

Abstract
Recent advances in spatial transcriptomics (ST) techniques provide valuable insights into cellular interactions within the tumor microenvironment (TME). However, most analytical tools lack consideration of histological features and rely on matched single-cell RNA sequencing data, limiting their effectiveness in TME studies. To address this, we introduce the Morphology-Enhanced Spatial Transcriptome Analysis Integrator (METI), an end-to-end framework that maps cancer cells and TME components, stratifies cell types and states, and analyzes cell co-localization. By integrating spatial transcriptomics, cell morphology, and curated gene signatures, METI enhances our understanding of the molecular landscape and cellular interactions within the tissue. We evaluate the performance of METI on ST data generated from various tumor tissues, including gastric, lung, and bladder cancers, as well as premalignant tissues. We also conduct a quantitative comparison of METI with existing clustering and cell deconvolution tools, demonstrating METI’s robust and consistent performance.
Similar content being viewed by others

Introduction
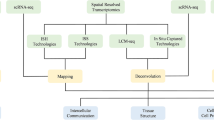
Spatial transcriptomics (ST) measures gene expression while preserving the spatial information that is not available in conventional single-cell RNA sequencing (scRNA-seq)1. The spatial location sheds light on TME’s cellular composition and organization, facilitating investigations into spatial gene expression patterns and cellular interactions at different tumor regions2,3,4. Commonly used ST platforms can be broadly categorized into two types: next-generation sequencing (NGS)-based, such as Visium5, GeoMx6, Slide-Seq7, and hybridization-based approaches such as MERFISH8, seqFISH9, and CosMx10. NGS-based ST methods cover the entire transcriptome but not at single-cell resolution, while in situ hybridization-based methods offer superior spatial resolution but are limited to a small portion of the genome, restricting their potential in discovery-based studies.
Many ST platforms allow a high-resolution scan of the Hematoxylin and Eosin stained (H&E) image of the same tissue section, which is valuable in downstream analysis. Various cell types, distinguished by their unique cell morphologies, can be identified through close examination of the H&E image without the need for cell deconvolution analysis using gene expression. Additionally, potential technical and analytical artifacts can be addressed more effectively by incorporating histological features into ST data analysis. Several state-of-the-art methods have been developed to integrate spatial gene expression and images for various tasks. For example, MUSE11 characterizes tissue composition through the combined analysis of morphologies and transcriptional states in ST data using a deep learning approach; BayesSpace12 employs a fully Bayesian statistical approach to leverage spatial neighborhood information for the enhancement of resolution in ST data and to conduct clustering analysis. SpaGCN13 utilizes a graph convolutional network (GCN) to integrate gene expression and histology to identify spatial domains; TESLA14 uses a convolutional neural network to integrate gene expression and histology to map tumor core, edge, and different cell types at image pixel level. Robust Cell Type Decomposition (RCTD)15 utilizes cell type profiles derived from single-cell RNA sequencing data(scRNA-seq) to decompose cell types and adjusts for variations between sequencing technologies. CytoSPACE16 is designed to align individual cells from a scRNA-seq atlas with spatial expression profiles. Despite the outstanding performance of these methods, they share several limitations. Firstly, they are general analytic tools that work on data generated from any tissue type and are not specifically tailored for studying cancer cells and the tumor microenvironment (TME). Without the ability to incorporate domain knowledge of cancer genomics, these methods may overlook crucial features specific to cancer cells and other key components in the TME. For instance, scRNA-seq may fail to capture certain cell types and states within tissue environments, and a significant cell type might not align with any cluster identified by clustering algorithms. Additionally, certain methods depend on the availability of scRNA-seq data and cell annotations, limiting the flexibility and utility of these approaches in various research contexts. These aforementioned limitations restrict our characterization of the key components and complex interactions in the TME.
In this study, we present an analytic framework that systematically analyzes cancer cells and cells of the TME by incorporating spatial gene expression, tissue histology, and prior knowledge of cancer and TME cells. Our methodology starts with the identification of key cellular components and their states within the TME, including various immune cells and their transcriptional states, tumor stromal components such as cancer-associated fibroblasts (CAFs), and the epithelial compartment. Morphology-Enhanced Spatial Transcriptome Analysis Integrator (METI) also offers complementary information on cell morphology for various cell types from the H&E images. The combined results from gene expression and histological features provide a comprehensive understanding of the spatial cellular composition and organization within the tissue. The evaluation of METI shows robust and consistent performance across ST datasets generated from diverse cancer types, including gastric cancer, lung cancer, and bladder cancer (Supplementary Table 1).
Results
Overview of METI’s workflow
METI analyzes the TME in a systematic, step-by-step manner, focusing on the progression from normal to premalignant cells and then to malignant cells, while also examining the lymphocytes within each tissue section. METI takes standard ST data as input, including a spot-by-gene matrix for gene expression data, an H&E image of the corresponding tissue section, and X, Y coordinates that map the location of each spot onto the image. The goal of METI is the precise identification of various cell types and their respective states within the TME. Each module in METI is tailored to address a particular cell type, enabling focused analysis leveraging domain-specific knowledge (Fig. 1). In the first module, METI identifies normal and premalignant cells, such as goblet cells in the stomach17,18,19. In module 2, METI identifies tumor cell-enriched regions and characterizes their cell states heterogeneity. Module 3 focuses on spatial mapping of T cells including CD4+ and CD8+ T cells, and various T cell states such as regulator T cells (Treg) and exhausted T cells (Tex). In addition to T cells, METI identifies other immune cells including neutrophils, B cells, plasma cells, and macrophages in module 4. In the last module, METI focuses on a comprehensive analysis of CAFs, a subset of activated stromal cells that play a crucial role in cancer progression and therapy resistance20,21,22. This module maps CAFs and their subtypes, including myCAFs, iCAFs, and apCAFs23,24,25,26,27. The outputs generated by METI are comprehensive, including specific cell-type segmentation results, gene expression data, and integrated segmentation-gene expression results that offer a holistic view of the tissue sample, as well as 3D-density plots for the spatial visualization of cell density. We have demonstrated that METI achieves more accurate cell type identification compared to existing methods. Additionally, it remains robust in instances where one modality may be of low quality, as high-quality data from the other modality can compensate, ensuring the reliability and effectiveness of our analysis.

METI takes 10x Visium Spatial Transcriptomics (ST) data, with a spot-by-gene matrix for gene expression data, Hematoxylin and Eosin (H&E) images, and XY coordinates that map the location of each spot onto the image as input. With METI algorithm, METI offers cell type identification, nuclei segmentation and the functionality of generating 3D cell density plots in five distinct modules. Module 1 is dedicated to mapping normal and premalignant cells through the integration of gene expression (GE) data and H&E images. Module 2 focuses on identifying cancer cell domains and characterizing their heterogeneity. Module 3 is dedicated to T cell mapping and phenotyping. Module 4 involves in-depth analysis of other immune cells. Lastly, Module 5 pertains to the analysis of Cancer-Associated Fibroblasts (CAFs).
Mapping normal and premalignant cells
The first module of METI focuses on dissecting the normal and premalignant cells within the epithelial cell compartment. Here, we used goblet cells as an example because they display a distinctive morphological appearance in H&E-stained images. Goblet cells are shaped like wine goblets, with pale, almost white vesicles at the top and oval nuclei at the base. Goblet cells are commonly found in the respiratory, digestive, and reproductive tracts, including the small intestine, colon, and bronchi. They play a critical role in maintaining homeostasis in these tissues. In the context of disease, the abnormal presence of goblet cells in the gut is a key characteristic of a precancerous condition known as intestinal metaplasia17,19,28,29,30.
To showcase the capabilities of METI’s module 1, we applied it to identify goblet cells in a human stomach adenocarcinoma (STAD) sample, labeled as G1, annotated by our gastrointestinal pathologists (Fig. 2a). METI first combines canonical goblet cell markers reported in previous publications31,32,33,34,35 into a meta gene signature, including MS4A10, MGAM, CYP4F2, XPNPEP2, SLC5A9, SLC13A2, SLC28A1, MEP1A, ABCG2, and ACE2 (Supplementary Table 2). This meta gene visually represents the overall expression levels of the goblet cell molecular signature across the whole section. As shown in Fig. 2b, this meta gene was then used to annotate goblet cell-enriched regions using a machine learning model, TESLA14. Notably, cell type annotation in TESLA mainly relies on gene expression, which may lead to false negative annotation due to the regional variation and high level of noise for some marker genes. For instance, as shown in Fig. 2c, TESLA fails to identify goblet cells in region 4. Further examination shows that this false negative in detection was due to the overall low unique molecular identifier (UMI) counts captured in region 4, as illustrated in Fig. 2d. To address the limitations caused by low-quality gene expression data, METI simultaneously performed goblet cell identification on the H&E image. METI employed a K-mean-based segmentation method to detect different morphological components, such as background, nuclei, fiber, gland, and necrosis. Next, by filtering on the color, shape, and size of these components (see Methods), METI is able to accurately detect individual goblet cells characterized by their morphology signature, i.e., their round hollow centers (Fig. 2e). This morphological analysis by METI enabled the identification of goblet cells in region 4 (Fig. 2f), an area overlooked by transcriptomic data alone. Conversely, goblet cells in regions 1 and 3 were not detected through image analysis because the tissue was fragmented, exhibiting discontinuous and fragile shapes. By integrating the gene expression and image analysis results, METI successfully identified all four regions enriched with goblet cells, as shown in Fig. 2g. This integrative approach for goblet cell detection overcomes the limitations posed by low UMI counts and provides a more accurate characterization of goblet cells within the analyzed samples. A detailed examination of goblet cell detection, utilizing both gene expression data and imaging, reveals that accurate detection can only be achieved through the integration of these two modalities. Such detection cannot be achieved by popular spatial clustering methods alone and the necessity of combining both modalities for comprehensive analysis is depicted in the Supplementary Information.

a Pathology annotation depicting goblet cell enriched regions in STAD G1. b Goblet meta gene expression plot at pixel-level. c Spot annotation indicating regions of high goblet cell gene expression on the H&E image. d Total UMI counts for individual spots. e Identification of four distinct goblet-enriched regions on the left side, accompanied by zoomed-in views of goblet regions of the H&E image and segmentation outcomes for regions 2 and 4. f Spot annotation using segmentation results. g METI combined result by integrating gene expression and segmentation. h Pathology annotation highlighting tumor cell-enriched spots of STAD G2 (left), pixel depiction of EPCAM highly expressed regions (middle), and EPCAM+ region annotation on the H&E image (right). i Pixel-level gene expression plots for tumor subtypes, MKI67, MSLN, SOX9, and CLDN18. j Overlay of regions expressing tumor-related genes and SOX9-positive regions. k Nuclei segmentation (left) and 3D cell density plots (right).
Identification of cancer cell domains and heterogeneity
The majority of solid tumors originate from epithelial cells, known as carcinomas, including gastric, lung, bladder, breast, prostate, and colon cancers, while some other solid tumors start in other types of tissues including sarcoma and melanoma. Regardless of their cell of origin, understanding the molecular features and cellular heterogeneity of malignant cells is crucial for unraveling the mechanisms underlying tumor growth, invasion, metastasis, and therapeutic response. Therefore, METI’s second module focuses on the analysis of malignant cells. This module starts by identifying cancer cells using cancer cell markers that are curated by the authors such as cytokeratins (CK), EPCAM, and trefoil factors. As depicted in Fig. 2h, METI effectively identifies all tumor regions in STAD sample G2, in strong agreement with annotations made by our experienced pathologists. Next, METI incorporates additional markers to characterize cancer cell states and heterogeneity, including markers of cell proliferation such as MKI67 to map proliferative cancer cells, stemness-related markers such as SOX9 to identify stem-like cancer cells in STAD, and therapeutic targets like CLDN18 and MSLN to further characterize tumor subtypes36,37,38,39. These aforementioned marker genes exhibit distinct expression patterns within the tumor region of sample G2, as illustrated in Fig. 2i. They can be utilized to characterize different states of cancer cell states. For example, as shown in Fig. 2j, METI is not only able to identify the SOX9+ tumor region but also can illustrate the co-localization or exclusivity of different cancer cell states in Supplementary Fig. S1. This module offers a flexible and customizable approach, allowing users to input their genes of interest for tumor state identification. Additionally, users can employ genes associated with critical pathways such as KRAS, EGFR, and factors like hypoxia to conduct a comprehensive exploration of cancer cell states and spatial heterogeneity across diverse cancer types.
Quantifying the distribution and density of cells spatially within biological tissues is crucial for diverse applications, particularly in the field of pathology and oncology. While gene expression provides a molecular lens, the associated H&E images can be leveraged to measure spatial cell distribution and density. Following a parallel process in module 1, METI next conducted tumor cell nuclei segmentation, and then generated 3D tumor cell density plots (Fig. 2k), visually depicting the spatial distribution and density of cancer cells. This function serves to convey the spatial distribution, density, and pattern of cell types of interest.
T cell mapping and phenotyping
Module 3 in METI is dedicated to characterizing T cells and their various states within the TME. Initially, we utilize specific T cell markers, including CD3D and CD3E, to map T cell-enriched regions. Within the identified T cell regions, we further discern the different states of T cells. By adding specific cell lineage markers such as CD4, CD8A, and CD8B40, we can further distinguish CD4+ T cells, CD8+ T cells, and their various states including CD4+ Tregs (e.g., FOXP3, IL2RA) and CD8+ Tex cells by incorporating known immune checkpoint genes (e.g., PD-1, TIM-3, and LAG-3, CTLA-4, TIGIT) and Tex related transcription factors (e.g., TOX)40. Furthermore, this module provides function of overlaying two or more different T cell states within defined cancer cell regions directly on the same tissue section, allowing us to visualize their spatial relationships. Given that the level and spatial distribution of infiltrated T cells are critical factors influencing tumor immune phenotypes and immunotherapy responses, METI’s 3D module creates cell density plots for the entire image, serving to visually depict the spatial distribution of T cells within the TME.
To showcase the capability of this module, we applied METI to analyze a STAD sample G3 and a lung adenocarcinoma (LUAD) sample L1. The pathology annotations for both samples are presented in Supplementary Fig. S2. METI identified regions characterized by elevated T cell gene expression levels, as illustrated in Fig. 3a. Next, to delineate regions enriched in CD8+ T cells, we restrict our analysis to T cell-enriched regions only. The regions enriched in CD8+ T cells are shown in Fig. 3b, and different states including CD4+ Tregs and CD8+ Tex cells are shown in Fig. 3c, d. Mapping distinct T cell states aids in elucidating their spatial landscape and relationships within the analyzed STAD and LUAD samples, as well as cellular interactions, fostering the generation of insightful hypotheses.

a Pixel-level visualization of T cell marker gene expression in STAD G3 and LUAD L1 (left), accompanied by annotation indicating regions of T cell marker gene expression on the H&E image. b Pixel-level representation of CD8+ T cell marker gene expression (left), along with annotation of CD8+ T cell marker gene-expressing regions on the H&E image (right). c Pixel-level representation of CD4+ Treg marker gene expression. d Pixel-level depiction of CD8+ Tex marker gene expression. e Overlay displaying the intersection of tumor+ region and CD4+ Treg-positive region. f Overlay illustrating the overlap between tumor+ region and CD8+ Tex-positive region. g 3D cell density plots for STAD G3 and LUAD G1. h Overlay demonstrating the spatial relationship between CD4+ Treg and CD8+ Tex-positive regions.
As the relative locations of CD4+ Tregs and CD8+ Tex cells to cancer cells impact tumor immune phenotypes41,42 and immunotherapy responses, we have overlaid regions with cancer cells with those enriched with CD4+ Tregs and CD8+ Tex cells, respectively, as depicted in Fig. 3e, f. Based on the overlay results, we observe distinct enrichment patterns in CD4+ Tregs and CD8+ Tex cells across different samples. Specifically, in sample G3, CD4+ Tregs are slightly less abundant than CD8+ Tex cells. Conversely, in the sample L1, CD8+ Tex cells are less abundant than CD4+ Tregs (Fig. 3e, f). This highlights the variability in T cell states among different tumor types. To better illustrate the spatial cell distribution of the whole image, METI provides 3D cell density plots (Fig. 3g) based on the nuclei density segmented from the H&E image. For the STAD sample, a region in the upper left displays higher cell density, whereas the LUAD sample shows relatively homogeneous cell density throughout. Furthermore, we conducted an overlay of CD4+ Treg and CD8+ Tex signals to study their spatial co-localization patterns (Fig. 3h). Notably, CD4+ Tregs and CD8+ Tex cells tend to co-localize at the bottom left of the LUAD sample, while the rightmost part of the LUAD sample solely comprises CD4+ Treg cells, indicating the heterogeneity in spatial distribution and cellular composition of T cells. This co-localization analysis provides a better understanding of the coexistence and potential interplay between these two T cell states. Moreover, METI can assist researchers in studying various types of T cells, such as naïve T cells, memory T cells, follicular helper T cells, and their transcriptional states. Users can customize it to plot specific T cell types and states of interest. This flexibility allows researchers to explore T cell state composition and distribution within TME.
In-depth analysis of other immune cells
METI’s module 4 is capable of detecting immune cell types other than T cells, including neutrophils, macrophages, B cells, and plasma cells, which are critical components in the TME. METI utilizes validated gene signatures to identify specific immune cell types/states40,43,44,45. We have applied this module to two bladder cancer samples B1 and B2 for neutrophil detection. The two H&E images are shown in Fig. 4a, c. The neutrophil-enriched regions in these two sections were verified by our experienced pathologists as ground truth for evaluation (Supplementary Fig. S3). As shown in Fig. 4b, d, METI identified regions exhibiting elevated gene expression levels of neutrophil marker genes in both sections. Subsequently, METI conducted corresponding annotation for neutrophil-enriched regions directly on H&E image, isolating regions expressing the neutrophil marker genes and providing a magnified view, as illustrated in Fig. 4e. Upon zooming in on these annotated regions, neutrophils, which exhibit characteristic multi-lobed nuclei, were easily distinguished in the image analysis. In Fig. 4f, four regions where neutrophils have been pathology-verified were circled out and subsequently segmented for neutrophil detection. The results correlated well with the annotation using gene expression. We also provided 3D cell density as shown in Fig. 4g to illustrate the spatial cell distribution in the vicinity of neutrophils within the TME.

a H&E image of bladder cancer sample B1. b Pixel-level visualization of neutrophil marker gene expression in BLCA-B1. c H&E image of bladder cancer sample BLCA-B2. d Pixel-level visualization of neutrophil marker gene expression in BLCA-B2. e Annotation indicating regions of high neutrophil gene expression on the H&E image for BLCA-B1 and BLCA-B2; Zoom-in display of three neutrophil-enriched regions of BLCA-B1 and BLCA-B2, and four yellow-circled regions where neutrophils present visually. f Zoomed-in view of four yellow-circled region in (e) and corresponding segmentation results. g 3D cell density plots for BLCA-B1 and BLCA-B2. h Pixel-level visualization of B cell marker gene expression in STAD G4 (left), accompanied by annotation indicating regions of B cell marker gene expression on the H&E image (right). i Pixel-level visualization of plasma cell marker gene expression in STAD G4 (left), accompanied by annotation indicating regions of plasma cell marker gene expression on the H&E image (right). j 3D cell density plots for STAD G4. k Pixel-level visualization of macrophage marker gene expression in STAD G4 (left), accompanied by annotation indicating regions of plasma macrophage marker gene expression on the H&E image (right). l Zoomed-in view of macrophage regions of the H&E image and segmentation.
Additionally, we demonstrate the capability of this module by mapping B cells and plasma cells in an STAD sample (Fig. 4h, i). The 3D cell density plot (Fig. 4j) aligns well with the lymphoid aggregates in sample STAD G4 annotated by our pathologists (Supplementary Fig. S2a). Similarly, macrophages can be correctly mapped in the LUAD sample based on pathology annotation (Fig. 4k and Supplementary Fig. S2b). Within the regions showing high macrophage marker gene expression, a randomly selected region was segmented, revealing a cluster of macrophages (Fig. 4l). In addition to the aforementioned immune cell types, this module maintains flexibility by allowing users to investigate other specific immune cell populations of interest using their curated gene signatures.
Analysis of cancer-associated fibroblasts (CAFs)
In Module 5, METI is designed to analyze stromal cell components including CAFs and various CAFs subtypes within the TME. CAFs are known for their exceptional heterogeneity, both phenotypically and functionally46,47,48. They are categorized as activated fibroblasts, representing an essential component of the TME with both tumor-promoting and tumor-restraining activities49,50,51. CAFs are phenotypically and functionally heterogeneous. Different subtypes of CAFs such as myofibroblastic CAFs (myCAFs), inflammatory CAFs (iCAFs), and antigen-presenting CAFs (apCAFs) have been identified and described23,24,25,26,27.
We applied module 5 to the gastric sample G2 which was annotated to contain abundant tumor stroma by our pathologists (Fig. 5a). METI first segmented CAFs and generated a fibroblast cell density plot as illustrated in Fig. 5b. Next, we found that the fibroblasts enriched region annotated by METI have diminished UMI counts, aligning with the notion that cancer cells tend to have higher UMI counts compared to other cell types (Fig. 5c). METI next effectively mapped CAFs within the sample using the CAF metagene (Fig. 5d) and annotated CAFs directly on H&E image, which was highly consistent with the pathology annotation. Within the annotated CAF regions, METI further delves into the characterization of CAF subtypes, including myCAFs, iCAFs, and apCAFs (Fig. 5e–g)23,24,25,26,27. To characterize the spatial co-localization of CAF subtypes, we overlayed the three CAF populations with the total CAF-positive regions (Fig. 5h). This approach allows us to better understand the spatial heterogeneity of CAFs within the TME. Likewise, METI can co-map CAFs, cancer cells, and any other immune cell subsets of interest to provide additional insights into cellular interactions among them. This module remains adaptable, enabling users to explore other subregions related to CAFs based on their specific interests.

a Pathology annotation of fibroblast-enriched spots in STAD G2. b Fibroblasts segmentation result (left), accompanied by 3D fibroblast density plots (right). c Total UMI counts for individual spots. d Pixel-level meta gene expression plot for CAF (left), with annotations highlighting regions of elevated CAF gene expression on the corresponding H&E image (right). e Pixel-level meta gene expression plot specifically for myCAF (left), accompanied by annotations indicating regions of elevated myCAF gene expression on the H&E image (right). f Pixel-level meta gene expression plot for iCAF (left), with annotations denoting regions of elevated iCAF gene expression on the H&E image (right). g Pixel-level meta gene expression plot for apCAF (left), with annotations indicating regions of elevated apCAF gene expression on the H&E image (right). h Overlay showcasing regions of high gene expression for myCAF, iCAF, apCAF, and general CAF.
Quantitative comparison with existing tools
We initiated our analysis by comparing METI’s performance against two spatial clustering methods, SpaGCN13 and BayesSpace12, specifically in the context of goblet cell annotation within a human STAD dataset. The pathologist’s annotations, serving as a benchmark, indicated the locations of goblet cells on the H&E image at the spot level, as depicted in Supplementary Fig. S4a, b. METI showcased high accuracy (ACC = 0.778) in identifying these spots, as demonstrated in Supplementary Fig. S4c.
To conduct a fair comparison with the unsupervised nature of SpaGCN and BayesSpace, where cluster identities are initially unknown, we established a criterion: a cluster is considered a goblet cluster if over 10% of its spots are true goblet cells. This approach allowed us to categorize clusters into goblet and non-goblet groups for a binary comparison and to calculate the accuracy for both BayesSpace and SpaGCN. The clustering results for BayesSpace are illustrated in Supplementary Fig. S4d. BayesSpace achieved its highest accuracy of 0.704 at n = 15 clusters, which is lower than METI. Further analysis using river plots in Supplementary Fig. S4e, revealed that BayesSpace struggled to isolate a single cluster exclusively composed of goblet cells, regardless of the cluster number chosen. SpaGCN encountered similar difficulties, as shown in Supplementary Fig. S4f, with its highest accuracy of 0.734 at n = 5 clusters. These findings emphasize METI’s advantage in cell type annotation over existing clustering methods. While both METI and the spatial clustering methods are reference-free, METI is capable of providing accurate cell type annotations by incorporating domain knowledge into its framework. In contrast, the spatial clustering methods primarily capture the overall tissue structure rather than cell type enrichment.
Furthermore, we utilized gastric single-cell RNA sequencing data as a reference to evaluate the performance of METI, RCTD, and CytoSpace in identifying goblet cells, CD4+ T cells, and CD8+ T cells within two distinct human gastric cancer spatial transcriptomics (ST) datasets, as detailed in Supplementary Fig. S5. For the first dataset, RCTD displayed limited accuracy of 0.525 in Supplementary Fig. S5a, notably struggling to accurately determine the location of goblet cells. In contrast, CytoSpace exhibited a significantly higher accuracy of 0.751 in Supplementary Fig. S5b, aligning closely with the findings presented by METI in Supplementary Fig. S5c.
Due to the lack of pathologist annotations in the second dataset, hence the accuracy cannot be quantified, we focused on comparing the capabilities of RCTD and CytoSpace in identifying CD4+ T cells and CD8+ T cells. RCTD has a limited ability to accurately identify CD4+ T cells and CD8+ T cells (Supplementary Fig. S5d, g), while CytoSpace outperforms RCTD (Supplementary Fig. S5e, h). Consequently, to further validate these findings, we analyzed the expression of marker genes for a visual assessment of the results, as depicted in Supplementary Fig. S5f, i, which further supports the presence of these cell types in the locations identified by METI and CytoSpace.
METI’s robustness with low-quality images
We illustrate METI’s robustness with low-quality images using another human STAD dataset. As depicted in Supplementary Fig. S6a, blurriness from loss of camera focus obscures cell boundaries, making it difficult for our pathologists to provide detailed annotation of lymphocytes due to rough boundaries between different regions, as noted in Supplementary Fig. S6b. In this dataset, we applied METI for B cell annotation. B cells, along with other lymphocytes, are distinguishable in H&E images by their small, dark-purple nuclei. Therefore, the initial step with METI is to identify lymphocytes on the H&E images. As illustrated in Supplementary Fig. S6c, the blurriness of the image led to numerous false positive detections of lymphocytes, which are sparsely distributed, particularly around the tissue border. Moreover, the H&E staining does not adequately differentiate B cells from other lymphocytes due to their similar morphological features. The subsequent step in the METI is to identify B cells using specific gene markers, including MS4A1 and CD19. Supplementary Fig. S6d showcases the B cell distribution identified by METI through gene expression only. Given that B cells are a specific subtype of lymphocytes, METI further refines the detection by overlaying B cell data with the lymphocyte regions identified from image analysis, as displayed in Supplementary Fig. S6e. The results are then translated into spot-level data, as depicted in Supplementary Fig. S6f. This case demonstrates METI’s capability to merge gene expression and image analysis in a knowledge-aware manner, ensuring robustness in cell-type annotation even when image quality is low.
Discussion
In this study, we present METI, a robust machine-learning framework developed to address the demand for comprehensive profiling of diverse cell types and their phenotypic states within the TME. By effectively integrating transcriptomic data with histological image information, METI reduces the risk of non-specific mapping of key cell types in the TME. Unlike certain existing methods whose performance depends on the quality and granularity of cell type annotations in their reference data, METI is not constrained by these factors, providing considerable advantages in terms of flexibility and applicability across various datasets. These features set METI apart from existing cell deconvolution tools, which primarily rely on gene expression data and overlook tissue histology.
While METI utilizes established tools such as TESLA and K-means segmentation for its analysis framework, these methods were originally developed as general-purpose tools, not specifically optimized for cancer ST data analysis. METI’s innovation lies in its ability to compensate for low-quality data in one modality by robustly integrating tissue morphology with transcriptomic profiles in a knowledge-driven manner. We have systematically benchmarked METI against multiple popular spatial clustering and cell-type deconvolution methods, and the results demonstrate METI’s superior accuracy, high resolution, and improved visualization in analyzing key components of the TME.
METI comprises five modules that characterize various cellular components, encompassing distinct types and states of tumor cells, immune cells, and stromal cells. By jointly analyzing transcriptomic and histological information, METI offers a more comprehensive approach to understanding cancer cells and the TME compared to analyzing gene expression and histology images separately. Notably, our results have shown that METI is able to mitigate the impact of low-quality data in one modality by leveraging high-quality information from the other modality. As described in the goblet cell detection case with regional low UMI counts, METI’s integrated results compensate for the low-quality gene expression data. Moreover, this integrative approach makes METI robust against issues such as low resolution or artifacts in H&E images.
Additionally, METI is uniquely capable of stratifying various cell states, including CD4+ Tregs, CD8+ Tex cells, iCAFs, myCAFs, apCAFs, among many others. Notably, this stratification is not restricted to the aforementioned subtypes; users have the flexibility to define and explore other cell subtypes to suit their research needs. METI is designed to be user-friendly and accessible. To this end, we have integrated a predefined set of cell-type markers and model parameters within METI’s framework. Based on recent pan-cancer studies40,52, the expression of cell lineage markers and markers characteristic of cell phenotypic states for immune and stromal cells are typically consistent across different tumor tissues and datasets, serving as a valuable reference. Hence, METI can be used effectively without the need for user-specified input, making it an accessible tool for users across different levels of bioinformatics expertise. Furthermore, recognizing the diversity of research needs and the specificities of different tissue types and diseases, METI offers flexibility for more advanced users. Researchers can customize METI by inputting their own markers of interest, tailored to specific tissues or diseases under study. This flexibility allows users to leverage METI’s framework to conduct highly specialized analyses. By combining both predefined markers and the ability for users to introduce their markers, METI bridges the gap between ease of use and customizability, catering a broad spectrum of research requirements.
METI has potential translational value and practical utility. By seamlessly integrating with existing clinical diagnostic and treatment planning tools, METI has the potential to complement and enhance current diagnostic workflows, facilitating more informed decision-making in clinical settings. While METI presents a useful framework for in-depth cancer cell and TME cell profiling, it is important to recognize its limitations. One notable challenge arises from the dependence on transcriptomics data, which can be susceptible to technical fluctuations, constrained sample quality, dropout occurrences, and dependence on user-provided gene signatures, possibly resulting in the generation of false negative outcomes. Moreover, the performance of METI’s segmentation function could exhibit variability contingent on the quality and resolution of the images being analyzed.
Methods
Human samples
All human data in this manuscript are deidentified to ensure patient privacy. All human LUAD and normal lung tissues were obtained from patients who provided informed consent and under institutional review board-approved protocols at The University of Texas MD Anderson Cancer Center. Written informed consent was given to all the subjects. Two patients with primary muscle-invasive bladder carcinoma (T2–T4) were evaluated at the MD Anderson Cancer Center and underwent trans urethral resection of bladder tumor (TURBT). All patients received no treatments at the time of surgery. Bladder tumor tissues at the MD Anderson Cancer Center were obtained via institutional IRB-approved research protocol PA17-0577 (PI: J.G.). Signed informed consent was obtained from all patients. The collection of gastric tumor tissues at Zhejiang Cancer Hospital was approved by the institutional Ethics Committee (no. IRB-2020-109), and all patients provided written informed consent.
ST data generation
ST on FFPE slides was performed with the Visium spatial technology from 10X Genomics. Two to three consecutive tissue sections of 5-μm thickness were collected for RNA extraction with the Qiagen RNeasy FFPE Kit. To assess the RNA quality of the tissue, the purified RNA was immediately processed to calculate the percentage of total RNA fragments >200 nucleotides (DV200) using the Agilent RNA 6000 Pico Kit. Based on DV200 evaluation, blocks with DV200 > 30% were selected for proceeding with sectioning. The area of interest (11 × 11 mm) on section was carefully placed within the allowable area to ensure compatibility with the Visium CytAssist instrument. The tissues were then deparaffinized, stained, and decross-linked, followed by probe hybridization, ligation, CytAssist-enabled RNA digestion, and oligo capture, release, and extension. The Visium spatial gene expression FFPE libraries were constructed using the Visium CytAssist Spatial Gene Expression for FFPE Human Transcriptome Probe Kit (PN-1000444) following the manufacturer’s guidance. Constructed libraries were sequenced on the Illumina NovaSeq 6000 platforms to achieve a depth of at least 75,000 mean read pairs per spot.
ST data alignment
There are two steps related with ST alignment. First alignment is during the sample generation, and second alignment is related with annotation including pathologist annotation and METI’s automatic annotation. Firstly, the spatial gene expression and H&E image are generated on the same tissue section. According to 10x CytAssist technology, Visium slide with two Capture Areas are placed in the CytAssist instrument so that the tissue sections on the standard slides can be aligned on top of the Capture Areas. During the preprocessing step, we use the Spaceranger Count alignment process to obtain the coordinates of each measured spot. These coordinates are then used to accurately map each spot back onto the image. Secondly, Manual annotations by pathologist are completed in Loupe Browser, which is a ST image visualization tool developed by 10x Genomics. METI annotates gene expression onto the H&E image based on spot coordinates.
Data processing
METI takes spatial gene expression and histology image data as input. The ST gene expression data contains an N \(\times\) M matrix of UMI counts, where N denotes the number of spots and M represents the number of genes. Each spot is associated with 2-dimensional spatial coordinates denoted as (x, y). The gene expression values for each spot are normalized by dividing the UMI count of each gene within that spot by the overall UMI count of all genes in the same spot. The result is then scaled up by a factor of 10,000 and converted to a natural logarithm scale.
Meta gene plot generation
We utilized TESLA to get a pixel-level gene expression matrix. For a cell type which has \(K\) marker genes, we combine these \(K\) markers into a single meta-marker gene. Considering that not all marker genes may be expressed, the meta gene is designed to preserve the expression patterns for at least a subset of the marker genes. Given \(K\) marker genes and a predefined number \(k\) based on the specificity of the markers\(,{k}\le K\), for any given pixel \(i\), we first sort the relative expression of all markers in descending order as \(\{{e}_{1,i},\,{e}_{2,i},\,{e}_{3,i},\,{...,{e}}_{K,i}\}\). Next, we select the top \(k\) expression values and compute the meta gene’s relative expression at pixel \(i\) as:
If number of expressed marker genes less than \(k,\) which is \({k}^{{th}}\) marker has zero expression at a given pixel, the meta gene’s relative expression will be 0 at that pixel. This ensures that expression patterns present in less than \(k\) genes are excluded from the meta gene, preventing the generation of less representative patterns.
T cell states mapping
First, we employed CD3D and CD3E as markers to accurately annotate T cell-enriched regions using TESLA. The output of TESLA is a binary value array \({A}_{{T\; cell}}\) of the same size as the histology image, in which value of 1 indicates T cell enrichment while 0 indicates non-enrichment of T cells. Similarly, we used CD4 marker gene to identify rough CD4+ T cell distribution patterns using TESLA, and the results is stored as a binary array \({A}_{{{CD}4}^{+}{T}}^{{\prime} }\) indicating the enrichment of rough CD4+ T cells. Since CD4+ T cells are a subset of T cells, we then filter \({A}_{{{CD}4}^{+}{T}}^{{\prime} }\) based on \({A}_{{T\; cell}}\) to get a more accurate enrichment matrix of CD4 T cell, \({A}_{{{CD}4}^{+}{T}}\), as below:
This operation filtered out false positive detected CD4+ T cells by TESLA which do not express general T cell markers. Using a similarly pipeline, METI is able to identify CD8+ T cells using CD8A and CD8B. We obtained array \({A}_{{{CD}8}^{+}{T}}^{{\prime} }\) where 1 denotes CD8A or CD8B positive and 0 denotes negative. By intersecting \({A}_{{T\; cell}}\) and \({A}_{{{CD}8}^{+}{T}}^{{\prime} }\), we obtained \({A}_{{{CD}8}^{+}{T}}\) storing pixel with CD8+ T cell positive expression.
Next, to identify the CD4+ Treg cells, we input specific marker genes for Tregs, including FOXP3 and IL2RA, to delineate the corresponding regions and generate an array \({A}_{{Treg}}^{{\prime} }\). Given that CD4+ Treg cells is a subset of CD4+ T cells, we construct a new binary array, \({A}_{{{CD}4}^{+}{Treg}}\), by intersecting \({A}_{{{CD}4}^{+}{T}}\) and \({A}_{{Treg}}^{{\prime} }\). By visualizing \({A}_{{Treg}}\), we obtained the annotation that outlines the CD4+ Treg signature on the H&E image.
The identical procedure is employed to identify CD8+ Tex cells. We input specific marker genes for Tex cells, including HAVCR2, LAG3, CTLA4, TIGIT, PDCD1, and LAYN. With prior knowledge53, any pixel expresses any 2 marker genes out of these 6 marker genes will be set to 1. To annotate the corresponding regions, we generate an array \({A}_{{Tex}}^{{\prime} }\). Like the CD4+ Treg identification process, we create a new binary array \({A}_{{{CD}8}^{+}{Tex}}\) for CD8+ Tex cells by intersecting \({A}_{{{CD}8}^{+}{T}}\) and \({A}_{{Tex}}^{{\prime} }\). This array annotates the CD8+ Tex cell signature on the H&E image. In this case, we have \(K\) = 6 and set \(k\) = 2 based on previous publication, we derived the meta gene as formula (1), in which pixels with at least 2 marker genes expression out of the 6 have non-zeros expression. Then \({A}_{{{CD}8}^{+}{Tex}}\) can be obtained as:
For other immune cell annotations, the markers and choice of \(k\) can be found in Supplementary Table 3.
Nuclei segmentation
We first divided the image into multiple patches of size 2000 \(\times\) 2000 pixels, and supplement incomplete patches on the border with black blocks to 2000 \(\times\) 2000 pixels. We next converted the image from the default blue (B), green (G), red (R) color format to RGB format using OpenCV and then reshaped the 3D image array into a 2D array, where each row represents a pixel in the image, and three columns corresponds to the RGB color values of each pixel. Subsequently, we employed OpenCV K-means clustering algorithm to 2D image array with 10 channels, yielding two crucial outcomes, “centers” storing the coordinates of 20 centroids representing the color of each centroid, and “labels” storing the labels for each pixel indicating which centroid it belongs to. By assigning colors from the “centers” array to each pixel in the image based on their cluster labels stored in the “labels” array, segmented images were created. To better visualize the K-means clustering results, we set all the cluster centers to black as initial values and plot each color channel separately. To identify the cluster corresponding to the target region, such as nuclei, we performed a color mapping. Users first need to input the color of target region in RGB format based on the prior knowledge, for example:
For each cluster derived from K-means segmentation, we calculate its average color value as:
Next we calculate the Euclidean distance of each centroid color to \({c}_{{target}}\):
for each \(i\in \{1,\,2,\ldots,\,10\}\). Then we look for the cluster with the minimum Euclidean distance:
where \({i}^{*}\) represents the index of the cluster that is closest in color to \({c}_{{target}}\).
For example, in immune cell nuclei detection, we can use (50, 0, 100) as they appear dark blue/purple on H&E image, while for goblet, we used (240, 230, 230) as they look pale and clear. It is possible for a channel contains similar-colored noisy elements. We then incorporated a closing morphological operation using “morphologyEx” in OpenCV on the segmented results as a refinement step. This operation involves a combination of dilation and erosion processes as below:
where \(\oplus\) and \(\ominus\) denote the dilation and erosion, respectively.
This step aims to eliminates noise points, while concurrently dilate the nucleus. Subsequently, the nucleus is returned to its normal size through the erosion process. For certain process such as filtering our goblet cells, we have one more step applying filters to refine the result which helps us to keep specific cells only. For instance, with filters such as an element’s area >40, solidity >0.5, and a length-to-width ratio <3, we were able to distinguish goblet cells from other irrelevant elements. Notably, these parameter values are not fixed while they are adaptable based on cell types and samples. We employed a similar segmentation process for immune cells and fibroblasts, with the key distinction being the use of different filters and parameter values tailored to each cell type.
Combined results from gene expression and image
In Module 1, focusing on goblet cell detection, we initially conducted separate analyses using gene expression and image. Because the marker genes for goblet cells are highly specific, and their morphology is distinctively different from other cell types, detections from each modality more prone to false negatives due to data quality issues rather than false positives. Therefore, the final detection results for goblet cells are combined as a union of detections from both gene expression and image analysis.
In Module 2, emphasizing on cancer cells, tumor regions are delineated through the combined analysis of gene expression and image. To distinguish tumor subtypes, including proliferative cancer cells, stem-like cancer cells, and other subtypes, we employed specific tumor markers such as MKI67, SOX9, CLDN18, and MSLN to identify the regions of tumor subtypes. Additionally, we applied distinct color overlays to these tumor subtype regions to visually represent the co-localization or exclusivity different cancer cells, enabling better understanding of tumor heterogeneity within the TME.
In Modules 3 and 4, which focuses on immune cells, we initially identify lymphocyte cells based on their morphological features, characterized by dark-purple nuclei in the H&E images. Differentiating among various immune cell types like B cells, CD4+ T cells, and CD8+ T cells based on morphology alone is challenging. Therefore, these distinct immune cell types are subsequently identified by examining their gene expression profiles within the lymphocyte areas delineated from the image. Afterward, T cell subtypes such as Treg and Tex cells can be identified using their specific marker genes within the areas where T cells have already been pinpointed. This hierarchical detection strategy, grounded in biological knowledge, ensures precise classification of immune cells,
In Module 5, our primary focus is on CAFs. Initially, we identify fibroblast cells through both imaging and gene expression analysis. Subsequently, we stratify CAF subtypes, including myofibroblastic CAFs (myCAFs), inflammatory CAFs (iCAFs), and antigen-presenting CAFs (apCAFs), based on the expression of specific cell markers. To further elucidate the interactions within the TME, we apply distinct color overlays to these CAF subtype regions, demonstrating the co-localization of CAF subtypes within the TME.
3D cell density visualization
Utilizing the nuclei detected from the previous step, we further performed a strong erosion to make the overlapped nuclei separable, follow by objective detection using function “findContours” in OpenCV. Then we computed the density of nuclei within a 20 × 20-pixel patch by using the spatial coordinates of each spot. These nuclei counts were served as the Z-coordinate values to construct our density plot. We generated a 3D surface map based on the complete set of Z-values.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
A detailed description of lung cancer and gastric Visium datasets including data sources and accession numbers RE described in our previous studies40,43. The lung cancer (LC_1) dataset54 is available under restricted access and can be downloaded from EGA under accession number EGAS00001005021. The gastric cancer and bladder cancer raw sequencing and imaging data have been uploaded to GEO. We have released all newly generated data on GEO under the accession code GSE246011. We confirm that all data analyzed in this manuscript are accessible via the links included in the Supplemental Table S1.
Code availability
All original code has been deposited at GitHub (https://github.com/Flashiness/METI) and is publicly available as of the date of publication. We have obtained a DOI for our Github repository by linking it to Zenodo. The Zenodo DOI is: https://doi.org/10.5281/zenodo.11247565 [55].
References
Larsson, L., Frisen, J. & Lundeberg, J. Spatially resolved transcriptomics adds a new dimension to genomics. Nat. Methods 18, 15–18 (2021).
Walker, B. L. et al. Deciphering tissue structure and function using spatial transcriptomics. Commun. Biol. 5, 220 (2022).
Baccin, C. et al. Combined single-cell and spatial transcriptomics reveal the molecular, cellular and spatial bone marrow niche organization. Nat. Cell Biol. 22, 38–48 (2020).
Hwang, W. L. et al. Single-nucleus and spatial transcriptome profiling of pancreatic cancer identifies multicellular dynamics associated with neoadjuvant treatment. Nat. Genet 54, 1178–1191 (2022).
Marx, V. Method of the year: spatially resolved transcriptomics. Nat. methods 18, 9–14 (2021).
Merritt, C. R. et al. Multiplex digital spatial profiling of proteins and RNA in fixed tissue. Nat. Biotechnol. 38, 586–599 (2020).
Rodriques, S. G. et al. Slide-seq: a scalable technology for measuring genome-wide expression at high spatial resolution. Science 363, 1463–1467 (2019).
Chen, K. H. et al. RNA imaging. Spatially resolved, highly multiplexed RNA profiling in single cells. Science 348, aaa6090 (2015).
Lubeck, E. et al. Single-cell in situ RNA profiling by sequential hybridization. Nat. Methods 11, 360–361 (2014).
He, S. et al. High-plex imaging of RNA and proteins at subcellular resolution in fixed tissue by spatial molecular imaging. Nat Biotechnol. 40, 1794–1806 (2022).
Bao, F. et al. Integrative spatial analysis of cell morphologies and transcriptional states with MUSE. Nat. Biotechnol. 40, 1200–1209 (2022).
Zhao, E. et al. Spatial transcriptomics at subspot resolution with BayesSpace. Nat. Biotechnol. 39, 1375–1384 (2021).
Hu, J. et al. SpaGCN: integrating gene expression, spatial location and histology to identify spatial domains and spatially variable genes by graph convolutional network. Nat. Methods 18, 1342–1351 (2021).
Hu, J. et al. Deciphering tumor ecosystems at super resolution from spatial transcriptomics with TESLA. Cell Syst. 14, 404 (2023).
Cable, D. M. et al. Robust decomposition of cell type mixtures in spatial transcriptomics. Nat. Biotechnol. 40, 517–526 (2022).
Vahid, M. R. et al. High-resolution alignment of single-cell and spatial transcriptomes with CytoSPACE. Nat. Biotechnol. 41, 1543–1548 (2023).
Correa, P., Piazuelo, M. B. & Wilson, K. T. Pathology of gastric intestinal metaplasia: clinical implications. Am. J. Gastroenterol. 105, 493 (2010).
Leung, W. & Sung, J. Intestinal metaplasia and gastric carcinogenesis. Aliment. Pharmacol. Ther. 16, 1209–1216 (2002).
Jencks, D. S. et al. Overview of current concepts in gastric intestinal metaplasia and gastric cancer. Gastroenterol. Hepatol. 14, 92–101 (2018).
Saw, P. E., Chen, J. & Song, E. Targeting CAFs to overcome anticancer therapeutic resistance. Trends Cancer 8, 527–555 (2022).
Asif, P. J. et al. The role of cancer-associated fibroblasts in cancer invasion and metastasis. Cancers 13, 4720 (2021).
Feng, B. et al. Cancer-associated fibroblasts and resistance to anticancer therapies: status, mechanisms, and countermeasures. Cancer Cell Int. 22, 166 (2022).
Ohlund, D. et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J. Exp. Med. 214, 579–596 (2017).
Elyada, E. et al. Cross-species single-cell analysis of pancreatic ductal adenocarcinoma reveals antigen-presenting cancer-associated fibroblasts. Cancer Discov. 9, 1102–1123 (2019).
Hu, B. et al. Subpopulations of cancer-associated fibroblasts link the prognosis and metabolic features of pancreatic ductal adenocarcinoma. Ann. Transl. Med 10, 262 (2022).
Sahai, E. et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nat. Rev. Cancer 20, 174–186 (2020).
Geng, X. et al. Cancer-associated fibroblast (CAF) heterogeneity and targeting therapy of CAFs in pancreatic cancer. Front Cell Dev. Biol. 9, 655152 (2021).
Jass, J. R. Role of intestinal metaplasia in the histogenesis of gastric carcinoma. J. Clin. Pathol. 33, 801–810 (1980).
Park, Y. H. & Kim, N. Review of atrophic gastritis and intestinal metaplasia as a premalignant lesion of gastric cancer. J. Cancer Prev. 20, 25 (2015).
Tatematsu, M., Tsukamoto, T. & Inada, K. Stem cells and gastric cancer: role of gastric and intestinal mixed intestinal metaplasia. Cancer Sci. 94, 135–141 (2003).
Gipson, I. K. Goblet cells of the conjunctiva: a review of recent findings. Prog. Retin Eye Res. 54, 49–63 (2016).
Van Landeghem, L. et al. Activation of two distinct Sox9-EGFP-expressing intestinal stem cell populations during crypt regeneration after irradiation. Am. J. Physiol. Gastrointest. Liver Physiol. 302, G1111–G1132 (2012).
Koulis, A. et al. CD10 and Das1: a biomarker study using immunohistochemistry to subtype gastric intestinal metaplasia. BMC Gastroenterol. 22, 197 (2022).
Hopkins, E. G. et al. Intestinal epithelial cells and the microbiome undergo swift reprogramming at the inception of colonic Citrobacter rodentium infection. mBio 10, e00062-19 (2019).
Wang, J. et al. Differential gene expression in normal esophagus and Barrett’s esophagus. J. Gastroenterol. 44, 897–911 (2009).
Takan, I. et al. “In the light of evolution:” keratins as exceptional tumor biomarkers. PeerJ 11, e15099 (2023).
Aguilar-Medina, M. et al., SOX9 stem-cell factor: clinical and functional relevance in cancer. J. Oncol. 2019, 6754040 (2019).
Cao, W. et al. Claudin18. 2 is a novel molecular biomarker for tumor-targeted immunotherapy. Biomark. Res. 10, 1–21 (2022).
Lv, J. & Li, P. Mesothelin as a biomarker for targeted therapy. Biomark. Res. 7, 1–18 (2019).
Chu, Y. et al. Pan-cancer T cell atlas links a cellular stress response state to immunotherapy resistance. Nat. Med. 29, 1550–1562 (2023).
Ostroumov, D. et al. CD4 and CD8 T lymphocyte interplay in controlling tumor growth. Cell. Mol. life Sci. 75, 689–713 (2018).
Echarti, A. et al. CD8+ and regulatory T cells differentiate tumor immune phenotypes and predict survival in locally advanced head and neck cancer. Cancers 11, 1398 (2019).
Hao, D. et al. The single-cell immunogenomic landscape of B and plasma cells in early-stage lung adenocarcinoma. Cancer Discov. 12, 2626–2645 (2022).
Xue, R. et al. Liver tumour immune microenvironment subtypes and neutrophil heterogeneity. Nature 612, 141–147 (2022).
Cheng, S. et al. A pan-cancer single-cell transcriptional atlas of tumor infiltrating myeloid cells. Cell 184, 792–809.e23 (2021).
Glabman, R. A., Choyke, P. L. & Sato, N. Cancer-associated fibroblasts: tumorigenicity and targeting for cancer therapy. Cancers 14, 3906 (2022).
Gascard, P. & Tlsty, T. D. Carcinoma-associated fibroblasts: orchestrating the composition of malignancy. Genes Dev. 30, 1002–1019 (2016).
Mhaidly, R. & Mechta-Grigoriou F. Fibroblast heterogeneity in tumor micro-environment: role in immunosuppression and new therapies. Semin. Immunol. 48, 101417 (2020).
LeBleu, V. S. & Kalluri, R. A peek into cancer-associated fibroblasts: origins, functions and translational impact. Dis. Models Mech. 11, dmm029447 (2018).
Mao, X. et al. Crosstalk between cancer-associated fibroblasts and immune cells in the tumor microenvironment: new findings and future perspectives. Mol. Cancer 20, 1–30 (2021).
Liu, T. et al. Cancer-associated fibroblasts: an emerging target of anti-cancer immunotherapy. J. Hematol. Oncol. 12, 1–15 (2019).
Zheng, L. et al. Pan-cancer single-cell landscape of tumor-infiltrating T cells. Science 374, abe6474 (2021).
Galletti, G. et al. Two subsets of stem-like CD8+ memory T cell progenitors with distinct fate commitments in humans. Nat. Immunol. 21, 1552–1562 (2020).
Han, G. et al. An atlas of epithelial cell states and plasticity in lung adenocarcinoma. Nature 627, 656–663 (2024).
Acknowledgements
This study was supported in part by the National Institutes of Health/NCI grants R01CA266280 (L.W.), U01CA264583 (H.K. and L.W.), U24CA274274 (A.M. and L.W.), the start-up research fund and the University of Texas MD Anderson Cancer Center (MDACC) Institutional Research Grant (IRG) Program (L.W.), and The Break Through Cancer Foundation (L.W., A.A.J., and A.M.), as well as the Cancer Prevention and Research Institute of Texas award RP220101 (H.K. and L.W.). L.W. and H.K. are Andrew Sabin Family Foundation Fellows at MDACC. A.S. is supported by an NCI T32CA217789 MDACC postdoctoral fellowship. This study was also supported by the NCI P30 Core Grant CA016672. In addition, A.M. acknowledges support from the Sheikh Khlaifa bin Zayed Foundation and the MDACC Moon Shots Program in Pancreas Cancer. J.A. acknowledges support from the Stupid Strong Foundation.
Author information
Authors and Affiliations
Contributions
L.W. conceived the study and designed the experiments. L.W. and J.H. jointly supervised the study. J.J.Q., J.W., J.C., M.P.P., X.C., A.S., Z.X., H.K., K.Y., J.A., and J.G. contributed to sample and information collection and data generation. R.L., J.W., A.L., reviewed the histology images and performed pathology annotation. L.W. supervised the bioinformatics data processing and analysis. J.J. performed bioinformatics data analysis and developed METI. Y.L., Y.C., F.P., G.H., G.P., K.S.C, X.Y., R.W., E.D., and Y.D. assisted with data processing and analysis. L.W., J.H., J.J., H.K., J.A., A.A.J., B.C., A.F., A.M., and J.G. contributed to data interpretation. J.J., J.H., and L.W. wrote and revised the manuscript and all co-authors reviewed the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous, reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Jiang, J., Liu, Y., Qin, J. et al. METI: deep profiling of tumor ecosystems by integrating cell morphology and spatial transcriptomics. Nat Commun 15, 7312 (2024). https://doi.org/10.1038/s41467-024-51708-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-51708-9
- Springer Nature Limited