

Abstract
The COVID-19 pandemic has led to the deaths of millions of people and severe global economic impacts. Small molecule therapeutics have played an important role in the fight against SARS-CoV-2, the virus responsible for COVID-19, but their efficacy has been limited in scope and availability, with many people unable to access their benefits, and better options are needed. EDP-235 is specifically designed to inhibit the SARS-CoV-2 3CLpro, with potent nanomolar activity against all SARS-CoV-2 variants to date, as well as clinically relevant human and zoonotic coronaviruses. EDP-235 maintains potency against variants bearing mutations associated with nirmatrelvir resistance. Additionally, EDP-235 demonstrates a ≥ 500-fold selectivity index against multiple host proteases. In a male Syrian hamster model of COVID-19, EDP-235 suppresses SARS-CoV-2 replication and viral-induced hamster lung pathology. In a female ferret model, EDP-235 inhibits production of SARS-CoV-2 infectious virus and RNA at multiple anatomical sites. Furthermore, SARS-CoV-2 contact transmission does not occur when naïve ferrets are co-housed with infected, EDP-235-treated ferrets. Collectively, these results demonstrate that EDP-235 is a broad-spectrum coronavirus inhibitor with efficacy in animal models of primary infection and transmission.
Similar content being viewed by others
Introduction
The global emergence of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) and its associated respiratory disease, coronavirus disease 2019 (COVID-19), has resulted in > 770 million reported cases and approximately 7 million deaths recorded to date worldwide1. The COVID-19 health crisis has led to the development of a diverse arsenal of therapeutics2 that have been critical in mitigating severe clinical outcomes3,4,5,6,7. Approved small molecule therapeutics directly targeting SARS-CoV-2 include the nucleoside inhibitors remdesivir and molnupiravir, and the protease inhibitors nirmatrelvir and ensitrelvir (the latter is only approved for use in Japan as of this writing). While these therapies have been crucial life-saving tools, there remains a strong need for new therapies with improved dosing regimens, efficacies, and patient eligibility.
Upon viral entry into a host cell, the positive-sense RNA genome of SARS-CoV-2 is translated by cellular ribosomes to produce two polyproteins, pp1a and pp1ab. Subsequently, two viral proteinases, papain-like cysteine protease and 3C-like protease (3CLpro), cleave pp1a and pp1ab into 16 non-structural proteins whereby the enzymatic activity of the 3CLpro is driven by a catalytic dyad (C145-H41)8. The conservation of structure and mechanism of action across coronaviruses as well as the indispensable nature of the 3CLpro for the virus’s life cycle make it an excellent therapeutic target9,10. Small molecule inhibitors targeting viral proteases have historically proven successful for the treatment of human immunodeficiency virus and hepatitis C virus11,12. Prior to the emergence of SARS-CoV-2, there was ongoing development of inhibitors against the 3CLpro of severe acute respiratory syndrome coronavirus (SARS-CoV)8,9. The sequence, structural identity, and mechanism of hydrolysis of the 3CLpro is highly conserved in SARS-CoV-2 as compared to SARS-CoV8,13,14,15, thereby providing a strong rationale for the development of SARS-CoV-2 3CLpro inhibitors.
To date, the FDA has approved the use of one anti-SARS-CoV-2 agent that inhibits the 3CLpro of SARS-CoV-2, ritonavir-boosted nirmatrelvir (Paxlovid)16,17. Ritonavir, an inhibitor of cytochrome P450 3A4, is co-administered with nirmatrelvir to increase nirmatrelvir’s pharmacokinetic half-life17,18,19. The EPIC-HR trial demonstrated that Paxlovid is typically well-tolerated in SARS-CoV-2 infected individuals20. However, there are adverse reactions and contraindications that may arise with the use of ritonavir, which has limited the approved populations for which this therapeutic is recommended19,21.
The development of antiviral resistance is a concern, particularly for RNA viruses22. Unfortunately, the continued emergence of SARS-CoV-2 variants increases the possibility of rendering current treatment measures less efficacious23,24,25,26,27,28. To reduce the medical impact of viral evolution and escape from immune and antiviral countermeasures, it is critical to continue developing novel therapeutics to combat SARS-CoV-2. To date, widespread transmission of nirmatrelvir-resistant variants has not been reported. However, it is possible that drug selective pressure could drive the development of clinically meaningful resistance. Indeed, several in vitro studies have determined that nirmatrelvir can select for variants bearing mutations in 3CLpro which may impact its efficacy27,29,30,31,32. A recent case report determined that two of these in-vitro-generated mutations, E166V and L50F, were isolated from an immunocompromised individual infected with SARS-CoV-2 and treated with Paxlovid28.
Climate change and continued human encroachment on wildlife natural habitats will continue to drive zoonotic spillover events33,34, and the possibility of a future viral zoonotic event stemming from the coronavirus subfamily Orthocoronavirinae within the Coronaviridae family should not be ignored. To date, nine coronaviruses have been identified that can infect and cause illness in humans: human coronavirus (HCoV) 229E, HCoV-NL63, canine coronavirus (Alphacoronaviruses), porcine deltacoronavirus (Deltacoronavirus), HCoV-OC43, HCoV-HKU1, Middle East respiratory syndrome coronavirus (MERS-CoV), SARS-CoV, and SARS-CoV-2 (Betacoronaviruses)35,36,37. Within the past two decades, three of these zoonotic spillover events have caused global epidemics or pandemics with the emergence of SARS-CoV in 2002 and MERS-CoV in 2012. It is likely that the 2019 coronavirus zoonotic spillover of SARS-CoV-2 into humans will not be the last.
EDP-235 is a 3CLpro inhibitor that is under development for the treatment of COVID-19 as a once-a-day oral medication without the need for ritonavir boosting38. Herein, we report the pre-clinical characterization of EDP-235, highlighting its pan-coronavirus activity, including efficacy against other zoonotic coronaviruses that could potentially cause future spillover events. We demonstrate EDP-235’s ability to inhibit SARS-CoV-2 3CLpro variants bearing mutations associated with nirmatrelvir resistance with equal or improved potency relative to nirmatrelvir. Furthermore, we establish that EDP-235 is highly efficacious in both Syrian hamsters and ferrets. Finally, therapeutically dosed EDP-235 is shown to prevent viral transmission among cohoused, untreated ferrets, underscoring the potential of EDP-235 to possibly prevent household transmission of SARS-CoV-2.
Results
EDP-235 reversibly binds and inhibits the 3CLpro of SARS-CoV-2
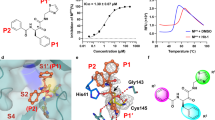
A fluorescence resonance energy transfer (FRET) assay using the recombinant ancestral SARS-CoV-2 (USA-WA1/2020) 3CLpro and a synthetic substrate that mimics the natural substrate of the 3CLpro was used to screen and identify di- and tri-peptidomimetic inhibitors of the SARS-CoV-2 3CLpro39,40. This led to the discovery of the nitrile-based peptidomimetic inhibitor EDP-235 (Fig. 1a inset). Kinetic studies were performed to evaluate the mechanism of inhibition of SARS-CoV-2 3CLpro. EDP-235 is a time-dependent, tight-binding, reversible covalent, substrate competitive inhibitor with a Kiapp of 3.0 nM and inhibits SARS-CoV-2 3CLpro with a half-maximal inhibitory concentration (IC50) of 4.0 nM (Fig. 1a and Supplementary Figs. 1–5, and Supplementary Table 1). To further confirm the mode of inhibition, a crystal structure of EDP-235 in complex with SARS-CoV-2 3CLpro was generated, from data collected to 2 Å (Fig. 1b, c and Supplementary Table 2). Under these conditions, EDP-235 is bound to the proteolytic active site of 3CLpro (Fig. 1b, c), where the sulfhydryl group of the catalytic residue C145 reacts with the nitrile group of EDP-235 to form a covalent thioimidate linkage. The spirolactam moiety of EDP-235 forms hydrogen bonds with several amino acids in the active site, including a potential interaction with the backbone amide of F140, the side chain of H163, and the side chain of E166. An H-π interaction is observed between leucine P2 of EDP-235 and H41. Additionally, the sidechain of Q189 may stabilize EDP-235 through weak hydrogen bonding with the trifluoroindole moiety of EDP-235. Other interactions include a pair of hydrogen bonds between the trifluoroindole moiety of EDP-235 and E166 and a possible F-amide n-π* interaction with P168. These observations are consistent with the kinetic studies and provide a structural rationale for the competitive mode of inhibition.

a Structure of EDP-235 and its FRET 3CLpro inhibition activity. Representative data are mean ± standard deviation of technical replicates from one experiment with non-linear regression fit (n = 6 independent experiments used to derive IC50 and Kiapp values). b Semi-transparent surface representation of the X-ray crystal structure of SARS-CoV-2 3CLpro dimer (shown in cyan and gray) in complex with EDP-235 solved to 2 Å. EPD-235 is represented by red stick structure. c Detailed cartoon representation of the binding interactions between EDP-235 and residues located in SARS-CoV-2 3 CLpro catalytic active site. Hydrogen bonds are depicted between the NH side chain of H136 with the spyrolactam C = O of EDP-235, the C = O side chain of E166 with the spyrolactam NH of EDP-235, the NH side chain of H41 with amide C = O of EDP-235, and the NH side chain of Q189 with the trifluoroindole F of EDP-235. The catalytic residue C145 is represented as a yellow stick configuration and monomer coloration is matched to (b, d). The surface map of 3CLpro and electron density of EDP-235 was rendered using UCSF Chimera. d A close-up semi-transparent surface representation of EDP-235 in the binding pocket. 3CLpro = 3C-like protease; FRET = fluorescence resonance energy transfer; IC50 = half-maximal inhibitory concentration; Kiapp = apparent inhibition constant; SARS-CoV-2 = severe acute respiratory syndrome coronavirus 2; UCSF = University of California San Franscisco.
EDP-235 has potent antiviral activity against SARS-CoV-2
We assessed the ability of EDP-235 to inhibit the replication of SARS-CoV-2 utilizing different cell systems and readouts described in Table 1. In a SARS-CoV-2 replicon system41,42, EDP-235 displayed a 4.5 nM half-maximal effective concentration (EC50). These results were recapitulated in infectious virus assays using cytopathic effect (CPE) and viral yield reduction (VYR) endpoints using various derivatives of Vero cells. Unlike human airway epithelial cells, monkey kidney-derived Vero E6 cells express high levels of the efflux transporter P-glycoprotein 1 (PGP)43. Given the possibility of 3CLpro inhibitors to serve as PGP substrates, antiviral assays utilizing Vero E6 cells were performed in the presence and absence of the PGP inhibitor (PGPi) CP-10035644. In Vero E6 cells infected with ancestral (A: SARS-CoV-2 USA-WA1/2020), B (Germany/BavPat1/2020), Delta (B.1.617.2), or Omicron (B.1.1.529) strains, EDP-235 inhibited CPE and virion production with EC50S ranging from 11–22 nM and 3–7.4 nM, in the absence and presence of PGPi, respectively. Using a VYR assay endpoint in the more permissive Vero E6-TMPRSS2 cells, EDP-235 inhibited multiple SARS-CoV-2 Omicron variants of interest with EC50s ranging from 10 to 73 nM (Table 1). Viral inhibition curves for these data are included in Supplementary Fig. 6. Comparable results were obtained for FRET biochemical assays with IC50s ranging from 1.9 to 5.7 nM for all 3CLpro variants assessed (Supplementary Table 3 and Supplementary Fig. 7).
Since SARS-CoV-2 predominately replicates in lung epithelial cells, we evaluated the potency of EDP-235 in a more relevant cellular architecture. We utilized primary human airway epithelial cells maintained at an air-liquid interface (pHAEC-ALI). EDP-235 inhibited viral replication, as quantified by 50% tissue culture infectious dose (TCID50), of SARS-CoV-2 lineage B with EC50/90 values of 29/33 nM, respectively. Near-identical potency values were derived when reverse-transcriptase-quantitative polymerase chain reaction (RT-qPCR) was used to monitor viral replication (Table 1).
Across all strains and readouts, EDP-235’s in vitro potencies were equivalent to or improved upon those observed for nirmatrelvir. No cytotoxicity was detected in Vero E6, HuH-7, Madin-Darby canine kidney, A549-ACE2, MRC5, or HeLa cells exposed to EDP-235 for 72 hours (h) at concentrations up to 50,000 nM (Supplementary Fig. 8). EDP-235 has weak inhibition (IC50 ≥ 2 µM) of mammalian cysteine proteases and an excellent selectivity index (SI) ≥ 500-fold compared to its inhibition of SARS-CoV-2 3CLpro (Supplementary Table 4). EDP-235 did not inhibit any other host protease families at tested concentrations (IC50 > 100 µM).
EDP-235 maintains activity against known SARS-CoV-2 nirmatrelvir-resistant variants
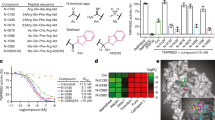
Since the development of SARS-CoV-2 antiviral resistance is a possibility, the activity of EDP-235 was assessed against 24 single-residue and 17 multi-residue 3CLpro variants associated with nirmatrelvir resistance. The most well-known and clinically documented of these were the 3CLpro L50F and E166V point mutations27,28,29,30. The potency of EDP-235 against these 3CLpro variants ranged from an IC50 of 3-184 nM, with nirmatrelvir subject to greater loss of potency (IC50 4 - >10,000 nM) (Table 2, Supplementary Fig. 7, and Supplementary Table 5 for expanded variant testing). EDP-235 showed a > 5-fold decrease in potency for 21 variants, with nirmatrelvir suffering even greater potency reductions against these variants. However, each of these variants had 7-125-fold reductions in enzymatic efficiency relative to ancestral 3CLpro, indicating a detrimental impact on enzymatic activity, and consequentially a possible decrease in both viral fitness and potential to replicate to levels associated with infection and transmission.
Circulating SARS-CoV-2 variants bearing mutations in 3CLpro within the EDP-235 binding site could potentially lead to emergence of clinical drug resistance. Analysis of the SARS-CoV-2 3CLpro co-crystal with EDP-235 revealed 22 residues within 5 Å of EDP-235. To identify circulating SARS-CoV-2 variants with mutations at these 22 sites, we conducted a search of the GISAID database for variants that had a prevalence ≥ 1% of sequencing reads. We identified 15 variants as of January 2022. When these mutations were engineered into SARS-CoV-2 3CLpro, we did not observe any EDP-235 resistance in the biochemical FRET assay. IC50s for these variants ranged from 2.2 to 13 nM with 4 variants having no detectable enzymatic activity (Supplementary Table 6 and Supplementary Fig. 7). Some inactivating mutations such as C145F (active site mutation) could have arisen due to sequencing errors for very low frequency variants or other inaccuracies in the GISAID database.
EDP-235 is a pan-coronavirus inhibitor
Beyond the current threat of SARS-CoV-2, several other coronaviruses are known to infect humans. We therefore evaluated the prospect of EDP-235 to serve as a pan-coronavirus inhibitor. EDP-235 maintained nanomolar to sub-nanomolar potency against all coronaviruses tested in antiviral and biochemical assays (Table 3). The 3CLpro amino acid sequences from the Toronto-2 and HKU-39849 strains of SARS-CoV are 100% conserved, and EDP-235 inhibited the biochemical activity of this protease with an IC50 of 1.9 nM. As expected, EDP-235 limited replication of both SARS-CoV clinical isolates with EC50 values ranging from < 0.5 to 24 nM. In addition, EDP-235 potently inhibited the replication of HCoV-229E ( < 10 nM EC50s in multiple assay formats), HCoV-OC43 (57 nM EC50), HCoV-HKU1 (3.8 nM IC50), and HCoV-NL63 (6.1 nM EC50). These data are summarized in Table 3 and Supplementary Fig. 9.
EDP-235 was also tested against a broad panel of other human RNA viruses that encode proteases using infectious cell culture assays, however no activity was observed beyond the coronaviruses.
Zoonotic CoVs are inhibited by EDP-235
As a zoonotic spillover event remains a constant threat, we sought to determine if EDP-235 could inhibit zoonotic CoVs. Viral species were selected based on percent sequence identity of the receptor binding domain (RBD) compared to that of SARS-CoV-245, on the assumption that a high RBD sequence identity is an important factor which may portend an increased likelihood of a spillover event. Three bat and two pangolin CoV species, as well as the porcine epidemic diarrhea virus (PEDV) (included as an outlier species based on lack of RBD sequence conservation) were tested in a FRET assay. EDP-235 exhibited an IC50 < 3 nM against all tested zoonotic 3CLpros (Table 4, and Supplementary Fig. 10). We also explored EDP-235’s ability to block viral replication of infectious zoonotic CoVs recombinantly engineered to express NanoLuc luciferase46,47,48. These viruses were previously isolated from pangolin and bat species and have a high degree of similarity to SARS-CoV, MERS, and SARS-CoV-2. In line with the biochemical assays, EDP-235 maintained activity against each of the infectious zoonotic viruses (EC50 6.9–21 nM) (Table 4).
EDP-235 is efficacious in a SARS-CoV-2 hamster model
Syrian hamsters can recapitulate many features of COVID-1949,50. Therefore, we evaluated the in vivo efficacy of EDP-235 using a Syrian hamster model (Fig. 2). Hamsters were grouped into four cohorts of either naïve dosed with vehicle (0.5% methylcellulose) or infected dosed with vehicle, EDP-235 at 200 mg/kg twice daily (BID), or EDP-235 at 500 mg/kg BID. Hamsters were intranasally infected with 6 × 103 TCID50 of SARS-CoV-2 (USA/WA1/2020) and compound dosing was oral. Prophylactic oral dosing commenced 1 h pre-challenge and lasted for 3.5 days, with sacrifice 4 days post-infection (dpi). While infected vehicle-treated animals exhibited rapid weight loss, EDP-235 prevented SARS-CoV-2-induced weight loss (Fig. 2b). Further evaluation with immunohistochemistry (IHC) staining of the lungs for viral nucleoprotein (N) revealed a remarkably reduced amount of staining in EDP-235-treated animals as compared to infected, vehicle-treated animals (Fig. 2c, d). Hematoxylin and eosin (H&E) lung staining revealed the majority of the lung tissue from infected vehicle-treated animals was affected by SARS-CoV-2 (abundant dark consolidations, Fig. 2e arrows), while EDP-235-treated animals did not have such findings (Fig. 2e). Cumulative histopathology scoring of alveolar hemorrhage, alveolar inflammation, interstitial inflammation, vascular inflammation, bronchial-alveolar hyperplasia, perivascular edema, and immunoreactivity showed significant decreases in EDP-235-treated animals relative to infected vehicle-treated animals (Fig. 2f). Lung viral loads displayed significant differences between EDP-235-treated and untreated animals at study endpoint, with the former demonstrating multi log10 reductions in live virus, genomic viral RNA (gRNA), and sub-genomic viral RNA (sgRNA) (Fig. 2g–i).

a Study design. Hamsters were orally dosed with EDP-235 or vehicle 1 h prior to intranasal inoculation with 6\(\times\)103 TCID50/animal SARS-CoV-2 USA/WA1/2020 or PBS for naïve. b Percent body weight change from 0-day baseline (n = 8 per cohort). c IHC staining of terminal lung samples for SARS-CoV-2 N protein (brown) (2X magnification). Minimal focal immunoreactivity seen in 200 mg/kg animal (arrow). Representative images shown (n = 8 per cohort). d Sections of panel C were scored from 1 (minimal/minor) to 5 (severe/overwhelming) (n = 8 per cohort). e H&E stain of terminal lung samples (1.25X magnification). SARS-CoV-2 effects characterized by dark consolidated lung regions (arrows). Representative images shown (n = 8 per cohort). f Left lung composite histopathology score. Microscopic finding categories were alveolar hemorrhage, alveolar/interstitial inflammation, vascular inflammation, bronchial-alveolar hyperplasia, perivascular edema, and immunoreactivity. Each variable scored from 1-5 as in (d) (n = 8 per cohort). g Infectious virus titer in terminal lung samples (n = 8 per cohort). h sgRNA (N protein) in terminal lung samples (n = 8 per cohort). i) gRNA (N protein) in terminal lung samples (n = 8 per cohort). All graphs are mean ± SEM; statistical significance determined versus infected VC by 2-way ANOVA with Tukey’s (b) or 1-way ANOVA with Dunnett’s (d, f, g, h, i) post hoc test. All P values were < 0.0001 unless indicated otherwise in the figure. ANOVA = analysis of variance; g/sgRNA = genomic/sub-genomic RNA; h = hour; H&E = hematoxylin and eosin; IHC = immunohistochemistry; LoD = Limit of Detection; N protein = nucleocapsid protein; PBS = phosphate buffered saline; sac. = sacrifice; SARS-CoV-2 = severe acute respiratory syndrome coronavirus 2; SEM = standard error of the mean; TCID50 = median tissue culture infectious dose; VC = vehicle control.
EDP-235 therapeutically blocks SARS-CoV-2 replication and transmission in ferrets
While Syrian hamsters recapitulate the pathology produced in human SARS-CoV-2 infection, they are not as well suited to evaluate viral transmission as ferrets. Ferrets replicate SARS-CoV-2 to high titers in their upper airways and have previously been employed in the study of SARS-CoV-2 transmission51,52. The ferret model was used to evaluate EDP-235’s ability to therapeutically halt viral replication and to prevent transmission from infected to naïve animals (Fig. 3). Prior to in vivo work, a 24 h oral single dose pharmacokinetic study was performed in ferrets, administering either 200 or 500 mg/kg. While no pharmacokinetic dose response was observed between the 200 and 500 mg/kg dosing groups (Supplementary Fig. 11), it was noted that plasma exposure levels were comparable to those seen in human phase 1 clinical studies. This was especially true when compared to the human 400 mg fed state, with the exception that EDP-235’s half-life in ferrets dosed at 200 mg/kg was ~2-3-fold reduced compared to that observed in humans (6.7 h versus 13-22 h geometric mean in humans)53. For these pharmacokinetic parameter reasons, 200 mg/kg ferret dosing was selected for the efficacy study, given either once per day (QD) or BID. Ferrets were inoculated intranasally with 1 × 105 plaque-forming units (PFU) of SARS-CoV-2 USA/WA1/2020. At 12 h post-infection (hpi), animals were dosed orally with either vehicle or EDP-235 for three days. At 60 hpi, uninfected and untreated naïve ferrets were co-housed 1:1 with infected ferrets and monitored for 5.5 days for viral transmission (Fig. 3a).

a Study design. Ferrets were infected with 1 × 105 PFU of SARS-CoV-2 USA/WA1/2020 intranasally. 12 hpi, animals were orally dosed with vehicle or EDP-235 QD or BID. 60 hpi uninfected and untreated ferrets were co-housed 1:1 with infected dosed animals. Originally infected animals were sacrificed 4 dpi while the contact ferrets were monitored for an additional 4 days. b Percent body weight change from baseline (0 days) (n = 6 per cohort). c Animal temperature (n = 6 per cohort) (d) Nasal lavage viral load determined by RT-qPCR (nsp9 gene) (n = 6 per cohort). e Nasal lavage infectious virus quantified by TCID50 (n = 6 per cohort). f Rectal swab viral load determined by RT-qPCR (nsp9 gene) (n = 6 per cohort). g Endpoint (day 4 or day 8) nasal turbinate viral load determined by RNA levels of nsp9 (n = 6 per cohort). h Infectious virus in endpoint nasal turbinates determined by TCID50 (n = 6 per cohort). All graphs are mean ± SEM. All P-values were < 0.0001 unless indicated otherwise in the figure. Statistics are for both QD and BID differences versus vehicle and were run using 2-way ANOVA with Tukey’s (b–f) or 1-way ANOVA with Dunnett’s (g, h) post hoc test. In (b–f), both QD and BID treatment resulted in the same statistical significance versus vehicle-treated animals in both primary challenged and contact animals except where noted in (e). ANOVA = analysis of variance; BID = twice daily; dpi = days post-infection; hpi = hours post-infection; LoD = limit of detection; nsp9 = non-structural protein 9; PFU = plaque-forming units; QD = once a day; RT-qPCR = reverse transcription quantitative polymerase chain reaction; sac. = sacrifice; SARS-CoV-2 = severe acute respiratory syndrome coronavirus 2; SEM = standard error of the mean; TCID50 = median tissue culture infectious dose; VC = vehicle control.
All ferrets were clinically scored daily as none/normal for lethargy, orbital tightening, diarrhea, respiratory distress, activity, nasal discharge, and neurological symptoms. Primary-infected animals maintained consistent weight and temperature (Fig. 3b, c). On each study day, rectal swab and nasal wash samples were obtained. Nasal turbinates were collected upon sacrifice, and viral replication was evaluated. Viral loads increased rapidly in primary infected vehicle-treated animals, peaking at 2–3 dpi (Fig. 3d–f). Across all readouts and dosing cohorts, EDP-235 rapidly reduced viral loads in initially infected ferrets (Fig. 3d–h), with significant virion reductions observed in the first samples obtained 12 h post-dosing (Fig. 3e). After 48 h of dosing, virion levels were below the LoD, representing a > 3 log10 drop relative to vehicle-treated animals.
Viral transmission occurred rapidly (within 12 h of co-housing), from vehicle-treated source animals to untreated contact animals, with peak viral loads observed 2.5 days post-co-housing. Both EDP-235 dosing regimens completely suppressed viral transmission, as ferrets co-housed with EDP-235-treated infected ferrets had undetectable viral loads (Fig. 3d–h). In contrast to primary-infected ferrets, contact ferrets co-housed with the infected vehicle-treated ferrets showed significant weight loss and increases in body temperature (Fig. 3b, c). This change was likely driven by a ferret-adaptive spike mutation in the RBD, N501T, which can emerge rapidly54, and was observed in sequenced virus collected from primary infected vehicle-treated ferrets on day 4. Shotgun metagenomic next-generation sequencing of day 4 viruses from EDP-235-treated animals was used to detect any potential resistance mutations but read-depths were insufficient to call any variants, likely due to the high efficacy of EDP-235.
Discussion
In this study, we present the in vitro and in vivo characterization of EDP-235, an oral antiviral, specifically designed to target the 3CLpro of SARS-CoV-2. While the real-world efficacy of targeting the 3CLpro has been established with nirmatrelvir, the need for additional therapeutics with a simpler dosing regimen, expanded patient eligibility through fewer drug-drug interactions, improved tolerability, enhanced potency, and reduced resistance liability would benefit those suffering from COVID-193,55. The in vitro data reported here demonstrate improvements by EDP-235 relative to nirmatrelvir regarding potency, resistance, and in preventing transmission in ferret studies. EDP-235 is currently in development with once-daily dosing without the use of any boosting agent such as ritonavir.
Across a range of orthogonal assays, EDP-235 demonstrated potent nanomolar efficacy against multiple SARS-CoV-2 lineages and variants. EDP-235 has a SI ≥ 500fold for the 3CLpro versus 30 human proteases and 50% cellular cytotoxicity concentration values of > 10 µM. These results indicate minimal off-target effects resulting in low toxicity. In the phase 2 SPRINT trial56, EDP-235 was generally safe and well tolerated with a low frequency of adverse events (1.3%, 6.4%, and 2.6% for 200 mg, 400 mg, and placebo arms respectively), with no serious adverse events or discontinuations observed, providing further evidence of the safety of EDP-235 in people.
The potent antiviral efficacy of EDP-235 extends beyond SARS-CoV-2 to other clinically relevant HCoVs. These endemic HCoVs are estimated to be responsible for 10–30% of all common cold infections57. Notably, EDP-235 also demonstrates activity against bat, pangolin, and pig zoonotic CoVs, effectively inhibiting the 3CLpros of these species as well as their ability to replicate in cell culture. While SARS-CoV-2 currently poses the greatest health threat to people, there remains a risk of zoonotic spillover events, especially given humanity’s widespread encroachment on natural animal habitats. EDP-235’s broad-spectrum pan-coronavirus activity, and thus utility against future emerging CoVs, highlights its significance in the context of pandemic preparedness. Additionally, the rare documented cases of SARS-CoV-2 cross-transmission occurring between humans and other mammals58,59 speaks to the ability of the virus to evolve undetected prior to re-infection in humans. These observations underscore the necessity of a broad-spectrum coronavirus inhibitor.
As SARS-CoV-2 mutates to better infect human cells and evade our adaptive immune system, real-world examples of emerging resistance present therapeutic challenges. Previously approved monoclonal antibody treatments have lost their effectiveness due to rapid and continual spike mutations24. Encouragingly, there has been limited development of resistance to the nucleoside analogs remdesivir and molnupiravir, as well as the 3CLpro inhibitor nirmatrelvir28,60,61. However, multiple in vitro studies have selected for 3CLpro mutations conferring resistance to nirmatrelvir27,29,30,31,32, so the possibility of clinical resistance remains.
In cross-resistance testing, EDP-235 matched or improved upon the antiviral potency of nirmatrelvir across all assessed variants. Variant EDP-235 resistance was inversely proportional to 3CLpro activity, with EDP-235 potency shifts > 5-fold suffering 7-125-fold reductions in enzymatic activity. These deficiencies may contribute to the lack of emergence of the most drug-resistant variants in patient populations, as they are likely fitness-impaired. Indeed, the 3CLpro’s lack of sequence-plasticity15 likely contributes to EDP-235’s broad activity across both human and zoonotic coronaviruses.
The translatability of the Syrian golden hamster model to human clinical benefit has been established with the 3CLpro inhibitors nirmatrelvir and ensitrelvir62,63. Pathophysiology in hamsters resembles the early pathology of SARS-CoV-2 in people, with histological changes, high viral loads achieved in the lungs, and associated clinical signs49,64. EDP-235 was protective against virus-induced weight loss, and significantly reduced viral burden and lung histopathology. Given the translatability of the hamster model, these results suggest that EDP-235 has the potential to not only reduce viral load but also improve clinical symptoms.
The ferret model’s utility as a predictor of clinical relevance for people has also been established with remdesivir, molnupiravir, and nirmatrelvir52,54,65. In ferrets, SARS-CoV-2 replicates to high titers primarily in the upper airways. While ferrets do not generally present with clinical signs, they do function well as a transmission model of disease51. Our ferret study was designed to evaluate the antiviral effect of EDP-235 when dosed therapeutically, as well as its ability to prevent SARS-CoV-2 contact transmission to treatment-naïve animals co-housed 48 h post-treatment of the infected animals. Indeed, not only did EDP-235 show potent antiviral effects in primary-infected ferrets, it also completely prevented transmission to treatment-naïve animals. The ability to prevent or reduce transmission has broad implications with regards to isolation of infected individuals and quarantine of household contacts.
Social isolation following infection with SARS-CoV-2 is disruptive to the lives of both the infected and their families66,67. This also carries broader economic and social burdens, from lost productivity with isolation to continued viral transmission enabled by a premature return to social settings. Both Pfizer’s phase 2/3 EPIC-PEP trial of Paxlovid and Merck’s MOVe-AHEAD phase 3 trial of Lagevrio explored post-exposure prophylactic treatment to prevent this transmission, but while positive trends were observed for both studies, neither was able to reach statistical significance over placebo68,69. Similar results were seen in a ferret model; nirmatrelvir administered with ritonavir was dosed to match human exposure levels. However, this failed to block transmission to untreated contact animals in a similar design to our own study, or when dosed prophylactically post-exposure52. Conversely, parallel testing of molnupiravir prevented such transmission in ferrets. While the methodologies of this molnupiravir ferret work differed from the Merck clinical trial design (infected-dosed ferrets prevented transmission to naïve versus patient prophylactic administration post-exposure did not), these results somewhat obscure the translatability of ferret transmission studies to human clinical benefit52,65. Despite this ambiguity, the ability of EDP-235 to prevent viral transmission between ferrets, while nirmatrelvir did not, suggests EDP-235 may offer a distinct advantage over Paxlovid in blocking transmission to close contacts. Having an antiviral agent which could enable not only a faster but also a safer return of infected individuals to social environments would be of great benefit.
In summary, EDP-235 is a potent oral inhibitor of SARS-CoV-2, with consistent potencies against all commonly circulating variants tested to date as well as against other human and non-human coronaviruses. EDP-235 is highly selective for the coronavirus 3CLpro active site and has no measurable cytotoxicity up to 10 µM. EDP-235 demonstrated an improved profile over nirmatrelvir in cross-resistance testing against known SARS-CoV-2 3CLpro variants found both naturally and through in vitro selection27,28. In animal testing, EDP-235 protected Syrian golden hamsters from viral-induced distress and histopathology, with multi-log10 reductions in viral titers and RNA levels. In the ferret model, therapeutic EDP-235 treatment rapidly decreased both viral titers and RNA below the LoD and prevented transmission to untreated co-housed ferrets. EDP-235’s potential as a treatment for COVID-19 should continue to be evaluated in human clinical trials.
Methods
Cell culture, viruses, and compounds
HuH-7 (JCRB0403), LLC-MK2 (CCL-7), HCT-8 (CCL-244), and MRC5 (CCL-171) cells were obtained from JCRB (HuH-7) or ATCC and cultured in Dulbecco’s Modified Eagle Medium (Corning), OPTI-MEM reduced serum media (Gibco), Roswell Park Memorial Institute 1640 medium (Gibco), or Eagle’s Minimum Essential Medium (Corning) respectively. All cell lines were supplemented with either 2% (LLC-MK2 cells) or 10% fetal bovine serum (Gibco) and 1X Anti/Anti or 1X penicillin and streptomycin (Gibco). HuH-7 cells were supplemented with 1X GlutaMAX (Gibco). All cell lines tested negative for mycoplasma contamination (MycoAlertTM Mycoplasma Detection Kit, Lonza). HCT-8 and MRC-5 cell lines were authenticated using STR profiling. HuH-7 cell line was authenticated using isozyme analysis. LLC-MK2 cell line was not authenticated by vendor.
HCoV-229E (University of Texas Medical Branch, and ATCC), was propagated in MRC-5 cells. HCoV-NL63 (BEI Resources) was amplified in LLC-MK2 cells. HCoV-OC43 (University of Texas Medical Branch) was grown in HCT-8 cells. HCoV-229E, HCoV-NL63 and HCoV-OC43 were propagated at an MOI of 0.1. When infected monolayers had 60–70% CPE, viruses were collected, pre-clarified, supplemented with cryoprotectant agents, and titered by TCID50 or plaque assays. Southern Research (SR) obtained and titered SARS-CoV-2 USA-WA1/2020, B.1.617.2, B.1.1.529 strains, and SARS-CoV Toronto 2. Viroclinics Biosciences (VCB) obtained and titered SARS-CoV-2 Germany/BavPat1/2020, B.1.351, B.1.1.7, SARS-CoV HKU-39849, and MERS-CoV EMC/2012 strains. Georgia State University (GSU) obtained and titered SARS-CoV-2 B.1.1.529, BA.2, BA.2.12.1, BA.4, BA.5.1, BQ.1.1, XBB.1.5, EG.5.1, EG.5.1.1, EG.5.2, and BA.2.86 strains. SR and VCB obtained Vero E6 cells from ATCC. GSU obtained Vero E6-TMPRSS2 cells from BBS Bioscience.
Unless otherwise specified, all cells, virus infections and cell-based assays were incubated at 37 °C in a humidified and 5% CO2 atmosphere.
All compounds were synthesized by Enanta Pharmaceuticals, Inc. EDP-235 and Nirmatrelvir had purity of 98.6% and 98.2%, respectively. Compounds were resuspended in dimethyl sulfoxide (DMSO) and added manually or with an automated liquid handler (ECHO 650 Beckman Coulter). CP-100356 was purchased from Axon Medchem with a purity of 98.7%. All compounds utilized in this study meet community criteria for chemical probes.
Biochemical assays
IC50s were determined using a previously established FRET assay40. Briefly, ancestral (USA-WA1/2020), variants described, or zoonotic SARS-CoV-2 3CLpro proteins were expressed and purified from E. coli (Shanghai ChemPartner Co.). Recombinant proteins were incubated at room temperature for 30 min with increasing concentrations of compound (EDP-235 or nirmatrelvir) in 50 mM HEPES pH 7.5, 5 mM NaCl, 1 mM EDTA, 0.1 mg/mL BSA, 1 mM DTT, and 0.01% volume per volume TritonX-100. The FRET substrate, Tamra-SITSAVLQSGFRKMK-Dabcyl-OH, (BACHEM Holding) was added 30 minutes post-incubation, and fluorescence was measured over 1 h at room temperature (Envision, PerkinElmer). Progress curves were used to determine the initial velocities with IC50s and hill coefficient values calculated as previously described in ref. 70. The final concentration of 3CLpro protein was 2.5-100 nM, and FRET substrate was 20–40 μM. Variations in concentrations were due to inherent differences in 3CLpro activity and FRET substrate affinity. For time-dependent inhibition experiments, progress curves obtained at various concentrations of EDP-235 against ancestral 3CLpro without preincubation were corrected for baseline by subtraction of the no enzyme progress curves. Individual baseline corrected progress curves were fitted to the time-dependent inhibition model using non-linear regression fitting in GraphPad Prism70. The reversibility of inhibition was evaluated through incubation with varying 3CLpro concentrations (0–50 μM) with 50 μM of EDP-235 for 2 h at RT, followed by jump-dilution with 20 μM FRET substrate as given above. The progress curves for activity recovery were then fitted to the time-dependent inhibition model. To determine the Kiapp of 3 nM, kinetic mechanism experiments were performed with FRET substrate concentrations ranging from 0 to 100 μM and the FRET substrate being added at time 0. Kiapp values were generated by fitting to substrate competitive inhibition with respect to the FRET peptide70.
X-ray crystallography and structure determination
X-ray crystallography work was performed at Evotec. Crystals were grown over two days in a 200 mM sodium thiocyanate, 20 percent weight per volume polyethylene glycol 3350 (MDL JCSG+ condition B2) with a drop ratio of 30:63:6 (protein:reservoir:seed suspension). Mass spectrometry was used to confirm the covalent attachment of EDP-235 and the 3CLpro. To collect diffraction data, the crystal was transferred to a drop of neat reservoir solution, to this was added 10% weight per volume polyethylene glycol 400 in reservoir solution. The resulting drop was stirred until homogenous with a final composition of 5% weight per volume of polyethylene glycol 400 in reservoir condition. The crystal was promptly cryo-cooled by dunking into liquid nitrogen. An X-ray diffraction dataset to 2.0 Å was recorded with the Advanced Photon Source (Argonne National Laboratory), using IMCA-CAT’s beamline at full transmission and an oscillation range of 0.2° and exposure time of 0.02 seconds per frame for 900 frames. The wavelength of the X-ray beam was 1.000 Å. Integration, scaling and merging of the diffraction data was performed with autoPROC STARANISO software (1.1.7 (20211020), 2.3.87 beta for linux64-ifort made on Jun 08 2022 at 02:21:31, Global Phasing Ltd.). Molecular replacement was performed using Phaser as implemented in the CCP4i suite (CCP4 Interface version 7.1.018). The ligand model and its crystallographic restraints were created using Grade2 software (version 1.2.0, Global Phasing Ltd.). Refinement of the structure, including generation of restraints for the covalent link between the ligand and C145, was performed using the crystallography software Buster (2.11.8 (22-FEB-2023), Global Phasing Ltd.) with manual rebuilding and water placement in COOT (0.9.7, Emsley, University of Cambridge). Additional crystallography details can be found in the Supplementary methods, Supplementary Table 2, and Supplementary Fig. 12.
SARS-CoV-2 replicon screening
A bacterial artificial chromosome encoding a SARS-CoV-2 (USA-WA1/2020) replicon was purchased (Telesis Bio) and a modified screening protocol for SARS-CoV-2 replicon system was developed in-house41,42. The replicon fragment and a codon-optimized SARS-CoV-2 N (GeneART, Invitrogen) were PCR amplified (Platinum SuperFi II PCR Master Mix, Invitrogen), in vitro transcribed with mMESSAGE mMACHINE T7 Ultra (Invitrogen) and purified using the Monarch kit (New England Biolabs) per manufacturers’ recommendations. 650 ng of replicon and N were electroporated using the Neon Transfection System (Invitrogen) at 1700V-20ms-1 pulse into HuH-7 cells (1\({{{\rm{\times }}}}\)107 cells/mL) following the manufacturers’ protocol. Cells were diluted to 800 cells/μL by adding complete HuH-7 media excluding penicillin and streptomycin, and compounds were added. Electroporated HuH-7 cells containing no RNA and treated with DMSO were used as a low control. High control contained SARS-CoV-2 replicon, N RNA, and was treated with DMSO. Plates were incubated for 20 h and luminescence was measured using the Renilla-Glo® Luciferase Assay Reagent (Promega). The percent residual activity of the replicon was determined after normalizing to DMSO-treated low and high controls.
Coronavirus infectious virus assays
BSL3-experiments were performed at multiple facilities: Georgia State University (GSU), Southern Research (SR) and Viroclinics Biosciences (VCB). Vero E6-TMPRSS2 cells were seeded 14 h pre-infection (GSU), or Vero E6 cells were seeded in suspension at the time of infection (SR and VCB). Monolayers were infected with an MOI of 0.1 (SARS-CoV-2 Omicron strains), 0.002 (SARS-CoV-2 lineage A USA-WA1/2020 and Delta strains, SARS-CoV Toronto-2 isolate), or 0.001 (SARS-CoV-2 lineage B Germany/BavPat1/2020 isolate, Alpha and Beta strains, SARS-CoV HKU-39849 isolate and MERS-CoV EMC/2012 isolate). EDP-235 in the presence or absence of 0.5–2 μM PGPi was added at the time of infection. Results were determined as follows: at 48 hpi, the supernatant was collected and titered by either TCID50 or plaque assay (GSU). At 72 hpi, cell health was determined using Cell-Titer-Glo (Promega) per manufacturer’s instructions and luminescence was measured (CLARIOstar, BMG LabTech Inc.) (SR). After 18–24 hpi, cells were immunostained for SARS-CoV-2 N protein and foci were counted (Immunospot Image Analyzer) (VCB). All experiments performed at GSU using infectious SARS-CoV-2 strains were approved by the Georgia State Institutional Biosafety Committee under protocol B20016 and performed in BSL-3/ABSL-3 facilities.
SARS-CoV-2 3D pHAEC experiments were performed at VCB, with Mucilair pHAEC tissues sourced from Epithelix. Tissues were pre-treated basolaterally with EDP-235 for 2 h prior to apical infection at MOI of 1 (SARS-CoV-2 lineage B Germany/BavPat1/2020 isolate) for 1 h. The inoculum was removed, tissues were washed and incubated for 48 h. At 24 and 48 hpi, 200 µL apical samples were harvested for analysis by TCID50 and RT-qPCR.
HCoV-229E, OC43 and NL63 antiviral assays were performed at Enanta Pharmaceuticals, Inc., and were assessed in MRC-5, HCT-8, and LLC-MK2 cells, respectively. MRC-5 cells were infected in suspension at an MOI of 0.05 and incubated at 34 °C for 6 days. HCT-8 and LLC-MK2 cells were seeded 24 h prior to infections and monolayers were infected at 100 TCID50 (OC43) or an MOI of 0.01 (NL63). OC43 and NL63 infections were incubated at 34 and 32 °C, respectively, for 7 days. For CPE-based assays, cell health was assessed using ATP-lite (Perkin Elmer), and luminescence was measured (Envision). For RT-qPCR-based endpoints, viral RNA was extracted from cells and supernatant (Quick-RNA Viral 96 Kit, Zymo Research) and quantified by RT-qPCR using TaqMan RNA-to-Ct-1Step (Applied Biosystems) on the POD Quant Studio 7 Flex Machine (Thermo Fisher).
For HCoV-229E infections in 3D pHAEC EpiAirway sourced from a 23-year-old Caucasian male (MatTek, AIR-100), apical surfaces were washed once with transepithelial electrical resistance buffer, and DMSO or compound was added to the basal media. Adsorptions were performed apically for 1.5 h with 2 × 103 PFU per tissue. After 1.5 h the adsorption inoculum was removed, the apical surface was washed once with transepithelial electrical resistance buffer, and plates incubated for 5 days. The basal media was changed once after 2-3 days, maintaining DMSO or compound concentration. Total RNA was extracted using RNAqueous Total RNA Isolation Kit (ThermoFisher), and viral RNA was quantified using RT-qPCR as described above.
Infectious zoonotic CoV antiviral assays were performed at University of North Carolina, Chapel Hill. Reporter sarbecoviruses or merbecoviruses expressing recombinant nanoluciferase were used to perform cell-based antiviral assays in either A549-hACE2 or Huh7.5 cells, respectively. Reporter viruses were generated from infectious clones as previously described in refs. 47,48,71. Briefly, 2 × 104 A549-hACE2 or 2.5 × 104 Huh7.5 cells were seeded 24 h before infection with DMSO normalized across the plate. Cells were infected at a MOI of 0.1 for 1 h after which the inoculum was removed, and monolayers washed one time. A dilution series of compound was added to the monolayer and replication was measured at 24 hpi by addition of NanoGlo Luciferase Assay System (Promega).
Cytotoxicity assays
Cytotoxicity assays were performed identically to their respective viral efficacy studies, but without addition of virus and using a cell viability readout.
Protease selectivity
Selectivity of EDP-235 was determined as described in the biochemical assays section of these Methods. Additional materials and conditions are listed in Supplementary Table 7. All work was performed at Reaction Biology.
In vivo studies
Studies with male golden Syrian hamsters 1-2 months of age were run at BIOQUAL, Inc. with eight animals per cohort. Hamster in vivo studies involving SARS-CoV-2 were performed in accordance with BIOQUAL standard procedures, the Association for Assessment and Accreditation of Laboratory Animal Care, the Animal Welfare Act, the Public Health Service Policy on Human Care and Use of Laboratory Animals, and Centers for Disease Control. Experiments with SARS-CoV-2 involving hamsters were performed under Institutional Animal Care and Use Committee-approved protocol #22-101 P and were sourced from Envigo. Intramuscular anesthesia was used for all procedures. Vehicle and EDP-235 doses (20 and 50 mg/mL) were prepared in 0.5% methylcellulose in deionized water and mixed for 30 min prior to and during oral dosing. Viral intranasal inoculation of SARS-CoV-2 USA/WA1/2020 (generated by BIOQUAL) was delivered as 3 × 104 PFU per nostril in 50 µL PBS, with the head tilted back for 20 s. Animal body weight symptoms were recorded throughout the study. At 4 dpi, animals were euthanized. The whole lung was collected during necropsy, with weight and physical observations recorded before sectioning and snap-freezing or storage in 10% neutral buffered formalin. Nares and tracheas were collected and snap frozen. TCID50 analysis was run on lung, nares, and trachea samples. RT-qPCR (N gene) analysis was run on lung and nares samples. Histopathology services were performed by Experimental Pathology Laboratories, Inc. Fixed lung was processed for IHC detection of SARS-CoV-2 nucleocapsid protein, using a SARS-CoV-2 (COVID-19) nucleocapsid antibody on formalin-fixed paraffin-embedded tissue. Anti-SARS-CoV-2, 0.02 μg/mL, primary antibody (ProSci, 9099; SARS-CoV-2 (COVID-19) Nucleocapsid Antibody) was applied to tissues overnight at 4 °C ( ± 2°C) followed by ImmPRESS Polymer Anti-Rabbit IgG Reagent (made in goat, RTU, Vector Labs, MP-7451) for 30 min, DAB solution (3 min), and counterstained with hematoxylin.
Studies with female ferrets 6–10 months of age sourced from Triple F farms were run at GSU with six animals per cohort. All in vivo studies involving ferrets were performed in compliance with the Guide for the Care and Use of Laboratory Animals, National Institutes of Health guidelines, and the Animal Welfare Act Code of Federal Regulations. Experiments with SARS-CoV-2 involving ferrets were approved by the Georgia State Institutional Animal Care and Use Committee under protocol A20031. Vehicle and EDP-235 doses (200 mg/kg BID or QD) were prepared in 0.5% methylcellulose in deionized water before each dosing. Ferrets received intranasal viral inoculation of 1 × 105 PFU of SARS-CoV-2 USA/WA1/2020, and oral dosing of vehicle or EDP-235 began at 12 hpi and continued for three days. At 60 hpi (48 h post-treatment initiation), each ferret was co-housed with an uninfected, untreated contact ferret until day 4. Source ferrets were euthanized on day 4, and contact ferrets were further monitored before being euthanized on day 8. Homogenized nasal turbinates were collected from all ferrets subsequent to euthanizing. On days 0-4, nasal lavages from source and contact ferrets were collected twice a day. On days 5–8, nasal lavages were collected from contact ferrets once a day. Weight and temperature were recorded from all ferrets once a day, and a rectal swab was taken. Lavage and turbinate samples were subject to TCID50 analysis to determine infectious viral load. RT-qPCR (nsp9 gene) was used to quantify RNA copies in lavage, turbinates, and rectal swab samples.
Pharmacokinetic studies
Ferret plasma exposure levels and pharmacokinetic calculations were determined as follows; performed at Enanta Pharmaceuticals, Inc. Following a single oral dose of EDP-235 in 3 female ferrets per arm, plasma samples were collected at 1, 2, 3, 4, 6, 8, 12, and 24 h. Subsequently, plasma samples were deproteinized with acetonitrile (Fisher) and an internal standard acetonitrile solution. Post centrifugation, quantitation of EDP-235 within the supernatant was determined by liquid chromatography-tandem mass spectrometric method. Calibrators were prepared with blank plasma and standards following the same procedure as sample preparation.
Materials and data analysis
Primer/probe sequences utilized in Methods are listed in Supplementary Table 8. Data analysis for all experiments was performed in GraphPad Prism (version 10.1.2 (324)) or XLfit (version 2405, Microsoft Excel). Binding constants and best-fit equations were determined using GraphPad Prism Software. Efficacy, and cytotoxicity concentrations were calculated with a variable slope four-parameter logistic model.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All source data used in this study are provided in the Supplementary Information/Source Data files. The EDP-235 bound 3CLpro X-ray crystal structure can be found at the protein data bank (https://www.rcsb.org/). PDB accession #: 8VDJ. Source data are provided with this paper.
References
World Health Organization. Coronavirus disease (COVID-19). (2023).
Li, G., Hilgenfeld, R., Whitley, R. & De Clercq, E. Therapeutic strategies for COVID-19: progress and lessons learned. Nat. Rev. Drug Discov. 22, 449–475 (2023).
Hammond, J. et al. Oral Nirmatrelvir for High-Risk, Nonhospitalized Adults with Covid-19. N. Engl. J. Med. 386, 1397–1408 (2022).
Jayk Bernal, A. et al. Molnupiravir for Oral Treatment of Covid-19 in Nonhospitalized Patients. N. Engl. J. Med. 386, 509–520 (2022).
Matrajt, L., Brown, E. R., Cohen, M. S., Dimitrov, D. & Janes, H. Could widespread use of antiviral treatment curb the COVID-19 pandemic? A modeling study. BMC Infect. Dis. 22, 683 (2022).
Khunte, M., Kumar, S., Salomon, J. A. & Bilinski, A. Projected COVID-19 Mortality Reduction From Paxlovid Rollout. JAMA Health Forum 4, e230046 (2023).
Mukae, H. et al. Efficacy and Safety of Ensitrelvir in Patients With Mild-to-Moderate Coronavirus Disease 2019: The Phase 2b Part of a Randomized, Placebo-Controlled, Phase 2/3 Study. Clin. Infect. Dis. 76, 1403–1411 (2023).
Pillaiyar, T., Manickam, M., Namasivayam, V., Hayashi, Y. & Jung, S.-H. An Overview of Severe Acute Respiratory Syndrome-Coronavirus (SARS-CoV) 3CL Protease Inhibitors: Peptidomimetics and Small Molecule Chemotherapy. J. Med. Chem. 59, 6595–6628 (2016).
Yang, H. et al. The crystal structures of severe acute respiratory syndrome virus main protease and its complex with an inhibitor. Proc. Natl. Acad. Sci. USA 100, 13190–13195 (2003).
Ziebuhr, J., Snijder, E. J. & Gorbalenya, A. E. Virus-encoded proteinases and proteolytic processing in the Nidovirales. J. Gen. Virol. 81, 853–879 (2000).
Lv, Z., Chu, Y. & Wang, Y. HIV protease inhibitors: a review of molecular selectivity and toxicity. HIV AIDS (Auckl.) 7, 95–104 (2015).
de Leuw, P. & Stephan, C. Protease inhibitors for the treatment of hepatitis C virus infection. GMS Infect. Dis. 5, Doc08 (2017).
Jin, Z. et al. Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature 582, 289–293 (2020).
Kandwal, S. & Fayne, D. Genetic conservation across SARS-CoV-2 non-structural proteins - Insights into possible targets for treatment of future viral outbreaks. Virology 581, 97–115 (2023).
Anand, K., Ziebuhr, J., Wadhwani, P., Mesters, J. R. & Hilgenfeld, R. Coronavirus Main Proteinase (3CLpro) Structure: Basis for Design of Anti-SARS Drugs. Science 300, 1763–1767 (2003).
Antiviral and Antibody Products Summary Recommendations. COVID-19 Treatment Guidelines https://www.covid19treatmentguidelines.nih.gov/therapies/antivirals-including-antibody-products/summary-recommendations/.
Owen, D. R. et al. An oral SARS-CoV-2 M(pro) inhibitor clinical candidate for the treatment of COVID-19. Science 374, 1586–1593 (2021).
Loos, N. H. C., Beijnen, J. H. & Schinkel, A. H. The Mechanism-Based Inactivation of CYP3A4 by Ritonavir: What Mechanism? Int. J. Mol. Sci. 23, 9866 (2022).
Eng, H. et al. Disposition of Nirmatrelvir, an Orally Bioavailable Inhibitor of SARS-CoV-2 3C-Like Protease, across Animals and Humans. Drug Metab. Dispos. 50, 576–590 (2022).
Pfizer Announces Additional Phase 2/3 Study Results Confirming Robust Efficacy of Novel COVID-19 Oral Antiviral Treatment Candidate in Reducing Risk of Hospitalization or Death | Pfizer. https://www.pfizer.com/news/press-release/press-release-detail/pfizer-announces-additional-phase-23-study-results.
Lam, C. & Patel, P. Nirmatrelvir-Ritonavir. in StatPearls (StatPearls Publishing, Treasure Island (FL), 2023).
Duffy, S. Why are RNA virus mutation rates so damn high? PLoS Biol. 16, e3000003 (2018).
Cox, M. et al. SARS-CoV-2 variant evasion of monoclonal antibodies based on in vitro studies. Nat. Rev. Microbiol. 21, 112–124 (2023).
Hoffmann, M. et al. The Omicron variant is highly resistant against antibody-mediated neutralization: Implications for control of the COVID-19 pandemic. Cell 185, 447–456.e11 (2022).
Zabidi, N. Z. et al. Evolution of SARS-CoV-2 Variants: Implications on Immune Escape, Vaccination, Therapeutic and Diagnostic Strategies. Viruses 15, 944 (2023).
Fernandes, Q. et al. Emerging COVID-19 variants and their impact on SARS-CoV-2 diagnosis, therapeutics and vaccines. Ann. Med. 54, 524–540 (2022).
Iketani, S. et al. Multiple pathways for SARS-CoV-2 resistance to nirmatrelvir. Nature 613, 558–564 (2023).
Zuckerman, N. S., Bucris, E., Keidar-Friedman, D., Amsalem, M. & Brosh-Nissimov, T. Nirmatrelvir resistance - de novo E166V/L50V mutations in an immunocompromised patient treated with prolonged nirmatrelvir/ritonavir monotherapy leading to clinical and virological treatment failure - a case report. Clin. Infect. Dis. https://doi.org/10.1093/cid/ciad494. (2023).
Flynn, J. M. et al. Systematic Analyses of the Resistance Potential of Drugs Targeting SARS-CoV-2 Main Protease. ACS Infect. Dis. 9, 1372–1386 (2023).
Jochmans, D. et al. The Substitutions L50F, E166A, and L167F in SARS-CoV-2 3CLpro Are Selected by a Protease Inhibitor In Vitro and Confer Resistance To Nirmatrelvir. mBio 14, e0281522 (2023).
Zhou, Y. et al. Nirmatrelvir-resistant SARS-CoV-2 variants with high fitness in an infectious cell culture system. Sci. Adv. 8, eadd7197 (2022).
Heilmann, E. et al. SARS-CoV-2 3CL pro mutations selected in a VSV-based system confer resistance to nirmatrelvir, ensitrelvir, and GC376. Sci. Transl. Med. 15, eabq7360 (2023).
Carlson, C. J. et al. Climate change increases cross-species viral transmission risk. Nature 607, 555–562 (2022).
Gebara, M. F., May, P. H. & Platais, G. Pandemics, conservation, and human-nature relations. Clim. Change Ecol. 2, 100029 (2021).
Human Coronavirus Types | CDC. https://www.cdc.gov/coronavirus/types.html (2021).
Vlasova, A. N. et al. Novel Canine Coronavirus Isolated from a Hospitalized Patient With Pneumonia in East Malaysia. Clin. Infect. Dis. 74, 446–454 (2022).
Lednicky, J. A. et al. Independent infections of porcine deltacoronavirus among Haitian children. Nature 600, 133–137 (2021).
Enanta Pharmaceuticals, Inc. NCT05616728. A Study to Evaluate EDP-235 in Non-hospitalized Adults With COVID-19 (SPRINT). (2022).
Chia, C. S. B. & See, Y. Y. Novel Coronavirus Main Protease Di- and Tripeptide Inhibitors for Treating COVID-19. ACS Med. Chem. Lett. 13, 1388–1389 (2022).
Hoffman, R. L. et al. Discovery of Ketone-Based Covalent Inhibitors of Coronavirus 3CL Proteases for the Potential Therapeutic Treatment of COVID-19. J. Med Chem. 63, 12725–12747 (2020).
He, X. et al. Generation of SARS-CoV-2 reporter replicon for high-throughput antiviral screening and testing. Proc. Natl. Acad. Sci. USA 118, e2025866118 (2021).
Khan, J. Q. et al. Generation of a SARS-CoV-2 Reverse Genetics System and Novel Human Lung Cell Lines That Exhibit High Virus-Induced Cytopathology. Viruses 15, 1281 (2023).
De Rosa, M. F., Sillence, D., Ackerley, C. & Lingwood, C. Role of multiple drug resistance protein 1 in neutral but not acidic glycosphingolipid biosynthesis. J. Biol. Chem. 279, 7867–7876 (2004).
Kalgutkar, A. S. et al. N-(3,4-dimethoxyphenethyl)-4-(6,7-dimethoxy-3,4-dihydroisoquinolin-2[1H]-yl)-6,7-dimethoxyquinazolin-2-amine (CP-100,356) as a ‘chemical knock-out equivalent’ to assess the impact of efflux transporters on oral drug absorption in the rat. J. Pharm. Sci. 98, 4914–4927 (2009).
Zhou, H. et al. Identification of novel bat coronaviruses sheds light on the evolutionary origins of SARS-CoV-2 and related viruses. Cell 184, 4380–4391.e14 (2021).
Martinez, D. R. et al. Chimeric spike mRNA vaccines protect against Sarbecovirus challenge in mice. Science 373, 991–998 (2021).
Hou, Y. J. et al. Host range, transmissibility and antigenicity of a pangolin coronavirus. Nat. Microbiol 8, 1820–1833 (2023).
Tse, L. V. et al. A MERS-CoV antibody neutralizes a pre-emerging group 2c bat coronavirus. Sci. Transl. Med. 15, eadg5567 (2023).
Sia, S. F. et al. Pathogenesis and transmission of SARS-CoV-2 in golden hamsters. Nature 583, 834–838 (2020).
Chan, J. F. et al. Simulation of the Clinical and Pathological Manifestations of Coronavirus Disease 2019 (COVID-19) in a Golden Syrian Hamster Model: Implications for Disease Pathogenesis and Transmissibility. Clin. Infect. Dis. 71, 2428–2446 (2020).
Richard, M. et al. SARS-CoV-2 is transmitted via contact and via the air between ferrets. Nat. Commun. 11, 3496 (2020).
Cox, R. M. et al. Comparing molnupiravir and nirmatrelvir/ritonavir efficacy and the effects on SARS-CoV-2 transmission in animal models. Nat. Commun. 14, 4731 (2023).
Del La Rosa, G. EDP-235, an Oral, Once Daily, Ritonavir-Free, 3CL Protease Inhibitor for the Treatment of COVID-19: Results from Phase 1 Study in Healthy Subjects. (2023).
Cox, R. M. et al. Oral prodrug of remdesivir parent GS-441524 is efficacious against SARS-CoV-2 in ferrets. Nat. Commun. 12, 6415 (2021).
Toussi, S. S., Hammond, J. L., Gerstenberger, B. S. & Anderson, A. S. Therapeutics for COVID-19. Nat. Microbiol 8, 771–786 (2023).
Enanta Pharmaceuticals, Inc. SPRINT Data Presentation. (2023).
Paules, C. I., Marston, H. D. & Fauci, A. S. Coronavirus Infections—More Than Just the Common Cold. JAMA 323, 707–708 (2020).
Feng, A. et al. Transmission of SARS-CoV-2 in free-ranging white-tailed deer in the United States. Nat. Commun. 14, 4078 (2023).
CDC. COVID-19 and Your Health. Centers for Disease Control and Prevention https://www.cdc.gov/coronavirus/2019-ncov/daily-life-coping/animals.html (2020).
Hirotsu, Y. et al. Multidrug-resistant mutations to antiviral and antibody therapy in an immunocompromised patient infected with SARS-CoV-2. Med 4, 813–824.e4 (2023).
Sanderson, T. et al. A molnupiravir-associated mutational signature in global SARS-CoV-2 genomes. Nature 1–3. https://doi.org/10.1038/s41586-023-06649-6. (2023)
Abdelnabi, R. et al. The oral protease inhibitor (PF-07321332) protects Syrian hamsters against infection with SARS-CoV-2 variants of concern. Nat. Commun. 13, 719 (2022).
Sasaki, M. et al. S-217622, a SARS-CoV-2 main protease inhibitor, decreases viral load and ameliorates COVID-19 severity in hamsters. Sci. Transl. Med. 15, eabq4064 (2023).
Braxton, A. M. et al. Hamsters as a Model of Severe Acute Respiratory Syndrome Coronavirus-2. Comp. Med. 71, 398–410 (2021).
Cox, R. M., Wolf, J. D. & Plemper, R. K. Therapeutically administered ribonucleoside analogue MK-4482/EIDD-2801 blocks SARS-CoV-2 transmission in ferrets. Nat. Microbiol. 6, 11–18 (2021).
Clair, R., Gordon, M., Kroon, M. & Reilly, C. The effects of social isolation on well-being and life satisfaction during pandemic. Humanit Soc. Sci. Commun. 8, 28 (2021).
Jeffers, A. et al. Impact of Social Isolation during the COVID-19 Pandemic on Mental Health, Substance Use, and Homelessness: Qualitative Interviews with Behavioral Health Providers. IJERPH 19, 12120 (2022).
Mullard, A. Pfizer’s small-molecule antiviral misses on COVID prevention. Nat. Rev. Drug Discov. 21, 406–406 (2022).
Alpizar, S. A. et al. Molnupiravir for intra-household prevention of COVID-19: The MOVe-AHEAD randomized, placebo-controlled trial. J. Infect. 87, 392–402 (2023).
Tonge, P. J. Quantifying the Interactions between Biomolecules: Guidelines for Assay Design and Data Analysis. ACS Infect. Dis. 5, 796–808 (2019).
Rappazzo, C. G. et al. Broad and potent activity against SARS-like viruses by an engineered human monoclonal antibody. Science 371, 823–829 (2021).
Acknowledgements
We thank Jiajun Zhang, the Enanta Medicinal Chemistry, and CMC teams for assistance with compound needs and manuscript review. We also thank Ian Cade and the Evotec team for manuscript assistance and their work generating the EDP-235-bound crystal structure of the 3CLpro as well as the PDB upload. The development of live reporter sarbecovirus NanoLuc luciferase drug assays was supported by an NIH AID grant to R.S.B. (AI171292).
Author information
Authors and Affiliations
Contributions
Assay development, execution, data generation, and analysis performed by M.H.J.R., A.C.R., A.B., N.B., N.M.K., J.S.G., J.L., M.V., T.C., M.C., N.M., J.C., R.E.L., D.L., T.Z., R.M.C., C.M.L., J.D.W., and T.S. Experimental design, project guidance and oversight performed by M.H.J.R., A.C.R., A.B., N.M.K., J.S.G., M.V., T.C., J.C., L.J., K.D., R.K.P., S.R.L., R.S.B., G.W., B.G., and Y.S.O.. EDP-235 compound design and generation performed by R.S., G.W., and Y.S.O.. Manuscript writing performed in chief by M.H.J.R., N.M.K., and J.S.G.. All authors contributed to manuscript review and editing.
Corresponding author
Ethics declarations
Competing interests
M.H.J.R., A.C.R., A.B., N.B., N.M.K., J.S.G., J.L., M.V., T.C., M.C., R.S., N.M., J.C., R.E.L., D.L., T.Z., L.J., K.D., G.W., B.G, and Y.S.O. are either current or former employees of Enanta Pharmaceuticals and received salary and stock compensation during this study. R.M.C., C.M.L, J.D.W, R.K.P., S.R.L., T.S., and R.S.B. received funding from Enanta Pharmaceuticals. R.K.P. reports contract testing from Atea Pharmaceuticals and research support from Gilead Sciences, outside of the described work.
Peer review
Peer review information
Nature Communications thanks Kris White, and the other, anonymous, reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Rhodin, M.H.J., Reyes, A.C., Balakrishnan, A. et al. The small molecule inhibitor of SARS-CoV-2 3CLpro EDP-235 prevents viral replication and transmission in vivo. Nat Commun 15, 6503 (2024). https://doi.org/10.1038/s41467-024-50931-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-50931-8
- Springer Nature Limited