

Abstract
White organic light-emitting diodes (WOLEDs) hold significant promise in illumination and displays, but achieving high efficiency while maintaining stability is an ongoing challenge. Here, we strategically combine a blue donor-acceptor thermally activated delayed fluorescence (TADF) emitter featuring rapid reverse intersystem crossing rate and a yellow multi-resonance TADF emitter renowned for the fast radiative transition process to achieve warm WOLEDs with exceptional power efficiency exceeding 190 lm W−1 and external quantum efficiency (EQE) of 39%, setting records for WOLEDs. Meanwhile, these devices also exhibit an extended operational lifetime (LT80) of 446 h at an initial luminance of 1000 cd m−2. Another group of blue and yellow emitters based on our strategy achieves a standard white emission and a high EQE of 35.6%, confirming the universality of our strategy. This work presents a versatile strategy to harmonize singlet exciton radiation and triplet exciton up-conversion, thus achieving a win-win situation of efficiency and stability.
Similar content being viewed by others

Introduction
White organic light-emitting diodes (WOLEDs) with high efficiency and excellent stability have garnered increasing attention1,2,3,4, especially those based on all thermally activated delayed fluorescence (TADF) emitters5,6,7,8. Without the participation of expensive noble metals, all-TADF WOLEDs can achieve comparable performance with phosphorescent devices, making them more cost-effective and potential for practical application9,10,11. Thanks to previous efforts, remarkable efficiencies have been realized in all-TADF WOLEDs with the records of power efficiency (PE) and external quantum efficiency (EQE) standing at 130 lm W−1 and 32.8%, respectively8,10. However, their device stability remains inferior, impeding the commercialization of all-TADF WOLEDs. There is an urgent need to propose effective strategies to improve stability while maintaining high efficiency in all-TADF WOLEDs.
As widely recognized, the primary contributor to the diminished stability of TADF devices is the accumulation of long-lived triplet excitons7,9,12,13. To alleviate this issue, a common approach involves selecting donor-acceptor (D-A) type TADF emitters that exhibit a high reverse intersystem crossing rate (kRISC) for fabricating all-TADF WOLEDs. This facilitates the fast conversion of “dark” triplet (T1) excitons into “bright” singlet (S1) excitons, suppressing exciton annihilation events such as the triplet-triplet annihilation and triplet-polaron annihilation14,15. However, the twisted structure5,6,16 inherent in D-A type TADF emitters poses constraints on their radiative transition rate (kr), resulting in a bottleneck for further boosting the efficiency of all-TADF WOLEDs. In contrast, multi-resonance TADF (MR-TADF) emitters, which induce the TADF property through the multi-resonance effect facilitated by ortho-substituted boron and nitrogen planar skeleton17,18,19,20,21,22, circumvent such torsional limitations and exhibit enhanced kr. Consequently, MR-TADF emitters not only offer a high kr comparable to that of conventional fluorescent materials but also surpass them in exciton utilization efficiency, favoring higher device efficiency. Nonetheless, a drawback of MR-TADF emitters is their tendency to exhibit much slower kRISC than D-A type TADF emitters, creating a trade-off scenario where efficiency may improve while device stability becomes a concern23,24. Consequently, achieving a harmonious balance between efficiency and stability remains a formidable challenge when solely relying on one type of emitter.
Here, to address the contradiction, we develop an attractive approach by employing D-A type TADF emitters for the blue emission component and MR-TADF emitters for the yellow emission component in a dual-color all-TADF WOLED configuration (Fig. 1). The blue D-A type TADF emitters, characterized by high kRISC, efficiently drive the up-conversion of triplet excitons. A fraction of these excitons undergo radiative transitions, leading to blue light emission, while the remaining excitons swiftly undergo Förster-type energy transfer (FET) to access the S1 state of yellow MR-TADF emitters. Subsequently, taking advantage of the higher kr of yellow MR-TADF emitters, they promptly engage in radiative transition, emitting the yellow component. This orchestrated process harmonizes triplet up-conversion and singlet radiation, ensuring efficient utilization of excitons to achieve dual objectives of enhancing both efficiency and stability. Specifically, we selected D-A type TADF molecule 2,3,4,5,6-pentakis(3,6-di-tert-butyl-9H-carbazol-9-yl)benzonitrile (5TCzBN)25,26 as the blue emitter for our dual-color all-TADF WOLEDs due to its high kRISC value of 6.1 × 105 s−1. As for the yellow emitter, we selected a typical MR-TADF molecule, 14,17-di-tert-butyl-5,5-bis(3,5-di-tert-butylphenyl)−5H−11b,18b,21b-triaza-8b-boraanthra[1,9-ab]fluoreno[1,9-jk]perylene (MR-Y)27, characterized by its high kr,S of 5.4 × 107 s−1 and remarkable quantum yield of 98%. Using these emitters, we successfully fabricated a dual-emission-layer warm WOLEDs8,9,28,29,30 with record-beating PE and EQE of exceeding 190 lm W−1 and 39%, respectively, accompanied by an improved efficiency roll-off compared to the device without D-A type TADF molecule as sensitizer. A detailed mechanistic analysis unveils the synergistic effect arising from the combination of D-A type TADF and MR-TADF emitters increases the rate of Förster-type energy transfer (kFET) from 2.7 × 107 s−1 to 5.6 × 107 s−1, thereby augmenting the population of radiative singlet excitons. Simultaneously, the rate of Dexter-type energy transfer (kDET) decreases from 2.0 × 107 s−1 to 2.4 × 105 s−1, mitigating the accumulation of dark triplet excitons at MR-TADF emitters. Consequently, the device lifetime improves by 4.5 times compared to the control device, achieving an extended LT80 (operational time to 80% of the initial luminance) of 446 h at an initial luminance of 1000 cd m-2, making it the most stable all-TADF warm WOLEDs8,13,31. Furthermore, utilizing the proposed strategy, another combination of D-A type TADF and MR-TADF emitters achieves standard white emission with CIE (x,y) coordinates of (0.33,0.34) and a high EQE of 35.6%, confirming the universality of our strategy. Additionally, through precise optimization of exciton recombination regions, we effectively regulated the CIE coordinates, transitioning from (0.28, 0.47) to (0.35, 0.53), thus accommodating diverse illumination requirements32,33,34. This work pioneers the integration of MR-TADF materials into all-TADF WOLEDs, thereby laying a solid foundation for the design of highly efficient and stable all-TADF WOLEDs.

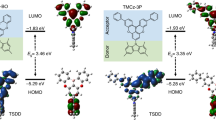
Schematic representation of the strategy developed in this work: employing D-A type TADF materials (lower left) with rapid kRISC as the blue emitter and MR-TADF materials (lower right) with fast kr as the yellow emitter. B, Y, D, A, kRISC, kISC, and kr refer to blue emitter, yellow emitter, donor, acceptor, reverse intersystem crossing rate, intersystem crossing rate, and radiative decay rate, respectively.
Results
Prior to fabricating WOLED devices, we investigated the performance of blue devices without doping MR-Y, using the following structure: indium-tin oxide (ITO)/1,4,5,8,9,11-hexaazatriphenylenehexacarbonitrile (HAT-CN, 5 nm)/1,1-bis[(di-4-tolylamino)phenyl]cyclohexane (TAPC, 30 nm)/tris(4-carbazolyl-9-ylphenyl)amine (TCTA, 15 nm)/3,3-di(9H-carbazol-9-yl)biphenyl (mCBP, 10 nm)/emitting layer (EML, 30 nm)/(1,3,5-triazine-2,4,6triyl)tris(benzene-3,1-diyl)(tris(diphenylphosphine oxide)) (POT2T, 20 nm)/1-(4-(10-([1,1’-biphenyl]−4-yl)anthracen-9-yl)phenyl)-2-ethyl−1H-benzo[d]imidazole (ANT-BIZ, 30 nm)/8-hydroxyquinolinato lithium (Liq, 2 nm)/alumina (Al, 100 nm). In this configuration, HAT-CN serves as the hole injection layer, while both TAPC and TCTA act as the hole transporting layers. ANT-BIZ functions as the electron transporting layer. To confine exciton diffusion, mCBP and POT2T are employed as the electron-blocking layer and hole-blocking layer, respectively. The emitting layer consists of blue emitter of 5TCzBN and host of 1,3-dihydro-1,1-dimethyl-3-(3-(4,6-diphenyl-1,3,5-triazin−2-yl)phenyl)indeno[2,1-b]carbazole (DMIC-TRZ). These blue devices exhibit an emission peak at 490 nm with EQEs ranging from 18.3% to 22.0%. Notably, the device featuring a 20 wt% doping of 5TCzBN demonstrates the highest efficiency and minimal efficiency roll-off, as depicted in Supplementary Fig. 1 and Supplementary Table 1.
Hence, we fabricated single-emission-layer WOLEDs labeled as devices A and B, with the doping concentration of 5TCzBN fixed at 20%. The device structure is depicted in Fig. 2a, in which the EMLs of devices A and B consist of 0.3 wt% and 0.5 wt% MR-Y doped into 20 wt% 5TCzBN:DMIC-TRZ (the chemical structures of the relevant materials are shown in Fig. 2b), respectively. The current density-voltage-luminance (J-V-L) characteristics and normalized electroluminescence (EL) spectra of devices A and B at different voltages are illustrated in Supplementary Figs. 2a-2c. Due to the well-aligned energy levels of all layers, both devices could be turned on at a voltage of 2.2 V, representing the lowest value among previously reported devices (Supplementary Table 2). In comparison to device A, device B exhibits better performance (Fig. 2c), whose PE and EQE are respectively 184 lm W−1 and 35.9%, emitting intense warm-white light with an ultra-high luminance (Lmax) of 127,385 cd m−2. This enhancement of performance in device B can be attributed to the higher MR-Y concentration. However, it is worth noting that device B emitted white light with an excess of the yellow component (Fig. 2d), resulting in unsatisfactory color coordinates of (0.31, 0.51). Although reducing the MR-Y concentration to 0.3 wt% (device A) aids in improving the color coordinate (0.27, 0.46), it also brings about a sacrifice in efficiency. Furthermore, diminishing the doping ratio to 0.2% does not yield substantial chromatic enhancements, while concurrently bringing challenges in the device preparation process (Supplementary Fig. 3). Therefore, to achieve synchronous improvement of color coordinates and efficiency, further optimization of the device structure is imperative.

a The energy diagram. b Molecular structures of materials used in the emission layers. c EQE (upper panel) and PE (lower panel) versus luminance characteristics. d Normalized electroluminescent spectra measured at 1000 cd m−2. Source data are provided as a Source Data file.
We further designed a dual-emission-layer device, denoted as devices C and D, with their performances depicted in Fig. 3 and Supplementary Figs. 2d-2f. In Fig. 3a, the EML of device C comprises a blue-emissive layer (B-EML) consisting of 20 wt% 5TCzBN:DMIC-TRZ and a yellow-emissive layer (Y-EML) consisting of 0.5 wt% MR-Y:20 wt% 5TCzBN:DMIC-TRZ, each with a thickness of 15 nm. This design improves CIE coordinates to (0.30, 0.49) while maintaining a remarkable PE of 190 lm W−1 (confirmed at 203 lm W−1 using a PR735 SpectraScan Spectroradiometer, Supplementary Fig. 4) and an unprecedented EQE of 39% (Figs. 3b and 3c). Both the EQE and PE far surpass those of previously reported white devices, including all-fluorescence WOLEDs (130 lm W−1, 32.8%, ref. 8), all-phosphorescence WOLEDs (105 lm W−1, 28.1%, ref. 35) and hybrid fluorescence-phosphorescence WOLEDs (71 lm W−1, 25.4%, ref. 36), as shown in Figs. 3e, f and Supplementary Table 2. Importantly, device C exhibits reduced efficiency roll-off, maintaining a comparatively high PE of 82 lm W−1 and an EQE of 24.1% even at the luminance of 1000 cd m-2. In contrast, device D, fabricated without 5TCzBN in the Y-EML, demonstrates a comparable efficiency but shows a steeper roll-off at high brightness (Fig. 3c and Table 1). Furthermore, device C exhibits fast transient EL decay at 490 nm, indicating efficient energy transfer from 5TCzBN to MR-Y (Supplementary Fig. 5). Device D, however, shows slower transient EL decay at 550 nm, suggesting less effective triplet-to-singlet exciton conversion and increased triplet exciton accumulation on MR-Y. Subsequently, we evaluated the operational stabilities of devices C and D at an initial luminance of 1000 cd m-2, as illustrated in Fig. 3d. Obviously, device C displays a significantly extended operational lifetime (LT80) of 446 h, nearly 4.5 times longer than that of device D (LT80 of only 99 h), which is attributed to the incorporation of 5TCzBN in the Y-EML. In addition, by extrapolating with a degradation acceleration factor (n) of 1.7, the LT80 of device C at an initial luminance of 100 cd m-2 can be estimated as 22,353 h, positioning it as one of the most stable among all-fluorescence WOLEDs (Supplementary Table 2).

a Device architectures. Y-EML and B-EML refer to yellow-emissive layer and blue-emissive layer, respectively. b Normalized electroluminescent spectra measured at 1000 cd m-2. c EQE (upper panel) and PE (lower panel) versus luminance characteristics. d Lifetimes of devices at the initial luminance of 1000 cd m−2; The insets show the photograph of device C. e, f EQE (e) and PE (f) versus CIEx coordinates for representative reports including all-fluorescence (black), all-phosphorescence (blue) and hybrid fluorescence-phosphorescence WOLEDs (green). Source data are provided as a Source Data file.
To elucidate the reasons underlying the different operational lifetimes observed in devices C and D, we delved into the photophysical properties of the materials used in the EML. As depicted in Fig. 4a, the absorption spectrum of MR-Y exhibits noticeable overlaps with the photoluminescent (PL) spectra of both DMIC-TRZ and 5TCzBN. The corresponding Förster radius (R0) is 5.48 nm and 5.54 nm (Supplementary Note 1), respectively, suggesting the potential for energy transfer from both DMIC-TRZ and 5TCzBN to MR-Y37. To decipher the energy transfer process, we fabricated four thin films: pristine DMIC-TRZ, 0.5 wt% MR-Y:DMIC-TRZ, 20 wt% 5TCzBN:DMIC-TRZ, and 0.5 wt% MR-Y:20 wt% 5TCzBN:DMIC-TRZ for collecting the transient PL decay curves (Figs. 4b–d and Supplementary Fig. 6). As illustrated in Fig. 4b, the film containing 0.5 wt% MR-Y in DMIC-TRZ displays two distinct emission peaks at 468 nm and 550 nm, corresponding to DMIC-TRZ and MR-Y emission, respectively. Comparing the transient PL decay curves at 468 nm, it is evident that the film with MR-Y displays a considerably shorter exciton lifetime (0.05 μs) in contrast to the film without MR-Y (6.6 μs), confirming energy transfer from DMIC-TRZ to MR-Y (Fig. 4c). Similarly, within the 20 wt% 5TCzBN:DMIC-TRZ matrix (Fig. 4d), the film containing MR-Y also demonstrates a notably reduced lifetime from 5.8 μs to 1.6 μs at the emission wavelength of 490 nm when compared to the film without MR-Y. This observation provides compelling evidence for concurrent energy transfer from 5TCzBN to MR-Y.

a Absorption spectra of MR-Y in the toluene solution and PL spectra of DMIC-TRZ (red) and 5TCzBN films (green). R0, Förster radius. b Normalized PL spectra of films with or without MR-Y doping in the DMIC-TRZ matrix (upper panel) or the 20 wt% 5TCzBN:DMIC-TRZ matrix (lower panel). c, d Transient PL decay curves of films with (left panel) or without (right panel) MR-Y doping in the DMIC-TRZ matrix at the emission wavelength of 468 nm (c); in the 20 wt% 5TCzBN:DMIC-TRZ matrix at the emission wavelength of 490 nm (d). τp and τd, lifetimes of prompt fluorescent and delayed components, respectively. e Energy transfer pathways for the system without (left panel) or with (right panel) 20 wt% 5TCzBN. kRISC, kFET, and kDET, rate constants of reverse intersystem crossing, Förster-type energy transfer, and Dexter-type energy transfer, respectively. Source data are provided as a Source Data file.
Following the methodology outlined in the literature (relevant formulas in Supplementary Note 2)38,39,40,41, we further conducted computations and summarized the kFET and kDET in Fig. 4e and Table 2. In the absence of 5TCzBN, the calculated kFET and kDET from DMIC-TRZ to MR-Y are 2.7 × 107 s−1 and 2.0 × 107 s−1, respectively. The larger kDET values compared to DMIC-TRZ’s intrinsic kRISC value (3.3 × 105 s−1) suggest that triplet excitons do not efficiently undergo up-conversion in the host DMIC-TRZ, instead of populating the dark triplet state of MR-Y through a DET pathway. Moreover, considering MR-Y’s relatively slow RISC process (kRISC = 1.6 × 104 s−1), a significant accumulation of T1 excitons occurs within the device, resulting in substantial efficiency roll-off under high brightness conditions and inferior device stability in device D. With the additional doping of 20 wt% 5TCzBN, the rapid transfer of the excitons from the DMIC-TRZ to 5TCzBN occurs because only the emission of 5TCzBN in the doping film was observed. Therefore, we simplified the ternary system’s energy transfer behavior into a binary system to focus on the rate-determining step of energy transfer between 5TCzBN and MR-Y. In this scenario, the calculated kFET and kDET values become 5.6 × 107 s−1 and 2.4 × 105 s−1, respectively. Notably, the kDET is significantly reduced by two orders of magnitude (from 2.0 × 107 s−1 to 2.4 × 105), which is even lower than the kRISC of 5TCzBN (6.1 × 105 s−1). Consequently, the majority of excitons within 5TCzBN undergo FET to the bright S1 state of MR-Y, while only a negligible fraction follows the DET path to the dark T1 state of MR-Y. This leads to a substantial decline in the population of long-lived triplet excitons. Hence, it is clear that the integration of the D-A type TADF molecule 5TCzBN into the Y-EML reconfigures energy transfer pathways for excitons to effectively mitigate detrimental long-lived T1 excitons in the device. This alleviates the annihilation process of long-lived T1 excitons, ultimately enhancing device stability.
To explore the spatial distribution of exciton recombination for improved control of exciton allocation in WOLEDs, we employed an ultrathin orange fluorescence emitter, 2, 8-di-tert-butyl-5,11-bis(4-tert-butylphenyl)-6,12-diphenyltetracene (TBRb), as a detector at various positions within the EML (Fig. 5a). Due to the fine overlap between the absorption spectrum of TBRb and the PL spectrum of 5TCzBN (Supplementary Fig. 7), efficient FET from 5TCzBN to TBRb could occur. The spatial distribution of excitons is characterized by the relative EL intensity of TBRb and 5TCzBN at different locations8. A graph illustrating relative intensity as a function of position and voltage is developed, as depicted in Fig. 5b. Our findings indicate that at lower operating voltages, the exciton recombination zone predominantly localizes near the electron-transporting layer. As the voltage increases, this recombination zone distributes more uniformly across the entire EML. Accordingly, we can infer that initially, hole-electron recombination primarily occurs within the Y-EML, adjacent to the electron-transporting side. With higher voltages, the recombination extends into the B-EML, consequently enhancing the emission intensity of both the blue and yellow light components (Supplementary Fig. 8). This deduction aligns with the results from single-carrier devices (Supplementary Fig. 9), where the hole transport is faster than the electron transport. Additionally, by modifying the sequence of the B-EML and Y-EML in the device architecture of device C, we fabricated device E as a control (Supplementary Fig. 10), which displays reduced efficiency and a relatively dominant blue emission. This observation aligns with the above discussed exciton distribution. Thus, precise control of the order and thickness ratio between the B-EML and Y-EML is essential to achieve the desired CIE coordinates in dual-color WOLEDs.

a Energy-level diagrams (in eV) of devices employed for detecting exciton recombination zone. b Relative intensity profiles of TBRb and 5TCzBN as a function of position and voltage. c Energy-level diagrams (in eV) of devices C-1 ~ 4, with varying thicknesses of the B-EML (25, 20, 10 and 5 nm, respectively). d Normalized electroluminescent spectra of devices C-1 ~ 4 measured at 1000 cd m-2 (Insets depict CIE coordinates). Source data are provided as a Source Data file.
To adjust the CIE coordinates, we fabricated four devices with varying thicknesses of the B-EML and Y-EML, ranging from 25 to 5 nm (Fig. 5c). The device structures are detailed in Supplementary Table 3. As anticipated, all four devices achieve similar PEs of approximately 190 lm W−1 (Supplementary Fig. 11 and Table 1). However, these devices exhibit markedly different CIE coordinates and color temperatures, ranging from (0.28,0.47)/6833 K for device C-1, (0.30,0.49)/6310 K for device C-2, (0.33,0.52)/5616 K for device C-3 to (0.35,0.53)/5218 K for device C-4, showing a transition from warm white emission to warmer white emission (Fig. 5d). Evidently, our device design strategy provides a universal method for achieving different illumination sources without sacrificing efficiency, making it suitable for diverse scenarios. For example, increasing the thickness of the B-EML produces bright, cool light, which can help keep people alert and active. In contrast, decreasing the thickness of the B-EML generates a softer, warm light, potentially beneficial for human health during nighttime usage1,32,33,34,42,43,44.
Building upon the effective strategy of combining blue emitters with high kRISC and yellow emitters with fast kr, we have extended this approach to enhance color quality. We incorporated the orange-yellow MR-TADF emitter named 6,9-di-tert-butyl-N16,N16,N19,N19-tetraphenyl-3b,10b,13b-triaza-20b-boradifluoreno[1, 9-ab:1’, 9’-jk]perylene-16,19-diamine (MR-O)27, which exhibits an emission peaking at 580 nm with a high kr,S of 7.2 × 107 s−1, in devices F-1 ~ 4, to replace MR-Y and compensate for the lack of red components. As presented in Table 1 and Supplementary Fig. 12, all these devices exhibit improved CIEs compared to the MR-Y emitter, with particularly notable enhancements observed in devices F-3 and F-4 based on double-emission-layer, achieving warm-white emission with CIE coordinates of (0.31, 0.42) and (0.37, 0.45), respectively. Although the efficiencies of these devices are relatively lower than that of device C, they still surpass 130 lm W−1, exceeding the highest value reported to date.
In pursuit of standard white emission targeting a CIE of (0.33, 0.33) and high color rendering index (CRI), we introduced the bluer D-A type TADF emitter of 5-(3, 11-dimethyl-5,9-dioxa-13b-boranaphtho[3,2,1-de]anthracen-7-yl)-10,15-diphenyl-10,15-dihydro-5H-diindolo[3, 2-a:3′, 2′-c]carbazole41 (mMDBA-DI), which features a faster kRISC of 1.5 × 106 s−1 (Supplementary Fig. 13, Supplementary Fig. 14 and Table 2), for the fabrication of devices G-1 and G-2. The device structure is ITO/HAT-CN (5 nm)/TAPC (30 nm)/TCTA (15 nm)/mCBP (10 nm)/EML (30 nm)/DBFPO (10 nm)/ANT-BIZ (30 nm)/Liq (2 nm)/Al (100 nm). In devices G-1 and G-2, the EML consists of 0.3 wt% and 0.5 wt% MR-O doped into 20 wt% mMDBA-DI:DBFPO, where DBFPO (Dibenzo[b,d]furan-2,8-diylbis(diphenylphosphine oxide)), with a higher triplet energy (T1 = 3.1 eV) than DMIC-TRZ (T1 = 2.8 eV), serves as a suitable host for the bluer emitter mMDBA-DI (T1 = 2.9 eV). As shown in Fig. 6a, b and Table 1, both devices exhibit a high maximum EQE of over 35%, indicating efficient exciton utilization in the devices. However, the maximum PE of 90 lm W−1 is deemed unsatisfactory due to the elevated turn-on voltages (from 2.2 V to 3.0 V) necessitated by the high triplet energy host (DBFPO). Particularly, device G-1 achieves a maximum EQE of 35.8% and a maximum PE of 78 lm W−1 (confirmed at 84 lm W−1 using a PR735 SpectraScan Spectroradiometer, Supplementary Fig. 15) at a voltage of 3.6 V (Fig. 6c), with the corresponding CIE coordinates of (0.33, 0.34) locating at blackbody radiation curve. Conversely, device G-2, under a drive voltage of 5.2 V (Fig. 6d), manages to approach standard white light emission with CIE coordinates of (0.33, 0.34) and an EQE of 25.9% at 1000 cd m−2, which is apparently the most efficient WOLEDs with a CIE of x and y values both close to 0.33 at the practical brightness level reported so far8,13,31 (Supplementary Fig. 16 and Supplementary Table 4). This demonstrates that standard white light emission can be attained through careful optimization of material selection, doping concentration, and voltage adjustments. Furthermore, both devices G-1 and G-2 achieve a CRI of approximately 70, indicating excellent color quality for two-color WOLEDs. These outcomes highlight the effectiveness of our approach by employing MR-TADF emitters with high kr as yellow-light components and D-A type TADF molecules with rapid kRISC as blue-light components, offering a robust platform for advancing WOLED technology.

a Current density and luminance versus voltage characteristics. b EQE (upper panel) and PE (lower panel) versus luminance characteristics. c, d Normalized electroluminescent spectra measured at 3.6 V (dotted line) and 5.2 V (solid line) for device G-1 (blue, c) and device G-2 (red, d) (Note: The devices exhibit peak efficiency at 3.6 V and reach a brightness of 1000 cd m-2 at 5.2 V). Source data are provided as a Source Data file.
Discussion
We have presented an effective strategy to harmonize triplet exciton up-conversion and singlet exciton radiation in WOLEDs by employing D-A type TADF emitter as the blue emission component and MR-TADF emitter as the yellow emission component. Capitalizing on the advantageous combination of a high kRISC in the D-A type TADF emitter and a high kr in the MR-TADF emitter, our warm WOLEDs have demonstrated exceptional performance with a substantial EQE of 39% and a PE exceeding 190 lm W−1, accompanied by a long device lifetime with LT80 of 446 h at an initial luminance of 1000 cd m−2. Using the strategy, we have demonstrated standard white emission with a CIE of (0.33, 0.34) and a high EQE of 35.6% by precise modulation of blue and yellow emitters. Furthermore, our detailed investigation of the exciton recombination region offers valuable insights for precisely tuning the chromaticity coordinates of WOLEDs, meeting a diverse range of illumination requirements. We believe that this strategy holds broad applicability and great potential in developing highly efficient and stable WOLEDs.
Methods
Materials
The materials used for fabricating the devices were either purchased from the company or synthesized by co-authors.
Photophysical characterization
UV-vis absorption spectra and photoluminescence (PL) spectra were measured by a Shimadzu UV-2700 spectrophotometer (Shimadzu, Japan) and a Hitachi F-7100 fluorescence spectrophotometer (Hitachi, Japan), respectively. Phosphorescence spectra were recorded on the Hitachi F-7100 fluorescence spectrophotometer at 77 K. Transient PL decay curves were obtained by FluTime 300 (PicoQuant GmbH) in conjunction with a Picosecond Pulsed UV-LASER (LASER375) serving as the excitation source. The photoluminescence quantum yields (PLQY) were measured with a Hamamatsu UV-NIR absolute PL quantum yield spectrometer (C13534, Hamamatsu Photonics) equipped with a calibrated integrating sphere. During the PLQY measurements, the integrating sphere was purged with dry argon to maintain an inert atmosphere.
WOLED fabrication and characterization
The ITO-coated glass substrates used in this study had a sheet resistance of 15 Ω square−1. Prior to deposition, these substrates underwent a meticulous cleaning process involving successive ultrasonication steps with acetone and ethanol, followed by thorough drying using a nitrogen gas flow. Subsequently, the cleaned substrates were subjected to a 20-minute treatment in a UV-ozone surface processor (PL16 series, Sen Lights Corporation). Once treated, the samples were transferred to the deposition system. In the deposition process, the electron injection layer, Liq and Al were both thermally evaporated at a pressure of 5 × 10−5 Pa. The organic layers were deposited at rates ranging from 0.2 to 3 Å s−1. Following the organic film deposition, the Liq and Al layers were deposited at rates of 0.1 and 3 Å s−1, respectively. The emitting area of each device in the study was approximately 0.09 cm2. To characterize the devices, various measurements were conducted using specialized equipment. The current density-voltage-luminance (J-V-L), EQE/PE-L curves and electroluminescence spectra were measured using a Keithley 2400 source meter and an absolute EQE measurement system (C9920-12, Hamamatsu Photonics, Japan). The stability test of the encapsulated WOLEDs was conducted using a luminance meter (SR-3AR, TOPCON, Japan) under constant current density driving conditions with an initial luminance of 1000 cd m−2. The transient electroluminescence characteristics of the devices were detected with a HAMAMATSU C16361 transient spectrometer, together with NF Corporation WF1968 to generate short-pulse excitation. The amplitude and baseline of the pulse are 4 and 0 V, respectively. All the above processes were carried out at room temperature under common laboratory conditions.
Data availability
Source data are provided with this paper. The full set of data generated in this study has been deposited in the Figshare database under the accession code https://doi.org/10.6084/m9.figshare.24279349. Source data are provided with this paper.
References
Shen, Z., Burrows, P. E., Bulović, V., Forrest, S. R. & Thompson, M. E. Three-color, tunable, organic light-emitting devices. Science 276, 2009–2011 (1997).
Kido, J., Kimura, M. & Nagai, K. Multilayer white light-emitting organic electroluminescent device. Science 267, 1332–1334 (1995).
Sun, Y. et al. Management of singlet and triplet excitons for efficient white organic light-emitting devices. Nature 440, 908–912 (2006).
Reineke, S. et al. White organic light-emitting diodes with fluorescent tube efficiency. Nature 459, 234–238 (2009).
Zhang, Q. et al. Efficient blue organic light-emitting diodes employing thermally activated delayed fluorescence. Nat. Photonics 8, 326–332 (2014).
Uoyama, H., Goushi, K., Shizu, K., Nomura, H. & Adachi, C. Highly efficient organic light-emitting diodes from delayed fluorescence. Nature 492, 234–238 (2012).
Wu, Z. et al. Strategic-tuning of radiative excitons for efficient and stable fluorescent white organic light-emitting diodes. Nat. Commun. 10, 2380 (2019).
Liu, H., Fu, Y., Tang, B. Z. & Zhao, Z. All-fluorescence white organic light-emitting diodes with record-beating power efficiencies over 130 lm W-1 and small roll-offs. Nat. Commun. 13, 5154 (2022).
Zhang, C. et al. A π-D and π-A exciplex-forming host for high-efficiency and long-lifetime single-emissive-layer fluorescent white organic light-emitting diodes. Adv. Mater. 32, 2004040 (2020).
Chen, J.-X. et al. Thermally activated delayed fluorescence warm white organic light emitting devices with external quantum efficiencies over 30%. Adv. Funct. Mater. 31, 2101647 (2021).
Liu, H., Fu, Y., Tang, B. Z. & Zhao, Z. Realizing high efficiency and high color quality for all-fluorescence white organic light-emitting diodes by interlayer-sensitizing configuration with electron-capturing agent. Adv. Funct. Mater. 33, 2309770 (2023).
Mao, M. et al. Stable, high-efficiency voltage-dependent color-tunable organic light-emitting diodes with a single tetradentate platinum(II) emitter having long operational lifetime. Adv. Mater. 33, 2004873 (2021).
Zhang, C. et al. Color-tunable all-fluorescent white organic light-emitting diodes with a high external quantum efficiency over 30% and extended device lifetime. Adv. Mater. 34, 2103102 (2022).
Nakanotani, H. et al. High-efficiency organic light-emitting diodes with fluorescent emitters. Nat. Commun. 5, 4016 (2014).
Chan, C. Y. et al. Stable pure-blue hyperfluorescence organic light-emitting diodes with high-efficiency and narrow emission. Nat. Photonics 15, 203–207 (2021).
Chen, Z. et al. Pivotal role of transition density in circularly polarized luminescence. Chem. Sci. 14, 6022–6031 (2023).
Kim, H. & Yasuda, T. Narrowband emissive thermally activated delayed fluorescence materials. Adv. Opt. Mater. 10, 2201714 (2022).
Kondo, Y. et al. Narrowband deep-blue organic light-emitting diode featuring an organoboron-based emitter. Nat. Photonics 13, 678–682 (2019).
Bian, J. et al. Ambipolar self-host functionalization accelerates blue multi-resonance thermally activated delayed fluorescence with internal quantum efficiency of 100. Adv. Mater. 34, 2110547 (2022).
Zou, Y. et al. High-performance narrowband pure-red OLEDs with external quantum efficiencies up to 36.1% and ultralow efficiency roll-off. Adv. Mater. 34, 2201442 (2022).
Huang, Z. et al. Charge transfer excited state promoted multiple resonance delayed fluorescence emitter for high-performance narrowband electroluminescence. J. Am. Chem. Soc. 145, 12550–12560 (2023).
Yang, M., Park, I. S. & Yasuda, T. Full-color, narrowband, and high-efficiency electroluminescence from boron and carbazole embedded polycyclic heteroaromatics. J. Am. Chem. Soc. 142, 19468–19472 (2020).
Hu, Y. et al. Peripherally heavy-atom-decorated strategy towards high-performance pure green electroluminescence with external quantum efficiency over 40 %. Angew. Chem. Int. Ed. 62, e202302478 (2023).
Hu, Y. et al. Efficient selenium-integrated TADF OLEDs with reduced roll-off. Nat. Photonics 16, 803–810 (2022).
Zhang, D. et al. Highly efficient full-color thermally activated delayed fluorescent organic light-emitting diodes: extremely low efficiency roll-off utilizing a host with small singlet-triplet splitting. ACS Appl. Mater. Interfaces 9, 4769–4777 (2017).
Zhang, D., Cai, M., Zhang, Y., Zhang, D. & Duan, L. Sterically shielded blue thermally activated delayed fluorescence emitters with improved efficiency and stability. Mater. Horiz. 3, 145–151 (2016).
Yang, W. et al. An effective approach toward yellow-to-orange multi-resonance TADF emitters by integrating strong electron donor into B/N-based polycyclic architecture: high performance OLEDs with nearly 40% EQE. Adv. Funct. Mater. 33, 2213056 (2023).
Wang, J.-J. et al. High efficiency warm-white light-emitting diodes based on copper-iodide clusters. Nat. Photonics 18, 200–206 (2024).
Wang, Y. et al. A warm-white light-emitting diode based on single-component emitter aromatic carbon nitride. Nat. Commun. 13, 6495 (2022).
Chen, H. et al. Efficient and bright warm-white electroluminescence from lead-free metal halides. Nat. Commun. 12, 1421 (2021).
Han, C. et al. Ladder-like energy-relaying exciplex enables 100% internal quantum efficiency of white TADF-based diodes in a single emissive layer. Nat. Commun. 12, 3640 (2021).
Zhang, Z. et al. Manipulation of Pt···Pt interaction in platinum complex by methyl group to achieve single-doped white OLEDs: an approach to simulation of daylight from dawn until dusk. ACS Mater. Lett. 5, 920–927 (2023).
Chen, K. & Wei, Z. High-efficiency single emissive layer color-tunable all-fluorescent white organic light-emitting diodes. Chem. Phys. Lett. 786, 139145 (2022).
Fröbel, M. et al. Get it white: color-tunable AC/DC OLEDs. Light Sci. Appl. 4, e247–e247 (2015).
Wu, S.-F. et al. White organic LED with a luminous efficacy exceeding 100 lm W−1 without light out-coupling enhancement techniques. Adv. Funct. Mater. 27, 1701314 (2017).
Zhang, H. et al. Novel deep-blue hybridized local and charge-transfer host emitter for high-quality fluorescence/phosphor hybrid quasi-white organic light-emitting diode. Adv. Funct. Mater. 31, 2100704 (2021).
Chen, Z. et al. High-performance circularly polarized electroluminescence with simultaneous narrowband emission, high efficiency, and large dissymmetry factor. Adv. Mater. 34, 2109147 (2022).
Liu, J., Liu, J., Li, H., Bin, Z. & You, J. Boron-dipyrromethene-based fluorescent emitters enable high-performance narrowband red organic light-emitting diodes. Angew. Chem. Int. Ed. 62, e202306471 (2023).
Chen, Z. et al. Cascade chirality transfer through diastereomeric interaction enables efficient circularly polarized electroluminescence. Adv. Funct. Mater. 33, 2215179 (2023).
Főrster, T. 10th spiers memorial lecture. transfer mechanisms of electronic excitation. Discuss. Faraday Soc. 27, 7–17 (1959).
Naveen, K. R. et al. Achieving high efficiency and pure blue color in hyperfluorescence organic light emitting diodes using organo-boron based emitters. Adv. Funct. Mater. 32, 2110356 (2022).
Joo, C. W. et al. Color temperature tunable white organic light-emitting diodes. Org. Electron. 15, 189–195 (2014).
Jiang, Y., Lian, J., Chen, S. & Kwok, H.-S. Fabrication of color tunable organic light-emitting diodes by an alignment free mask patterning method. Org. Electron. 14, 2001–2006 (2013).
Parthasarathy, G., Gu, G. & Forrest, S. R. A full-color transparent metal-free stacked organic light emitting device with simplified pixel biasing. Adv. Mater. 11, 907–910 (1999).
Acknowledgements
This work was funded by the National Natural Science Foundation of China (grant no. 52130308 to C.Y.), the Shenzhen Science and Technology Program (grant nos. JCYJ20220818095816036 and ZDSYS20210623091813040 to C.Y.), Research Team Cultivation Program of Shenzhen University (2023DFT004) and the Foundation for Basic and Applied Research of Guangdong Province (grant no. 2022A1515110445 to Z.C.).
Author information
Authors and Affiliations
Contributions
C.Y. supervised the projects. M.H., J.M., and C.Y. conceived the ideas. M.H., Z.C., and S.H. made the devices and carried out the characterizations. W.Y., Z.H., and Y.Z. synthesized the MR-Y, MR-O, and mMDBA-DI materials. S.G. and Y.T. characterized the device with spectroradiometers. M.H., Z.C., and C.Y. wrote the manuscript. M.H., Z.C., J.M., and C.Y. participated in discussions. All authors discussed the results and commented on the final manuscript.
Corresponding author
Ethics declarations
Competing interests
SZU has filed patent applications on luminescent compositions and devices. C.Y., M.H., J.M. and Z.C. are the authors of the invention. CN patent application no. 2023116829470 (pending). The other authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Zujin Zhao and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Huang, M., Chen, Z., Miao, J. et al. Harmonization of rapid triplet up-conversion and singlet radiation enables efficient and stable white OLEDs. Nat Commun 15, 8048 (2024). https://doi.org/10.1038/s41467-024-52401-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-52401-7
- Springer Nature Limited