis44. Nevertheless, clinical studies on p21 expression in various cancers have revealed conflicting implications45. A reduced p21 expression has been associated with a worse prognosis and survival in cancers, such as colorectal46, gastric47, and head-and-neck48. In contrast, increased p21 expression has been linked to higher tumor grade, aggressiveness, invasiveness, drug resistance, and poorer clinical outcomes in a range of cancers45, including breast49, glioma50, prostate51, and esophageal52. Although not fully understood, this two-faced nature of p21 may depend on its subcellular localization, where cytoplasmic localization is linked to its anti-apoptotic actions, while nuclear localization appears to favor its tumor-suppressor activities44. In addition, prolonged p21 expression may lead to replication stress and genomic instability due to the deregulation of the replication licensing machinery and by rewiring the global cellular DNA repair landscape towards predominately error-prone processes53,54. We therefore hypothesize that one of the mechanisms by which p53β-stop-lost exerts it pro-tumorigenic functions might be through elevated basal expression of p21. This could protect cells from apoptosis and fuel cancer development through genomic instability.
Corroborating the functional experiments, at least the protein expression of full-length p53β-stop-lost and Δ133p53β-stop-lost were detected in patient-derived B-cells using a p53β-stop-lost-specific antibody. We also observed an increased TP53β mRNA expression, suggesting an additional mechanism by which theTP53β-stop-lost variant affects p53 regulation. In addition, our findings indicates that loss of wild-type p53β expression is not required during tumorigenesis in carriers of the TP53β-stop-lost variant, as evidenced by the absence of LOH at the TP53 locus in most of the analyzed tumors samples. This observation contrasts with tumors harboring germline or somatic mutations affecting p53α, where LOH leading to the loss of the wild-type allele is a common early event in tumor development19,20. Future research will need to address the exact biological consequences and functional properties of each of the p53β-stop-lost isoforms in different cell types and cell contexts to further elucidate the mechanisms leading to cancer predisposition in TP53β-stop-lost carriers.
Thus far, the functional and clinical assessment of TP53 variants has mainly focused on the DNA-binding domain, however oligomerization domain and C-terminal p53α domain variants have also been associated with clinical disease (Fig. 6)55,56,57,58. In this study we investigated the pathogenicity of a coding variant outside the canonical TP53 coding sequence. The TP53β-stop-lost families do not fulfill the “classic” definition of Li-Fraumeni syndrome59, the revised Chompret criteria for Li-Fraumeni syndrome6 or Eeles’ and Birch’s Li-Fraumeni syndrome-like definitions60,61. This is mostly based on the absence of typical Li-Fraumeni syndrome cancers (including sarcomas, brain tumors, and adrenocortical carcinomas) and the presence of colorectal and thyroid cancer in TP53β-stop-lost carriers, which are both less common in other TP53 mutation carriers6,62. Furthermore, age of onset appears to be much later, illustrated by a 20 year difference in median age at first breast cancer diagnosis compared to other pathogenic TP53 mutation carriers63, while still being ~10 years younger than the population average (incidence data from the Dutch cancer registration). In addition, none of the TP53β-stop-lost carriers developed cancer before the age of 30 years compared to a 50% cancer penetrance in DNA-binding domain mutation carriers63.

Tumor distribution, median age of breast cancer diagnosis, and estimated penetrance’s are shown for carriers of pathogenic p53 DNA-binding domain variants (left) and the variant encoding p53α p.Arg337His (middle) compared to TP53-stop-lost (right). Germline TP53 variant data was obtained from the International Agency for Research on Cancer (IARC) TP53 database (Version R19) for the six most common DNA-binding domain variants (encoding p.Arg175His, p.Gly245Ser, p.Arg248Gln, p.Arg248Trp, p.Arg273His, and p.Arg282Trp), accounting for ~25% of all Li-Fraumeni syndrome families, and the Brazilian founder mutation encoding p53α p.Arg337His (6%)62. Penetrance of these two groups was calculated previously62,63. The clinical data of TP53β-stop-lost carriers is based on 14 cancer patients, 11 unaffected individuals at the end of follow-up, and 2 relatives lacking clinical data, however, calculations of accurate cancer risk estimates require additional carriers.
Although, co-segregation analysis provides supportive evidence for pathogenicity it is likely that additional factors contribute to phenotypic differences between the four families and the increased cancer risk observed in non-carriers, such as environmental risk factors and other genetic components, such as polymorphisms in TP53 and p53-response elements64,65,66 or undiscovered risk alleles. It will therefore be instrumental to collect data on additional carriers and to increase the follow-up of existing carriers for the establishment of an accurate tumor spectrum and risk estimates leading to an appropriate surveillance management, as well as treatment outcome and the potential presence of subsidiary pathologies associated with p53 dysfunction, such as immune-deficiencies.
It is plausible that TP53β variants, in general, are associated with a less severe phenotype and other tumor predispositions compared to variants affecting canonical p53, due to the expression of wild-type p53α and p53γ protein isoforms, the tissue-dependent expression of p53β isoforms42,67, the functional severity of TP53β variants7,9 and the variant-specific alterations in protein properties68.
Currently, the exact function of wild-type p53β isoforms remains largely elusive, probably because, like for p53β-stop-lost, its intrinsic activity is dependent on cell type, genetic background, and cell signals. To date, full-length p53β overexpression has been associated with increased cell death37,69,70 and replicative senescence71 in cultured cells and with a better prognosis for certain cancer types, such as breast cancer, acute myeloid leukemia and clear-cell renal cell carcinoma70,72,73 and a worse prognosis for other cancers, such multiple myeloma and melanoma74,75. On the other hand, the overexpression of the Δ133p53β isoform has been linked to increased migration, invasion, and cancer cell stemness76,77 in cultured cells and to a poorer clinical outcome in breast cancer76 and glioblastoma78 patients. Although the TP53 gene is highly conserved throughout evolution, the number and structure of the p53 isoforms differs between species12. For instance, the mouse C-terminal splicing variant Mp53AS is considered a homolog of human full-length p53β, however this isoform contains the complete oligomerization domain and a distinct 18 amino acid isoform-specific C-terminal sequence, thus hampering the functional investigation of human p53 isoforms using animal models.
In conclusion, we report the oncogenic effect of a variant affecting the isoform-specific protein sequence of p53β, albeit with a comparatively subtler effect than variants impacting the canonical p53α isoform. The p53β-stop-lost proteins modify p53 transcriptional activity, probably eliciting anti-apoptotic and pro-survival activities. Based on the functional data, in combination with the clinical presentation of carriers, we propose that the TP53β-stop-lost variant predisposes to various cancer types. At present, in the ClinVar database (Accession: VCV000372008.9; variation ID: 372008) this variant is classified as “Likely Benign” based on the ACMG guidelines (2015)79, and due to lack of functional evidence. Until now, few TP53 9β exon variants have been discovered and their clinical significance remained unknown, as TP53 research focused on the pathogenicity of coding variants affecting the canonical transcript13. Recent genetic studies have found a much higher prevalence of potentially pathogenicTP53 variants in populations unselected for the clinical criteria for Li-Fraumeni syndrome, than anticipated based on family studies80,81,82. In the majority of the familial and young-onset cancer patients no genetic cause can be identified2,3,4. Our findings provide an important rationale for investigating variants in the whole TP53 gene, including regions outside the canonical coding sequence, such as 9β exon, 9γ exon, internal promoter and other non-coding regions, in these patients.
Methods
The study was approved by the Medical Ethical Committee of the Leiden University Medical Center, The Netherlands (protocol P01.019 and P15.282). All patients of the whole-exome sequencing cohort and the patient-derived B-cells cohort provided written informed consent. Tissue samples from patients suspected of a hereditary cancer syndrome were obtained with informed consent from the patients themselves or from their next of kin. Consecutive series of anonymized tissue samples of colorectal cancer and breast cancer patients were handled according to the medical ethical guidelines described in the Code Proper Secondary Use of Human Tissue established by the Dutch Federation of Medical Sciences (https://www.federa.org/). All patients were informed about the potential secondary use of their tissues in translation research and provided implicit consent. The use of these samples was approved by the Medical Ethical Committee of the Leiden University Medical Center (P01.019). Animal experiments (p53β-stop-lost-specific antibody generation) were performed in full compliance with relevant Czech and EU animal welfare regulations.
Cohort
The whole-exome sequencing cohort comprised of 94 individuals suspected of carrying a genetic predisposition to colorectal cancer and consisted of: 49 individuals from 25 colorectal cancer families (including 1 unaffected parent), 11 early-onset (<40 years) mismatch-repair proficient colorectal cancer patients, 19 hyperplastic polyposis patients and 15 adenomatous polyposis patients. Known Mendelian cancer syndromes had previously been excluded in the clinical diagnostic setting. The c.1025A>C, p.*342Serext*18 variant in the TP53β transcript (NM_001126114.2/ ENST00000420246) was checked for its presence in four individuals from three breast cancer research families and in the in-house whole-exome sequencing database, containing 26 patients from 18 unexplained hereditary cancer families referred to the Department of Clinical Genetics. Clinicopathological data were either obtained from the national registry of The Netherlands Foundation for the Detection of Hereditary Tumors (http://www.stoet.nl) or during consultations at the Departments of Clinical Genetics at Leiden University Medical Center or University Medical Center Utrecht. Sex was self-reported and not considered in this study design.
The colorectal cancer cohort was comprised of 1289 patients: 259 unexplained early-onset/familial colorectal cancer patients83; 44 patients with multiple colorectal adenomas (10–50 polyps) negative for known disease causing variants84; 18 MUTYH-associated polyposis patients85; and a consecutive series of 968 colorectal cancer patients (partly described by Boot et al.86). The breast cancer series consisted of 671 patients87,88; 381 with unknown histological subtype; 116 ductal carcinomas in situ (DCIS); 115 invasive breast cancer no special type, and 59 with other histological subtypes.
Whole-exome sequencing
DNA derived from peripheral blood or normal fresh frozen colon mucosa was extracted using standard techniques. We performed exome enrichment with NimbleGen v2 exome capture on DNA from 22 samples and send the libraries for sequencing to the GenomeScan (Leiden, The Netherlands). Whole-exome sequencing of the rest of the samples was outsourced: 9 DNA samples were sent to Macrogen (Korea) who used TruSeq Exome Enrichment Kit, and 19 DNA samples were sent to GenomeScan (The Netherlands) who used Agilent SureSelect Human All Exon Kit (7 samples with v1 and 12 samples with v5, including the breast cancer research families). All the libraries were sequenced on Illumina HiSeq sequencers. Whole-exome sequencing of 48 DNA samples was outsourced to BGI (China); the library was construction with the BGI capture kit, followed by sequencing on the Complete Genomics’ Sequencing Platform (Complete Genomics Inc.). Sequencing data processing and variant calling was performed, see Supplementary Methods. Candidate variants were selected which were predicted to affect the coding sequence of any RefSeq transcript and had a population frequency below 0.01 (1000Genomes phase 3, ExAC 1.0, ESP6500SI-V2 or GoNL release 5). DNA of the patients in the in-house whole-exome sequencing database and family 4 were sent to and analyzed by Genome Diagnostics Nijmegen, Radboud UMC, The Netherlands, who performed exome capture with the Agilent SureSelect v5 Kit.
Screening TP53β-stop-lost variant
The candidate variant (rs764562217; NM_001126114.2: c.1025A>C; NM_000546.5: c.993+227A>C) was screened in available patient cohorts. DNA was derived from peripheral blood, fresh frozen tissues, or formalin-fixed paraffin-imbedded (FFPE) tissues using standard techniques. Genotyping of the variant was performed with KASPar allele-specific single nucleotide variant primers (Supplementary Table 9) following the manufacturer’s protocol (LGC Group). The variant was checked for its presence in next-generation sequencing data covering the TP53 9β exon generated with three different targeted sequencing panels; Oncomine Comprehensive panel89, custom DNA NuGEN Ovation Target Enrichement system panel, and a custom multiplex PCR panel (details available upon request). NuGEN libraries were sequenced by GenomeScan (The Netherlands) on Illumina HiSeq sequencers. The other panels were run in-house on Ion Torrent Sequencers (Life Technologies). BAM files were used for variant calling. Samples with a coverage lower than 20 reads of TP53 9β exon were excluded for all the panels.
In addition, TP53 variants were screen for in the public sequencing databases Genome aggregation database (gnomAD v2.1.1, https://gnomad.broadinstitute.org), Genomes of the Netherlands (GoNL release 5, https://nlgenome.nl) and Catalog of Somatic Mutations in Cancer (COSMIC v96, https://cancer.sanger.ac.uk).
Estimation of penetrance
Kaplan-Meier curves were plotted, and penetrance was calculated using GraphPad 8 (Prism).
Pairwise identity-by-descent (IBD) estimation
Genotypes at 7384 positions (single nucleotide variants with minor allele frequency (MAF) > 0.4 and linkage disequilibrium (LD) < 0.8 used for sample identification) were retrieved from the BAM files of 54 whole-exome sequencing sample using Samtools mpileup (version 1.6). Positions with missing genotypes (./.) or a depth of coverage (DP) < 10 were removed resulting in 3264 remaining positions. The merged VCF file was converted to PLINK format using VCFtools90. To further reduce the number of single nucleotide polymorphisms (SNPs) with high levels of LD, SNP pruning (indep-pairwise 50 5 0.5) was performed and SNPs in regions with high LD were removed using PLINK16 v1.07, as described in Anderson et al.91. IBD estimations were calculated with genotypes at 2865 positions for all pairs. IBD estimates Z0, Z1, and Z2 of pairs with documented family relationships (30 individuals from 12 families) were compared for quality assessment. PI-HAT values were used to estimate the proportion of shared alleles.
Co-segregation analysis
Co-segregation analysis based on the full-likelihood Bayes factor (FLB)17 was performed using the pedigrees of the four families, assuming an autosomal dominant model of inheritance and a population frequency of the variant of 0.000056% (gnomAD v2.1.1). Due to limited follow-up data, we considered as phenotype of interest any cancer diagnosis before 60 years of age, excluding lung cancer due to its strong environmental links. We obtained incidence data for any cancer (excluding lung cancer) below 60 years of age from the Dutch cancer registration (Nederlandse Kankerregistratie van IKNL), with a crude incidence rate of 7.5% in 2021. We estimated the penetrance in carriers before 60 years at 50%, based on literature regarding penetrance in Li-Fraumeni syndrome variant carriers92. The FLB scores were computed using the R package segregatr93. For analysis in independent families, individual FLB scores can be multiplied to produce a combined FLB score. We adapted FLB thresholds for at least two families from Jarvik and Browning18 to translate FLB scores to the ACMG-AMP pathogenicity evidence levels, which correspond to 4, 8, and 16 for supportive, moderate, and strong evidence, respectively.
Somatic mutation analysis
Nucleic acid was extracted using the Tissue Preparation System (Siemens Healthcare Diagnostics) from FFPE tissues (micro-dissected slides or tissue cores) of three precursor lesions and eleven cancerous lesions94. Five normal tissue samples were taken along for quality assessment. Sequencing was performed with the custom AmpliSeq Cancer Hotspot Panel and Ion AmpliSeq TP53 panel (Thermo Fisher Scientific) on the Ion Torrent Personal Genome Machine Sequencer (Life Technologies). For somatic mutation analysis we focused on driver mutations present in the tumor bulk and, therefore, removed mutations present in the normal tissue samples, non-coding mutations and mutations with a variant allele frequency (VAF) < 0.1. For samples with a poor quality (transition transversion ratio (Ts/Tv) > 25) mutations were removed with a VAF < 0.2. All remaining mutations were visually inspected for sequencing artefacts. The allelic imbalance factor (AIF) was assessed for the stop-lost variant in all tissue samples. AIF was calculated using the VAF of the mutation in tumor (T) and normal (N) tissues: (N × (1 − T)) / (T × (1 − N))95. AIF > 5 were classified as classical loss of heterozygosity (LOH), AIF = 2–5 as allelic imbalance, and AIF < 2 as experimental noise88.
Cell culture
Epstein-Barr virus immortalized B-lymphocyte cells were derived from peripheral blood mononuclear cells (PBMCs) of 9 colorectal cancer patients. Commercial cell lines were acquired from ATCC: H1299 (p53-null), HCT-116 (p53 wild-type) and MCF-7 (p53 wild-type) and HEK293T (generation of virus particles). Cells were cultured in DMEM (Gibco) or RPMI-1640 medium (Gibco), supplemented with 10% heat-inactivated fetal bovine serum (Sigma) and 50 units/ml penicillin-streptomycin (Gibco).
Targeted transcriptome sequencing
A custom Archer assay (ArcherDx, Inc.), covering the whole coding sequence of the TP53 gene, was designed and applied on 200 ng of mRNA according to the manufacturer’s instructions. Libraries were run on the Ion Torrent Proton system (Life Technologies) and analyzed using the ArcherDX pipeline (Suite_Analysis_v5.1.8). Soft-clipped reads, mapped to the TP53 locus, were extracted from the produced BAM file (SAMPLE.molbar.trimmed.dedupped.bam). This strategy allowed us to select all TP53, intron-spanning RNA reads, which were later realigned using the STAR aligner96. The number of reads covering each base in an exon was calculated and summed (sum of covered bases) for each exon of the full-length TP53β transcript (ENST00000420246, see Supplementary Table 10). The sum of covered bases per exon was normalized against the sum of covered bases of exon 9. In addition, the re-aligned BAM files were used to calculate the mutation counts of the stop-lost variant.
Quantitative real-time reverse transcriptase PCR and allele-specific mRNA analysis
mRNA was isolated from 10 µm fresh frozen tissue slices using TRIzol (Invitrogen) and rDNase treatment (Macherey-Nagel). For mRNA isolation from B-lymphocyte cells, B-cell suspensions (50.000 cells/well) were cultured in biological triplicates in 12-wells plates. mRNA of B-lymphocyte cells and cancer cell lines was isolated with the Nucleospin RNA kit (Macherey-Nagel). cDNA conversion of mRNA was performed with AMV Reverse Transcriptase (Roche) and OdT primers. Quantitative real-time reverse transcriptase PCR was performed using 1.0 pmol forward and reverse primer and 1x IQ SYBR Supermix (Bio-RAD). Gene expression was normalized to GAPDH. TP53α and TP53β isoform-specific primer pairs were designed manually. PCR primers for the p53-target genes were generated using the Universal Probe Library (Roche). All qPCR primer pairs are shown in Supplementary Table 11. Allele-specific expression analysis of the TP53β-stop-lost variant was performed using 2 µl 25x diluted cDNA as template for the KASPar assay. Paired DNA was analyzed to determine baseline amplification differences between the two alleles. Using the Cq values obtained for both alleles, the allelic dosage was calculated similarly to the Pfaffl method for relative gene expression97. All qPCR and KASPar reactions were performed in technical duplicate. Mean and standard deviations (SD) were plotted GraphPad.
Non-sense mediated decay inhibition
Non-sense mediated decay was inhibited by cycloheximide (Sigma-Aldrich). B-cell suspensions (50.000 cells/well) were cultured in 12-wells plates in biological triplicates. Cells were treated for 6 h with cycloheximide (1:5.000), DMSO (1:5.000), or growth medium prior to cell lysis.
Immunoblotting
Protein lysates were made in NP40 buffer76 (50 mM Tris (pH 7.5), 10% glycerol, 0.1% NP-40 Alternative (Calbiochem), 100 mM NaCl, 0.2 mM EDTA, supplemented with 1X Complete Protease Inhibitor Cocktail (Roche)), or Giordano buffer (50 mM Tris-HCl (pH 7.4), 5 mM EDTA, 150 mM NaCl, 0.1% Triton-X100 and 10% glycerol, supplemented with protease and phosphatase inhibitor cocktails (Roche)) or Hot SDS buffer (1% SDS, 10 mM Tris/EDTA (pH 7.4), supplemented with protease and phosphatase inhibitor cocktails). For the NP40 lysates, 20 µg protein extracts were supplemented with 0.1 M DTT and 1X LDS buffer (Invitrogen), loaded on NuPAGE 10% Bis-Tris polyacrylamide gels, separated by MOPS Running Buffer supplemented with antioxidant (Invitrogen) and transferred at 12 V for 16 h onto a nitrocellulose membrane (Protran 0.2 μm from GE Healthcare) in Tris-glycine buffer supplemented with 20% methanol (3 g Tris and 14.4 g glycine for 1 l). For the Giordano buffer lysates and hot SDS buffer lysates, 20 µg of the protein lysates were run on TGX Stain-Free FastCast Acrylamide 10% gels (Bio-RAD) and blotted using Trans-Blot Turbo RTA Transfer Kit, PVDF (Bio-Rad) in the Trans-Blot Turbo Transfer system (Bio-Rad). Blots were blocked for 30 min in 0.1% TBS-Tween20, 5% low-fat milk before being incubated overnight at 4 °C with the following primary antibodies: anti-full-length p53 antibody DO-7 (Agilent Dako; 1:500), anti-full-length p53 antibody DO-1 (1 µg/ml), pan-p53 polyclonal antibody SAPU (1 µg/ml), anti-p53β antibody KJC8 (1 µg/ml), anti-p53β stop-lost (p53β-SL; 1 µg/ml), p53α-domain-specific antibodies 421 and BP53.10 (1 µg/ml), anti-p21 (clone EA10; Calbiochem; 1 µg/ml or #2947; CST; 1:1000), anti-PARP (#9532, CST, 1:1000), anti-Caspase-3 (#9665; CST; 1:1000), anti-phospho-Histone H2A.X (Ser139; #9718; CST; 1:1000), anti-α-tubulin (#3873; CST; 1:30,000) and anti-Ku80 (ab80592; Abcam; 1:10,000), diluted in 0.1% TBS-Tween20, 5% low-fat milk. HPR-linked secondary antibodies were used for protein detection: anti-rabbit (#7074; CST; 1:3.000 or Jackson ImmunoResearch; 1:10,000), anti-mouse (#7076; CST; 1:2000 or Jackson ImmunoResearch; 1:10,000) and anti-sheep (HAF016, R&D systems; 1:2000 or Jackson ImmunoResearch; 1:10,000). Blots were activated with SuperSignal West Pico PLUS Chemiluminescent Substrate kit (Thermo Fisher Scientific) or with ECL Western Blotting Detection System (Amersham) and visualized using the Chemidoc Imaging system (Bio-Rad) or detected on Hyperfilm™ ECL (Amersham). The antibody recognition sites for the different anti-p53 antibodies are indicated in Supplementary Fig. 1.
Cellular stress was induced in cultured cells using doxorubicin (Sigma-Aldrich). B-cells were continuously treated with 0.5 µM doxorubicin or medium controls, and protein lysates were made at different timepoints (0 h, 3 h, 6 h, and 9 h). For the transduced cell lines experiments, 200,000 cells/well were seeded in 6-wells plates. After overnight attachment, cultures were continuously treatment with 1.0 µM doxorubicin (or medium control). Protein lysates were made at different timepoints (1 h, 2 h, 3 h, 4 h, 8 h, 24 h, and 48 h). Experiments were performed in duplicate.
p53β-stop-lost-specific antibody generation
The p53β-stop-lost-specific antibody was generated by Moravian-Biotechnology Ltd, (Czech Republic) according to their protocol in full compliance with Czech and EU animal welfare regulations. The peptide CSREHENGSMTLPDTDAT, corresponding to the last 18 amino acids of p53β-stop-lost protein, was conjugated to the carrier protein KLH and inoculated into one rabbit, as described in Harlow and Lane (1988)98. The p53β stop-lost-specific (p53β-SL) antibody was then peptide-affinity purified using the above immunizing peptide conjugated to Sulfolink coupling resin (Thermo Fisher Scientific). The antibody was eluded in 1 ml of 0.1 M glycine (pH 2.5) and immediately neutralized in 20 µl 1.5 M Tris-HCl (pH 7.4).
Transient transfections
In brief, 500,000 cells were seeded in 10 cm culture dishes 24 h prior to plasmid transfections with 0.5 µg (pcDNA3-p53β-stop-lost) and/or 0.5 µg (pcDNA3-p53α) with 3 µl Turbofect (Thermo Fisher Scientific). Cells were harvested 24 h later. Of note, empty pcDNA3 vector (Invitrogen) was used to balance DNA quantity as required.
Lentivirus construction and transduction
The coding sequence of full-length wild-type TP53β was sequenced by Integrated DNA Technologies (IDT). The variant encoding p53β-stop-lost was generated using site-directed mutagenesis by primer extension (primer sequences shown in Supplementary Table 9) and Q5 Hot Start High Fidelity 2x mix (NEB; details available upon request). Inserts were cloned into the multi-cistronic expression vector pUltra (EGFP-P2A-gene1-T2A-gene2; addgene plasmid #24129) using restriction enzymes (NheI and EcoRI, NEB). The viral particles were generated in HEK293T cells by co-transfection of pUltra vector (p53β wild-type, p53β-stop-lost or empty vector) with third-generation lentiviral vectors (pMD2g, pRSV/REV, pGAG/POL, pMDLg/pRRE) using polyethyenimine (PEI). The next day, the culture supernatant was harvested (500 × g for 5 min). HCT-116 and MCF-7 cancer cell lines were transduced with the different lentiviruses. 1.0 ml medium (RPMI-1640, 10% FCS), 500 µl lentivirus containing supernatant, polybrene (Sigma-Aldrich 10768-9; final concentration 8 µg/ml) and 5 × 105 cells were plated in 6-wells plates. 24 h post-transfection the virus-containing medium was replace with fresh complete medium (RPMI-1640, 10% FCS, Pen/Strep). When sufficient cells were obtained (>20 × 106), cells were sorted on EGFP-positive cells (gated for the lowest 10–15%) using FACS Aria 3 cell sorter (Biosciences; LUMC FACS facility) using 85um nozzle size and 45 PSI pressure.
Cell cycle assay
Transduced cell lines were seeded in 6-wells plates (200,000 cells/well). After overnight attachment, cultures were treated with 1.0 µM doxorubicin (or medium control) for 24 h. Adherent cells and supernatant were collected and fixed, see Supplementary Methods. Cells were stained with 5 µM DAPI in 0.1% PBS-Tween20, 1%BSA and incubated at room temperature for 30 min, followed by overnight at 4 °C. Cell cycle analysis was performed with NucleoCounter NC-250 (Chemometec), Winlist 3D v9.0.1 and Modfit v5.0 (Verity Software House). Each analysis measured 2000–10,000 single cell events. Using Sync Wizard, the S-phase was fitted with a statistical model: trapezoid, 3 compartments, and equal spacing (reduced chi-square (RCS) between 1 and 5). Experiments were performed in triplicate.
Whole-transcriptome analysis
Library preparation and sequencing was performed at Macrogen Europe (Amsterdam, The Netherlands). In brief, RNA-sequencing library preparation was performed with TruSeq Stranded Total RNA with Ribo-Zero Gold. Sequencing was performed on an Illumina NovaSeq 6000 sequencer. Quality control of the RNA-sequencing reads as well as adapter clipping, read alignment, and gene expression quantification was done using the BIOWDL RNAseq pipeline v4.0.0 (https://doi.org/10.5281/zenodo.3975552). In short, reads were aligned to the human reference genome (hg38 build) using STAR96 v2.7.5a, followed by gene expression quantification by htseq-count99 v0.12.4. For expression quantification gencode v35 (corresponding to Ensembl 101) was used for gene annotation. Differential expression analysis was performed in R (v4.0.2) using the tidybulk R package v1.2.0 (https://doi.org/10.5281/zenodo.4312265) with default values. In summary, expression values were normalized using the TMM method, and differential expression was calculated with quasi-likelihood estimation method in edgeR100,101 v3.32.0. Genes with an absolute logFC > 0.6 in both cell lines were selected. Gene-set enrichment analysis (GSEA) was performed making use of the Molecular Signatures Database (MSigDB v7.4; https://www.gsea-msigdb.org/gsea/msigdb/annotate.jsp) using hallmark gene sets102.
Structural analysis
The secondary protein structure of wild-type p53β and p53β-stop-lost were predicted using the intensive mode of Phyre238. In PyMOL Molecular Graphics System (Version 2.0 Schrödinger, LLC), the predicted protein structures were modeled into the crystal structure of p53α dimer of the DNA bound p53α tetramer (PDB 3TS8)103, and the C-terminal domains (protein sequence after amino acid 324) were visualized.
Co-immunoprecipitation
Cells were transfected with plasmids as described earlier. Cells were harvested 48 h post-transfection in 500 µl NP40 lysis buffer. 1 mg protein extract was incubated with 1 µg primary antibody on a wheel for 4 h at 4 °C. In the meantime, Dynabeads Protein G (Thermo Fisher Scientific) were blocked for 30 min at 4 °C on a wheel with 1 mg/ml IgG free BSA before being re-suspended in PBS. 150 µg blocked beads were then added to samples and incubated altogether on a wheel for 12 h at 4 °C. Beads were then washed three times with ice-cold NP40 before being re-suspended in 1X LDS buffer (Invitrogen) and heated for 10 min at 80 °C. Protein samples were recovered and supplemented with 0.1 M DTT before being loaded on NuPAGE 10% Bis-Tris polyacrylamide gels. 1% of protein extract used as input for the immunoprecipitation (10 µg) was loaded as a whole cell lysate control (WCL).
Immunofluorescence
H1299 cells were transfected with p53β-stop-lost expression vector. 24 h after transfection the cells were fixed with 1% formaldehyde-PBS for 15 min. Cells were then incubated for 1 h with rabbit p53β-SL antibody (1 µg/ml) and stained with Alexa-488-conjugated donkey anti-rabbit antibody (Jackson ImmunoResearch, 0.2 µg/ml). DNA was stained with DAPI (Thermo Fisher Scientific). Slides were visualized using fluorescent microscope (Zeis).
Luciferase reporter gene assay
H1299 cells were transfected at a density of 15,000 cells/well in 24-well plates. First, to determine the 50% maximum fold p53α-mediated induction by p53α on the CDKN1A, BAX and IGFBP3 promoters, 250 ng of p21-Luc, Bax-Luc or IGFBP3-Luc plasmid were mixed with 2.5 ng pSV-Renilla luciferase control reporter, increasing amounts of pcDNA3-p53α expression vector (0 ng, 50 ng, 100 ng, 200 ng or 400 ng) and 1 µl Turbofect. Transfected DNA and CMV promoters were kept in constant quantity by adding the empty pcDNA3 vector as needed. 24 h post-transfection, cells were washed twice with PBS and incubated for 15 min with shaking at room temperature with 100 µl Passive Lysis Buffer (Promega). 10 µL of each cell lysate was transferred in a 96-well plate, and the plate was then analyzed on luminometer (Microplate luminometer LB 96 V, EG&G Berthold) using the Dual-Luciferase Assay System (Promega). The 50% maximum fold p53α-mediated induction was reached with 100 ng of p53α vector for all tested promoters. Second, the maximum induction on these promoters by p53β or p53β-stop-lost was determined by transfecting increasing amounts of vector (pcDNA3-p53β or pcDNA3-p53β-stop-lost; 0 ng, 25 ng, 50 ng, 100 ng or 500 ng) instead of p53α, as described above. The promoters were not induced by any amount of p53β or p53β-stop-lost plasmid. Third, the effect of p53β or p53β-stop-lost on the 50% maximum fold p53α-mediated induction was measured for increasing amounts of pcDNA3-p53β or pcDNA3-p53β-stop-lost (0 ng, 50 ng,100 ng, 200 ng or 500 ng), as described for p53α, with 100 ng of p53α vector for all promoters. In parallel, to take account of the transfection efficiency and assess comparable expression of p53α, p53β, and p53β-stop-lost proteins, luciferase reporter gene assay protein extracts were analyzed by immunoblotting (LICOR) after normalization for Renilla activity using the pan-p53 SAPU antibody. To obtain comparable levels of protein expression, the following transfection conditions were selected: vector (empty pcDNA3 500 ng); p53α only (100 ng plasmid); p53β alone (50 ng plasmid); p53β-stop-lost alone (500 ng plasmid); p53α (100 ng plasmid) + p53β (50 ng plasmid); p53α (100 ng plasmid) + p53β-stop-lost (500 ng plasmid). Transfected DNA and CMV promoters was kept in constant quantity by adding the empty pcDNA3 vector as needed.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The authors declare that the experimental data supporting the findings of this study are available within the paper and its supplementary information files. The raw and processed sequencing data of RNA-sequencing are publicly available in the Gene Expression Omnibus (GEO) under accession number GSE250000. Restrictions are in place for access to raw whole-exome sequencing (WES) data because of lack of consent for data sharing. Filtered WES data will be made available for non-commercial research purposes upon request (TvW) after establishing a data transfer agreement and approval of the data access committee of the LUMC biobank. The response time is expected to take up to 6 weeks. Upon approval, access to the data will be granted for a period of 1 year. Source data are provided with this paper.
References
Mucci, L. A. et al. Familial risk and heritability of cancer among twins in Nordic countries. JAMA 315, 68–76 (2016).
Wendt, C. & Margolin, S. Identifying breast cancer susceptibility genes—a review of the genetic background in familial breast cancer. Acta Oncol. 58, 135–146 (2019).
Law, P. J. et al. Association analyses identify 31 new risk loci for colorectal cancer susceptibility. Nat. Commun. 10, 2154 (2019).
Chubb, D. et al. Genetic diagnosis of high-penetrance susceptibility for colorectal cancer (CRC) is achievable for a high proportion of familial CRC by exome sequencing. J. Clin. Oncol. 33, 426–432 (2015).
Hincza, K., Kowalik, A. & Kowalska, A. Current knowledge of germline genetic risk factors for the development of non-medullary thyroid cancer. Genes 10, 482 (2019).
Bougeard, G. et al. Revisiting Li-Fraumeni syndrome from TP53 mutation carriers. J. Clin. Oncol. 33, 2345–2352 (2015).
Fischer, N. W., Prodeus, A., Tran, J., Malkin, D. & Gariepy, J. Association between the oligomeric status of p53 and clinical outcomes in Li-Fraumeni syndrome. J. Natl. Cancer Inst. 110, 1418–1421 (2018).
Olivier, M. et al. Li-Fraumeni and related syndromes: correlation between tumor type, family structure, and TP53 genotype. Cancer Res. 63, 6643–6650 (2003).
Petitjean, A. et al. Impact of mutant p53 functional properties on TP53 mutation patterns and tumor phenotype: lessons from recent developments in the IARC TP53 database. Hum. Mutat. 28, 622–629 (2007).
Kratz, C. P. et al. Analysis of the Li-Fraumeni spectrum based on an international germline TP53 variant data set: an international agency for research on cancer TP53 database analysis. JAMA Oncol. 7, 1800–1805 (2021).
Schneider, K., Zelley, K., Nichols, K. E. & Garber, J. Li-Fraumeni Syndrome 1999 [Updated 2019 Nov 21], (GeneReviews® [Internet]. (University of Washington, 1993–2024).
Joruiz, S. M. & Bourdon, J. C. p53 isoforms: key regulators of the cell fate decision. Cold Spring Harb. Perspect. Med. 6, a026039 (2016).
Leroy, B. et al. Recommended guidelines for validation, quality control, and reporting of TP53 variants in clinical practice. Cancer Res. 77, 1250–1260 (2017).
Lek, M. et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 536, 285–291 (2016).
Papaemmanuil, E. et al. Clinical and biological implications of driver mutations in myelodysplastic syndromes. Blood 122, 3616–3627 (2013). quiz 3699.
Purcell, S. et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 81, 559–575 (2007).
Thompson, D., Easton, D. F. & Goldgar, D. E. A full-likelihood method for the evaluation of causality of sequence variants from family data. Am. J. Hum. Genet. 73, 652–655 (2003).
Jarvik, G. P. & Browning, B. L. Consideration of cosegregation in the pathogenicity classification of genomic variants. Am. J. Hum. Genet. 98, 1077–1081 (2016).
Kandoth, C. et al. Mutational landscape and significance across 12 major cancer types. Nature 502, 333–339 (2013).
Light, N. et al. Germline TP53 mutations undergo copy number gain years prior to tumor diagnosis. Nat. Commun. 14, 77 (2023).
Birch, J. M. et al. Cancer phenotype correlates with constitutional TP53 genotype in families with the Li-Fraumeni syndrome. Oncogene 17, 1061–1068 (1998).
Varley, J. M. et al. A detailed study of loss of heterozygosity on chromosome 17 in tumours from Li-Fraumeni patients carrying a mutation to the TP53 gene. Oncogene 14, 865–871 (1997).
Giacomelli, A. O. et al. Mutational processes shape the landscape of TP53 mutations in human cancer. Nat. Genet. 50, 1381–1387 (2018).
Cowen, L. E. & Tang, Y. Identification of nonsense-mediated mRNA decay pathway as a critical regulator of p53 isoform beta. Sci. Rep. 7, 17535 (2017).
Allen, M. A. et al. Global analysis of p53-regulated transcription identifies its direct targets and unexpected regulatory mechanisms. Elife 3, e02200 (2014).
Nguyen, T. T. et al. Revealing a human p53 universe. Nucleic Acids Res. 46, 8153–8167 (2018).
Wang, T. et al. Species-specific endogenous retroviruses shape the transcriptional network of the human tumor suppressor protein p53. Proc. Natl. Acad. Sci. USA 104, 18613–18618 (2007).
Cui, F., Sirotin, M. V. & Zhurkin, V. B. Impact of Alu repeats on the evolution of human p53 binding sites. Biol. Direct 6, 2 (2011).
Tatavosian, R. et al. Cell differentiation modifies the p53 transcriptional program through a combination of gene silencing and constitutive transactivation. Cell Death Differ. 30, 952–965 (2023).
Sullivan, K. D., Galbraith, M. D., Andrysik, Z. & Espinosa, J. M. Mechanisms of transcriptional regulation by p53. Cell Death Differ. 25, 133–143 (2018).
Pant, V., Sun, C. & Lozano, G. Tissue specificity and spatio-temporal dynamics of the p53 transcriptional program. Cell Death Differ. 30, 897–905 (2023).
Chinnadurai, G., Vijayalingam, S. & Gibson, S. B. BNIP3 subfamily BH3-only proteins: mitochondrial stress sensors in normal and pathological functions. Oncogene 27(Suppl 1), S114–S127 (2008).
Croessmann, S. et al. NDRG1 links p53 with proliferation-mediated centrosome homeostasis and genome stability. Proc. Natl. Acad. Sci. USA 112, 11583–11588 (2015).
Cheriyath, V., Leaman, D. W. & Borden, E. C. Emerging roles of FAM14 family members (G1P3/ISG 6-16 and ISG12/IFI27) in innate immunity and cancer. J. Interferon Cytokine Res. 31, 173–181 (2011).
Jia, H. et al. Interferon-α inducible protein 6 (IFI6) confers protection against ionizing radiation in skin cells. J. Dermatol. Sci. 100, 139–147 (2020).
Cheriyath, V. et al. G1P3 (IFI6), a mitochondrial localised antiapoptotic protein, promotes metastatic potential of breast cancer cells through mtROS. Br. J. Cancer 119, 52–64 (2018).
Marcel, V., Fernandes, K., Terrier, O., Lane, D. P. & Bourdon, J. C. Modulation of p53beta and p53gamma expression by regulating the alternative splicing of TP53 gene modifies cellular response. Cell Death Differ. 21, 1377–1387 (2014).
Kelley, L. A., Mezulis, S., Yates, C. M., Wass, M. N. & Sternberg, M. J. The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 10, 845–858 (2015).
Clore, G. M. et al. Refined solution structure of the oligomerization domain of the tumour suppressor p53. Nat. Struct. Biol. 2, 321–333 (1995).
Jeffrey, P. D., Gorina, S. & Pavletich, N. P. Crystal structure of the tetramerization domain of the p53 tumor suppressor at 1.7 angstroms. Science 267, 1498–1502 (1995).
Lee, W. et al. Solution structure of the tetrameric minimum transforming domain of p53. Nat. Struct. Biol. 1, 877–890 (1994).
Bourdon, J. C. et al. p53 isoforms can regulate p53 transcriptional activity. Genes Dev. 19, 2122–2137 (2005).
Saller, E. et al. Increased apoptosis induction by 121F mutant p53. EMBO J. 18, 4424–4437 (1999).
Georgakilas, A. G., Martin, O. A. & Bonner, W. M. p21: a two-faced genome guardian. Trends Mol. Med. 23, 310–319 (2017).
Abbas, T. & Dutta, A. p21 in cancer: intricate networks and multiple activities. Nat. Rev. Cancer 9, 400–414 (2009).
Bukholm, I. K. & Nesland, J. M. Protein expression of p53, p21 (WAF1/CIP1), bcl-2, Bax, cyclin D1 and pRb in human colon carcinomas. Virchows Arch. 436, 224–228 (2000).
Ogawa, M. et al. A combination analysis of p53 and p21 in gastric carcinoma as a strong indicator for prognosis. Int J. Mol. Med. 7, 479–483 (2001).
Kapranos, N. et al. p53, p21 and p27 protein expression in head and neck cancer and their prognostic value. Anticancer Res. 21, 521–528 (2001).
Winters, Z. E. et al. Subcellular localisation of cyclin B, Cdc2 and p21(WAF1/CIP1) in breast cancer. association with prognosis. Eur. J. Cancer 37, 2405–2412 (2001).
Korkolopoulou, P., Kouzelis, K., Christodoulou, P., Papanikolaou, A. & Thomas-Tsagli, E. Expression of retinoblastoma gene product and p21 (WAF1/Cip 1) protein in gliomas: correlations with proliferation markers, p53 expression and survival. Acta Neuropathol. 95, 617–624 (1998).
Aaltomaa, S., Lipponen, P., Eskelinen, M., Ala-Opas, M. & Kosma, V. M. Prognostic value and expression of p21(waf1/cip1) protein in prostate cancer. Prostate 39, 8–15 (1999).
Sarbia, M. et al. Expression of p21WAF1 predicts outcome of esophageal cancer patients treated by surgery alone or by combined therapy modalities. Clin. Cancer Res. 4, 2615–2623 (1998).
Galanos, P. et al. Chronic p53-independent p21 expression causes genomic instability by deregulating replication licensing. Nat. Cell Biol. 18, 777–789 (2016).
Galanos, P. et al. Mutational signatures reveal the role of RAD52 in p53-independent p21-driven genomic instability. Genome Biol. 19, 37 (2018).
Kamada, R., Nomura, T., Anderson, C. W. & Sakaguchi, K. Cancer-associated p53 tetramerization domain mutants: quantitative analysis reveals a low threshold for tumor suppressor inactivation. J. Biol. Chem. 286, 252–258 (2011).
Lang, V. et al. Tetramerization-defects of p53 result in aberrant ubiquitylation and transcriptional activity. Mol. Oncol. 8, 1026–1042 (2014).
Laptenko, O. et al. The p53 C terminus controls site-specific DNA binding and promotes structural changes within the central DNA binding domain. Mol. Cell 57, 1034–1046 (2015).
Toki, T. et al. De novo mutations activating germline TP53 in an inherited bone-marrow-failure syndrome. Am. J. Hum. Genet. 103, 440–447 (2018).
Li, F. P. et al. A cancer family syndrome in twenty-four kindreds. Cancer Res. 48, 5358–5362 (1988).
Birch, J. M. et al. Prevalence and diversity of constitutional mutations in the p53 gene among 21 Li-Fraumeni families. Cancer Res. 54, 1298–1304 (1994).
Eeles, R. A., Bartkova, J., Lane, D. P. & Bartek, J. The role of TP53 in breast cancer development. Cancer Surv. 18, 57–75 (1993).
Garritano, S. et al. Detailed haplotype analysis at the TP53 locus in p.R337H mutation carriers in the population of Southern Brazil: evidence for a founder effect. Hum. Mutat. 31, 143–150 (2010).
Amadou, A., Waddington Achatz, M. I. & Hainaut, P. Revisiting tumor patterns and penetrance in germline TP53 mutation carriers: temporal phases of Li-Fraumeni syndrome. Curr. Opin. Oncol. 30, 23–29 (2018).
Marcel, V. et al. TP53 PIN3 and MDM2 SNP309 polymorphisms as genetic modifiers in the Li-Fraumeni syndrome: impact on age at first diagnosis. J. Med. Genet. 46, 766–772 (2009).
Macedo, G. S. et al. p53 signaling pathway polymorphisms, cancer risk and tumor phenotype in TP53 R337H mutation carriers. Fam. Cancer 17, 269–274 (2018).
Whibley, C., Pharoah, P. D. & Hollstein, M. p53 polymorphisms: cancer implications. Nat. Rev. Cancer 9, 95–107 (2009).
Tang, Y. et al. Downregulation of splicing factor SRSF3 induces p53beta, an alternatively spliced isoform of p53 that promotes cellular senescence. Oncogene 32, 2792–2798 (2013).
DiGiammarino, E. L. et al. A novel mechanism of tumorigenesis involving pH-dependent destabilization of a mutant p53 tetramer. Nat. Struct. Biol. 9, 12–16 (2002).
Silden, E. et al. Expression of TP53 isoforms p53beta or p53gamma enhances chemosensitivity in TP53(null) cell lines. PLoS ONE 8, e56276 (2013).
Anensen, N. et al. Correlation analysis of p53 protein isoforms with NPM1/FLT3 mutations and therapy response in acute myeloid leukemia. Oncogene 31, 1533–1545 (2012).
Fujita, K. et al. p53 isoforms Delta133p53 and p53beta are endogenous regulators of replicative cellular senescence. Nat. Cell Biol. 11, 1135–1142 (2009).
Avery-Kiejda, K. A., Morten, B., Wong-Brown, M. W., Mathe, A. & Scott, R. J. The relative mRNA expression of p53 isoforms in breast cancer is associated with clinical features and outcome. Carcinogenesis 35, 586–596 (2014).
Zhang, H. et al. p53beta: a new prognostic marker for patients with clear-cell renal cell carcinoma from 5.3 years of median follow-up. Carcinogenesis 39, 368–374 (2018).
Rojas, E. A. et al. Expression of p53 protein isoforms predicts survival in patients with multiple myeloma. Am. J. Hematol. 97, 700–710 (2022).
Groen, K., Steffens Reinhardt, L., Bourdon, J. C. & Avery-Kiejda, K. A. It is not all about the alpha: elevated expression of p53β variants is associated with lower probability of survival in a retrospective melanoma cohort. Cancer Cell Int. 23, 228 (2023).
Gadea, G. et al. TP53 drives invasion through expression of its Delta133p53beta variant. Elife 5, e14734 (2016).
Arsic, N. et al. The p53 isoform Delta133p53beta promotes cancer stem cell potential. Stem Cell Rep. 4, 531–540 (2015).
Kazantseva, M. et al. Elevation of the TP53 isoform Delta133p53beta in glioblastomas: an alternative to mutant p53 in promoting tumor development. J. Pathol. 246, 77–88 (2018).
Richards, S. et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405–424 (2015).
Yurgelun, M. B. et al. Germline TP53 mutations in patients with early-onset colorectal cancer in the colon cancer family registry. JAMA Oncol. 1, 214–221 (2015).
de Andrade, K. C. et al. Variable population prevalence estimates of germline TP53 variants: a gnomAD-based analysis. Hum. Mutat. 40, 97–105 (2019).
de Andrade, K. C. et al. Higher-than-expected population prevalence of potentially pathogenic germline TP53 variants in individuals unselected for cancer history. Hum. Mutat. 38, 1723–1730 (2017).
Middeldorp, A. et al. Enrichment of low penetrance susceptibility loci in a Dutch familial colorectal cancer cohort. Cancer Epidemiol. Biomark. Prev. 18, 3062–3067 (2009).
Hes, F. J. et al. Colorectal cancer risk variants on 11q23 and 15q13 are associated with unexplained adenomatous polyposis. J. Med. Genet. 51, 55–60 (2014).
Middeldorp, A. et al. High frequency of copy-neutral LOH in MUTYH-associated polyposis carcinomas. J. Pathol. 216, 25–31 (2008).
Boot, A. et al. Allelic switching of DLX5, GRB10, and SVOPL during colorectal cancer tumorigenesis. Int J. Genom. 2019, 1287671 (2019).
Vos, C. B. et al. Genetic alterations on chromosome 16 and 17 are important features of ductal carcinoma in situ of the breast and are associated with histologic type. Br. J. Cancer 81, 1410–1418 (1999).
Cleton-Jansen, A. M. et al. Loss of heterozygosity mapping at chromosome arm 16q in 712 breast tumors reveals factors that influence delineation of candidate regions. Cancer Res. 61, 1171–1177 (2001).
Hovelson, D. H. et al. Development and validation of a scalable next-generation sequencing system for assessing relevant somatic variants in solid tumors. Neoplasia 17, 385–399 (2015).
Danecek, P. et al. The variant call format and VCFtools. Bioinformatics 27, 2156–2158 (2011).
Anderson, C. A. et al. Data quality control in genetic case-control association studies. Nat. Protoc. 5, 1564–1573 (2010).
Fischer, N. W. et al. TP53 Variant Clusters Stratify the Li-Fraumeni Spectrum and Reveal an Osteosarcoma-Prone Subgroup. medRxiv, 2024.01.06.23300162 (2024).
Ratajska, A., Vigeland, M. D., Wirgenes, K. V., Krohg-Sørensen, K. & Paus, B. The use of segregation analysis in interpretation of sequence variants in SMAD3: a case report. Mol. Genet. Genom. Med. 11, e2107 (2023).
van Eijk, R., Stevens, L., Morreau, H. & van Wezel, T. Assessment of a fully automated high-throughput DNA extraction method from formalin-fixed, paraffin-embedded tissue for KRAS, and BRAF somatic mutation analysis. Exp. Mol. Pathol. 94, 121–125 (2013).
Devilee, P., Cleton-Jansen, A. M. & Cornelisse, C. J. Ever since Knudson. Trends Genet 17, 569–573 (2001).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013).
Pfaffl, M. W. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 29, e45 (2001).
Harlow, E. & Lane, D. P. Antibodies: a Laboratory Manual (Cold Spring Harbor Laboratory, Cold Spring Harbor, 1988).
Anders, S., Pyl, P. T. & Huber, W. HTSeq—a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169 (2014).
Robinson, M. D., McCarthy, D. J. & Smyth, G. K. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140 (2009).
McCarthy, D. J., Chen, Y. & Smyth, G. K. Differential expression analysis of multifactor RNA-Seq experiments with respect to biological variation. Nucleic Acids Res. 40, 4288–4297 (2012).
Liberzon, A. et al. The molecular signatures database (MSigDB) hallmark gene set collection. Cell Syst. 1, 417–425 (2015).
Emamzadah, S., Tropia, L. & Halazonetis, T. D. Crystal structure of a multidomain human p53 tetramer bound to the natural CDKN1A (p21) p53-response element. Mol. Cancer Res. 9, 1493–1499 (2011).
Acknowledgements
We thank Frederik J. Hes, Carli M. Tops, Juul T. Wijnen, Hans F.A. Vasen, and Anne-Marie Cleton-Jansen for collecting clinical data and samples, and Arnoud Boot, Marina Ventayol Garcia, Alexandra Diot, Tessa Wilpshaar, Sanne Venneker, Wim Corver, Jaap van Eendenburg, Julia van Hees, George Janssen and Peter van Veelen provided technical assistance. This article is based upon work from COST Action CA17118, supported by COST (European Cooperation in Science and Technology). This project was funded by the Dutch Digestive Foundation (MLDS FP13-13), Stichting Sacha Swarttouw-Hijmans, and Leiden University Fund/Nypels-van der Zee Fonds awarded to T.v.W. Fight Colorectal Cancer-Michael’s Mission-AACR Fellowship in Young Onset, Late-Stage Colorectal Cancer Research 2015 (15-40-1645-DEMI), the KWF Bas Mulder Award UL (2015-7664) and the ZonMw Veni grant (016.176.l44) financed N.F.C.C.d.M. We are thankful to the UK-association of Li-Fraumeni patients The Georges Pantziarka TP53 Trust (https://tp53.co.uk), founded by Dr. Pan and Irene Pantziarka in memory of their son Georges Pantziarka, for financing S.M.J.
Author information
Authors and Affiliations
Contributions
T.v.W., N.F.C.C.d.M., J.C.B., and S.A.S. conceived and designed the study. D.R. performed next-generation sequencing analyses. N.F.C.C.d.M. and S.A.S. performed analysis and interpretation of whole-exome sequencing data. M.R.G. performed co-segregation analysis. S.A.S. and D.R. performed and interpreted other data analysis. A.R.M. provided whole-exome sequencing data. D.Q.C.M.B.S., M.N., B.P.M.v.N., and M.E.v.L. performed patient counseling and/or clinical data acquisition. H.M. performed the pathology review of the samples. J.S. performed genotyping. S.A.S., L.M.P., and N.G. performed molecular cloning. S.A.S. generated transduced cell lines and performed all mRNA analyses. L.M.P. and S.M.J. purified or validated stop-lost-specific antibody. S.M.J., N.G., A.M., J.S., and S.A.S. performed p53 protein analyses. S.M.J. and A.M. performed luciferase assays. S.A.S. and T.H.S. modeled the secondary protein structure. T.v.W., N.F.C.C.d.M., J.C.B., and H.M. supervised the work. S.A.S., J.C.B., N.F.C.C.d.M., and T.v.W. wrote the manuscript. All authors read and approved the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous, reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Schubert, S.A., Ruano, D., Joruiz, S.M. et al. Germline variant affecting p53β isoforms predisposes to familial cancer. Nat Commun 15, 8208 (2024). https://doi.org/10.1038/s41467-024-52551-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-52551-8
- Springer Nature Limited