

Abstract
Myosin1D (Myo1D) has recently emerged as a conserved regulator of animal Left-Right (LR) asymmetry that governs the morphogenesis of the vertebrate central LR Organizer (LRO). In addition to Myo1D, the zebrafish genome encodes the closely related Myo1G. Here we show that while Myo1G also controls LR asymmetry, it does so through an entirely different mechanism. Myo1G promotes the Nodal-mediated transfer of laterality information from the LRO to target tissues. At the cellular level, Myo1G is associated with endosomes positive for the TGFβ signaling adapter SARA. myo1g mutants have fewer SARA-positive Activin receptor endosomes and a reduced responsiveness to Nodal ligands that results in a delay of left-sided Nodal propagation and tissue-specific laterality defects in organs that are most distant from the LRO. Additionally, Myo1G promotes signaling by different Nodal ligands in specific biological contexts. Our findings therefore identify Myo1G as a context-dependent regulator of the Nodal signaling pathway.
Similar content being viewed by others

Introduction
Left-Right (LR) asymmetries in the positioning and shape of different tissues are found in both protostome and deuterostome lineages and are critically required for human organ function1. In spite of the importance of LR asymmetry, our understanding of the mechanisms that govern this third body axis remains fragmentary. A particularly striking feature of LR asymmetry is the fact that an evolutionary conserved mechanism of symmetry breaking has long remained elusive. Although Nodal proteins of the Transforming Growth Factor β superfamily have long been known to control LR asymmetry in all deuterostome and some protostome species2,3, it is only recently that the unconventional type 1 Myosin Myosin1D (Myo1D) has emerged as a potentially universal regulator of animal LR asymmetry4,5,6,7. Here, we identify the closely related protein Myosin1G (Myo1G) as a positive regulator of the Nodal signaling pathway.
Seminal studies in the mouse revealed the existence of a central LR Organizer (LRO) in which the Planar Cell Polarity (PCP)-dependent orientation of motile cilia promotes the generation of a directional symmetry-breaking fluid flow8,9,10. Symmetry-breaking cilia-driven fluid flows are also present in other species, including fish and frogs11,12. Already within the vertebrate phylum, the LROs of birds and reptiles do, however, lack motile cilia and rely - at least in chick - on lateralized cell flows to trigger symmetry breaking13,14. Additional mechanisms implicated in LR asymmetry include ion flows15 and Actin-dependent chiral cell remodeling16,17. While an increasing number of studies indicate that Actin- and PCP-dependent pathways lie at the core of a symmetry-breaking toolbox4,5,18,19,20,21,22, our understanding of the evolutionary conservation of the mechanisms controlling LR asymmetry remains fragmentary.
In vertebrates, Nodal ligands convey laterality information from the central LRO to target tissues1,2. Nodal ligands propagate on the left side of the embryo by inducing their own expression, allowing them to propagate from the posteriorly located LRO to more anterior target tissues23. In species with an LRO bearing motile cilia, Nodal is expressed initially in a bilaterally symmetric fashion at the LRO, together with the TGFβ signaling antagonist Dand524. Upon establishment of a ciliary LRO flow, dand5 transcripts are degraded on the left side of the LRO25,26,27, allowing Nodal to travel to the left lateral plate mesoderm and propagate by autoinduction.
Nodal ligands induce cellular responses through ligand/receptor complexes that comprise TGFβ type I and II receptors and the co-receptor Cripto/Oep28. Nodal ligand binding causes type II receptors to phosphorylate and activate their type I counterpart. A population of endosomes positive for the TGFβ signaling adapter Smad Anchor for Receptor Activation (SARA) promotes signal transduction by allowing Activin/Nodal receptors to recruit their transcriptional downstream mediators SMAD2 and 329. Upon phosphorylation by activated type I receptors, SMAD2 & 3 associate with SMAD4 to enter the nucleus and activate target genes23. As Nodal ligands are highly potent, tight regulation of Nodal signaling is essential not only for embryonic development but also to avoid tumorigenesis23,30. Lefty proteins act as feedback inhibitors of Nodal signaling that prevent the formation of productive ligand/receptor complexes23,31. In LR asymmetry, Lefty expression at the embryonic midline is important to form a barrier that prevents the spreading of left-sided Nodal ligands to the contralateral side32,33.
The requirement of Nodal ligands for LR asymmetry is, however, not universally conserved1, and a number of protostomian species, including the fruitfly Drosophila, altogether lack nodal homologs. Studies in Drosophila identified Myo1D as a master regulator of LR asymmetry34,35. In contrast to the central LRO of vertebrate organisms that governs LR asymmetry of all lateralized organs, Drosophila myo1d acts in a local, tissue-autonomous fashion to control genital and visceral laterality18,35. Of particular interest, studies in frogs, fish and humans showed that Myo1D is also required for vertebrate LR asymmetry4,5,6,7.
Zebrafish Myo1D is required for the establishment of a functional symmetry-breaking ciliary LRO flow5. In addition to myo1d, the fish genome harbors the closely related gene myosin1g (myo1g). Although myo1g mutations impair laterality and enhance the defects of myo1d mutants, we show in this work that Myo1G acts independently of the LRO flow, through an entirely different mechanism. We provide evidence that Myo1G represents a positive regulator of the Nodal signaling pathway whose function is essential for the Nodal-mediated transfer of laterality information.
Results
Myosin1G mutants present tissue-specific left-right asymmetry defects
Myo1D controls cilia orientation in the LRO to promote the generation of a symmetry-breaking LRO flow4,5,6. The closely related protein Myo1G (79% amino acid similarity) is also required for zebrafish LR asymmetry but has no detectable effect on the LRO flow5, suggesting that different type I Myosins regulate LR asymmetry through distinct mechanisms. To address this issue, we performed a detailed characterization of Maternal Zygotic (MZ) myo1g single and MZ myo1d; MZ myo1g double mutants.
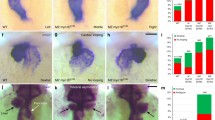
The asymmetric morphogenesis of the zebrafish heart becomes first apparent during the process of cardiac jogging, which transforms the cardiac disc that is initially located at the embryonic midline into a leftward point cardiac tube36,37,38. MZ myo1g single mutants present defects in the leftward jogging of cardiac progenitors (Fig. 1a and Supplementary Fig. 1), the penetrance of which is further enhanced in MZ myo1d; MZ myo1g double mutants (Fig. 1a). To study the effect of myo1g on brain laterality, we analyzed the expression of the Nodal ligand cyclops/nodal related 2 (cyc/ndr2), its feedback antagonist lefty1 (lft1) and its transcriptional effector pitx2 which display predominantly left-sided expression in the dorsal epithalamus of wild-type embryos33,39,40. In contrast to the mild defects observed in MZ myo1d mutants (Fig. 1b and Supplementary Fig. 2a, b), MZ myo1g single mutants displayed a significantly higher proportion of brain laterality defects (Fig. 1b and Supplementary Fig. 2a, b). In contrast to the effect on brain laterality, MZ myo1g mutants display normal expressions of floating head in the epiphysis (Supplementary Fig. 2c) and of otx5 in the pineal complex (Supplementary Fig. 2d), demonstrating thereby that myo1g loss of function impairs specifically the laterality but not the specification of dorsal forebrain structures. The penetrance of brain laterality defects in MZ myo1d; MZ myo1g double mutants is similar to the one observed in MZ myo1g single mutants, confirming the predominant role of myo1g in brain laterality (Fig. 1b and Supplementary Fig. 2a).

a, a′ Quantification of cardiac jogging indicates that MZ myo1g mutants present laterality defects that are enhanced in MZ myo1d; MZ myo1g double mutants (a). Concomitant inactivation of the LRO flow (through dnaaf1 mutation) reveals that MZ myo1d/g mutations enhance the cardiac jogging defects of flow-deficient animals (a′). b Brain asymmetry is impaired in MZ myo1g single and MZ myo1d; MZ myo1g double mutants. Frontal views of pitx2 expression at 30 somites, dorsal up. c MZ myo1g mutants do not show visceral LR defects. L liver, G gut, P pancreas. Dorsal views of foxa1 expression at 48 h, anterior up. d MZ myo1d/g inactivation enhances the brain laterality phenotypes of LRO flow-deficient dnaaf1 mutants. e, f Visceral laterality phenotypes of dnaaf1 mutants are unaffected by myo1d/g inactivation. f Dorsal views of foxa1 expression at 48 h, anterior up. Pictures are derived from the data set quantified in (e). Scale bars: 50 µm. All p values were obtained using non-directional statistical tests. Complete numerical and statistical information for all experiments are provided in the Source Data files.
In contrast to the effect of myo1g on brain laterality, analysis of liver, pancreas and gut laterality using the endodermal marker foxa1 failed to reveal visceral LR asymmetry defects in MZ myo1g single mutants (Fig. 1c). The observation that MZ myo1d; MZ myo1g double mutants present visceral laterality defects that are similar to MZ myo1d single mutants (Fig. 1c) confirms that myo1g is dispensable for the establishment of visceral laterality.
Myo1D is required for LRO morphogenesis and the generation of a ciliary fluid flow4,5,6. Accordingly, MZ myo1d mutants present defects at the level of all lateralized organs (Fig. 1a–c and Supplementary Fig. 2a, b). myo1g loss of function yields no discernable LRO flow defects5 and affects only a subset of organs (Fig. 1a–c and Supplementary Fig. 2a, b), raising the question of whether myo1g may control LR asymmetry through a flow-independent and potentially tissue-specific regulation of organ laterality, similar to the situation described for Drosophila myo1d18,35.
Myosin1G controls left-right asymmetry independently of the left-right organizer flow
To directly test if Myosin1 proteins exert LRO flow-independent functions in LR asymmetry, we investigated whether the LR asymmetry defects of animals that lack an LRO flow could be further modified by myosin1 inactivation. To this aim, we generated double and triple mutants to simultaneously inactivate myo1d & g and the essential regulator of ciliary motility dnaaf1/lrrc5041. Embryos that completely lack a LRO flow, as is the case for dnaaf1 mutants, display a distinctive randomization of cardiac, brain, and visceral laterality where LR asymmetry is properly established in roughly one-half of the population (situs solitus) but inverted in the other (situs inversus, Fig. 1a’, d, e, f). Only a small fraction of the embryos that lack an LRO flow display an altogether loss of LR asymmetry (i.e., absence of cardiac jogging and brain laterality markers, visceral situs ambiguus, Fig. 1a’, d, e).
In contrast, animals that lack both an LRO flow and myo1 function display a different phenotype, where the heart primordium fails to jog to either the left or the right side of the animal in most embryos (Fig. 1a’). dnaaf1; MZ myo1g double mutants additionally present a lack of asymmetric pitx2 expression in the dorsal epithalamus that contrasts with the randomization of lateralized gene expression observed in dnaaf1 single mutants (Fig. 1b, d). In contrast to the effect observed at the levels of the heart and brain, the visceral phenotypes of dnaaf1 mutant animals are unaffected by the loss of myo1g (Fig. 1e, f), confirming that myo1g is dispensable for visceral organ laterality.
These findings provide evidence for a LRO flow-independent function of Myosin1 proteins in LR asymmetry. The observations that (i) dnaaf1; MZ myo1g double mutants present a more pronounced loss of brain laterality than dnaaf1; MZ myo1d mutants (Fig. 1d) and that (ii) dnaaf1; MZ myo1d; MZ myo1g triple mutants are generally similar to dnaaf1; MZ myo1g double mutants (Fig. 1a’, d, e) suggest that Myo1G exerts a predominant role in the flow-independent control of LR asymmetry.
Myosin1G is required for Nodal pathway gene expression
Myo1 proteins could act in different ways to ensure a tissue-specific control of embryonic LR asymmetry. First, zebrafish myo1d & g could act in an organ-intrinsic fashion to promote chiral morphogenesis as in Drosophila18,35. Second, Myo1 activity could be required for the Nodal-mediated propagation of laterality information from the central LRO to different target tissues.
Already prior to the first morphological manifestations of asymmetric cell movement, the heart primordium displays asymmetries in gene expression in response to Nodal signaling from the left lateral plate mesoderm (LLPM)36,37. Of particular interest, the cardiac primordia of MZ myo1g single and MZ myo1d; MZ myo1g double mutants present a reduced left-sided expression of the Nodal downstream target and feedback inhibitor lefty2 (lft2) that could reflect impaired Nodal signaling (Fig. 2a). Accordingly, the expression of southpaw (spaw), the zebrafish Nodal ligand responsible for left-sided Nodal signaling, is reduced and extends less anteriorly in the LLPM of MZ myo1g single and MZ myo1d; MZ myo1g double mutants, while being affected to a lesser degree in MZ myo1d single mutants (Fig. 2b and Supplementary Fig. 3a). In further accordance with impaired Nodal signaling, myo1-deficient animals display a reduced expression of the Nodal-targets pitx2 and elovl6 in the LLPM (Fig. 2c and Supplementary Fig. 3b, c) and a reduced extension of the Nodal feedback inhibitor lft1 in the notochord that provides a molecular midline barrier for lateralized Nodal signaling (Fig. 2d).

a MZ myo1g single and MZ myo1d; MZ myo1g double mutants fail to display asymmetric lft2 expression in the cardiac primordium. Dorsal views at 22 somites, anterior up. b–d MZ myo1d/g mutants display a reduced anterior propagation of the expression of the Nodal ligand spaw (b, see also Supplementary Fig. 3a), the Nodal effector pitx2 (c, see also Supplementary Fig. 3b) and the Nodal feedback inhibitor lft1 (d). Lateral views at 18 somites, anterior left, dorsal up. Box plots in (b–d) indicate mean values ± SD. e Left-sided expression of Spaw RNA partially rescues the cardiac jogging defects of MZ myo1g mutants (see also Supplementary Fig. 4a, b). f Enhancing Spaw signaling through furinA RNA injection similarly rescues the cardiac jogging defects of MZ myo1g mutants. Scale bars: a 50 µm, b–d 100 µm. All p values were obtained using non-directional statistical tests. Complete numerical and statistical information for all experiments are provided in the Source Data files.
Promoting Nodal signaling restores cardiac laterality in MZ myosin1g mutants
If the LR asymmetry defects of MZ myo1g mutant animals are due to impaired Nodal signaling, augmenting left-sided Nodal signaling should allow for rescue cardiac laterality. To test this hypothesis, Spaw and GFP RNAs were co-injected into a single blastomere at the two-cell stage. By the end of gastrulation, the GFP tracer allowed to select animals in which the progeny of the injected blastomere was restricted to either the left or the right side of the embryo. In accordance with a potential requirement for myo1g in Nodal signaling, left-sided Spaw expression allowed to significantly decrease the number of MZ myo1g mutants for which the primordium fails to jog and stays at the midline and increase the percentage of MZ myo1g mutants that present a proper leftward cardiac jogging (Fig. 2e). In contrast, the chirality of cardiac looping, a process that subsequently generates the atrial and ventricular chambers and occurs largely independently of Nodal signaling42 was not restored by left-sided Spaw expression (Supplementary Fig. 4b). Animals in which Spaw-injected cells ended up on the right side of the embryo display aggravated cardiac jogging and looping defects compared to non-injected MZ myo1g mutants (Supplementary Fig. 4a, b).
Spaw activity is controlled by the proprotein convertase FurinA which promotes the cleavage of the Spaw prodomain to allow the formation of a mature ligand43. We, therefore, used furinA overexpression, which has previously been shown to extend the Spaw signaling range43, as an alternative strategy to increase Spaw signaling in MZ myo1g mutants. furinA-injected MZ myo1g mutants present a significantly reduced number of cardiac jogging defects compared to non-injected siblings (Fig. 2f), indicating again that MZ myo1g mutants can be partially rescued by promoting Nodal signaling. Finally, genetic analysis reveals that the cardiac jogging phenotypes of animals that are already devoid of Maternal and Zygotic spaw function (MZ spaw mutants) are not further modified by the loss of myo1g in MZ spaw; MZ myo1g double mutants (Supplementary Fig. 4c), in accordance with the hypothesis that Myo1G exerts a Spaw-dependent control of LR asymmetry.
The left-right organizer flow and myosin1 genes control Nodal propagation
Through its ability to promote the unilateral degradation of transcripts encoding the Nodal signaling antagonist Dand5, the LRO flow enables the left-sided propagation of nodal expression25,26,27. Our observation that Myo1G and (to a lesser degree) Myo1D act to promote the propagation of spaw expression (Fig. 2b and Supplementary Fig. 3a) raises the question of whether the enhanced laterality defects of embryos that lack both an LRO flow and myo1 gene function (Fig. 1) could be due to cumulative effects on nodal gene expression? To address this issue, we performed a comparative quantitative analysis of spaw expression in the Lateral Plate Mesoderm (LPM) of embryos that lack an LRO flow (due to dnaaf1 inactivation) as well as myo1d & g activities.
In wild-type control embryos, spaw extends anteriorly up to the level of the heart and brain primordia in the left LPM, while its expression is either entirely absent or only restricted to the posterior-most LPM on the right side of the embryo (Fig. 3a). Morpholino-mediated knock-down of dnaaf1 (Fig. 3a) or its genetic inactivation (Fig. 3b) cause a reduction in the left-sided extension of spaw which is likely due to a failure to downregulate dand5 on the left side of the LRO. Additionally, dnaaf1-deficient animals present a roughly symmetric expression of spaw in the right LPM (Fig. 3a, b). Simultaneous inactivations of myo1d/g and dnaaf1 cause a further reduction of the anterior extension of spaw expression on both the left and the right side of the animal (Fig. 3a, b), demonstrating thereby that Myosin1 proteins exert a flow-independent control of nodal ligand expression. Similar results were obtained using dnaaf1 morphants or mutants, although quantitatively stronger effects are observed upon the use of stable genetic mutants compared to transient morpholino knock-down. In accordance with our morphological analysis of embryonic laterality that suggested a predominant role of Myo1G in the LRO flow-independent control of LR asymmetry (Fig. 1), the inactivation of myo1g has a stronger effect on spaw expression in the LPM of dnaaf1-depleted animals than the loss of function of myo1d (Fig. 3a, b).

a, b Quantification of spaw extension in the Left (green dots) and Right (red dots) LPM of 18 somites stage LRO flow-deficient dnaaf1 morphant (a) or dnaaf1 mutant (b) embryos. myo1d/g loss of function causes a significant reduction of the antero-posterior extension of spaw expression in both the Left and the Right LPM. To allow direct comparison, mutant animals in a, b are derived from the same genetic background. Box plots in a, b indicate mean values ± SD. c, c′ Double in situ hybridization for spaw and the cardiac marker cmlc2/myl7 (see Supplementary Fig. 5 for pictures) reveals that spaw expression reaches the cardiac primordium in most WT control and LRO flow-deficient dnaaf1 morphant (c) or dnaaf1 mutant (c′) embryos, but fails to do so upon inactivation of myo1g. All p values were obtained using non-directional statistical tests. Complete numerical and statistical information for all experiments are provided in the Source Data files.
Nodal expression fails to reach the cardiac primordium in MZ myosin1g mutants
Spaw-mediated Nodal signaling is required to transmit laterality information from the LRO to target tissues. The zebrafish LRO, Kupffer’s Vesicle11 is located at the posterior tip of the notochord. Among the different tissues undergoing chiral morphogenesis, the visceral organ primordia are closest to the LRO, while heart and brain primordia are located more anteriorly at increasing distances. As our experiments show that MZ myo1g mutants present no defects in visceral LR asymmetry but increasingly severe phenotypes in the more anterior heart and brain (Fig. 1a–c), we wondered whether the reduced extension of left-sided spaw expression (Fig. 2b) may result in a failure to reach more anteriorly located organ primordia. To test this hypothesis, we performed two color in situ hybridization to simultaneously visualize spaw expression and the cmlc2/myl7-positive cardiac primordium.
Our analysis reveals that by the 22 somites stage, spaw expression has reached the cardiac primordium in most wild-type embryos (Fig. 3c, c’ and Supplementary Fig. 5a, a’). Similarly, spaw extends up to the level of the heart primordium on either the left, the right, or both sides of the embryo in most animals that are mutant or morphant for the LRO flow regulator dnaaf1 (Fig. 3c, c’ and Supplementary Fig. 5a, a’). In contrast, spaw expression fails to reach the cardiac primordium in a significant fraction of MZ myo1g mutants (Fig. 3c, c’ and Supplementary Fig. 5a, a’), providing thereby a potential explanation for their cardiac jogging defects. Compound inactivations of dnnaf1 and myo1g result in near complete failure of spaw expression to reach the cardiac primordium (Fig. 3c, c’ and Supplementary Fig. 5a, a’), in accordance with the predominant lack of cardiac jogging that is observed in these animals (Fig. 1a’).
Our experiments show that in MZ myo1g mutants, spaw expression frequently fails to extend anteriorly to reach the cardiac (Fig. 3c and Supplementary Fig. 5) and, therefore, necessarily also the even more anterior brain primordium, providing an explanation for the laterality defects that are observed in these two tissues (Fig. 1a, b). Conversely, the residual spaw expression of MZ myo1g mutants still extends up the level of the posterior gastrointestinal tract (Supplementary Fig. 6), a fact that may explain the observation the visceral laterality is correctly established in these animals (Fig. 1c).
MZ myosin1g mutants display a temporal delay in spaw expression
To investigate the mechanism through which myosin1 genes contribute to the LRO flow-independent regulation of Nodal signaling, we performed a time-course analysis of spaw expression during development. As myo1d contributes to both the regulation of the LRO flow5 and the flow-independent control of nodal expression (Fig. 3), we focused our analysis on myo1g, which plays a predominant role in the flow-independent control of Nodal signaling (Figs. 1, 3).
In wild-type embryos, spaw expression is initiated bilaterally in the cells that surround the LRO by the six somites stage (Fig. 4a). As development proceeds, spaw LRO levels increase until at around the 12 somites stage expression also becomes detectable in the left LPM where the ligand then propagates through autoinduction to reach more anterior target tissues (Fig. 4a). Analysis of spaw expression in MZ myo1g mutants revealed a temporal delay in the emergence of spaw expression at the level of the LRO and the subsequent propagation to the LPM (Fig. 4a). In contrast to the loss of myo1g function, a lack of LRO flow upon depletion of dnaaf1 is without effect on the initial induction of spaw expression at the LRO (Supplementary Fig. 7a).

a Time-course analysis of spaw expression indicates that initiation of spaw expression at the LRO and propagation to the Left LPM (black arrowhead) are delayed in MZ myo1g mutants. Vegetal views of the LRO region, anterior up. b, b′ qPCR analysis of spaw expression confirms that spaw levels are significantly reduced in MZ myo1g mutants (b). Conversely, spaw expression increases in Maternal Zygotic myo1cb (MZ myo1cb) mutants (b′, see also Supplementary Fig. 7b). c MZ myo1g mutants present a significantly reduced (p = 2E-05) rate of anterior-ward propagation of lft1 expression in the notochord (see also Supplementary Fig. 9a). d, e MZ myo1g mutants display a weaker induction of the nodal target gene lft1 in response to Spaw overexpression. d Animal pole views of germ ring stage embryos. While high amounts (20 pg) of Spaw RNA induce a similar lft1 induction in MZ myo1g mutants and wild-type controls, myo1g-deficient embryos present a reduced response to moderate amounts (10 pg) of Spaw RNA (arrow indicates ectopic expression, see Supplementary Fig. 9b for quantification). e qPCR analysis confirms that equal amounts of Spaw RNA induce a reduced lft1 induction response in MZ myo1g mutants compared to WT Control embryos from the same genetic background. f Conversely, the overexpression of Myo1G potentiates the capacity of low amounts (5 pg) of Spaw RNA to induce ectopic lft1. Scale bars: 100 µm. Box plots in b, b′, c, e, f indicate mean values ± SD. All p values were obtained using non-directional statistical tests. Complete numerical and statistical information for all experiments are provided in the Source Data files.
The observation that MZ myo1g mutants present a reduced spaw expression at the LRO was confirmed by quantitative qRT-PCR (Fig. 4b). Studies in Drosophila and zebrafish revealed that Myosin1C (Myo1C) proteins can act as Myo1D/G antagonists5,44. Although our analysis failed to reveal any morphological LR asymmetry defects in Maternal Zygotic myo1Cb mutants, gene expression analysis uncovered a mild upregulation of spaw at the LRO (Fig. 4b’ and Supplementary Fig. 7b), supporting the functional relationship between Myo1D/G agonists and their Myo1Cb antagonist.
myosin1g is dispensable for left-right organizer formation
The finding that zebrafish myo1d is required for LRO morphogenesis5,6 raises the question of whether the loss of myo1g may similarly cause general defects in LRO morphogenesis that would ultimately result in reduced Nodal signaling at the LRO. Our analysis of different markers genes involved in LRO specification and function does, however, not support this hypothesis. Analysis of the endodermal markers sox17 and sox32 indicates that the specification and clustering of LRO precursor cells occur normally in MZ myo1g mutants (Supplementary Fig. 8a, b). In accordance with the fact that myo1g controls LR asymmetry independently of the LRO flow, myo1g loss of function has no effect on the expression of the ciliary motility genes foxj1a, dnah9, and odad1 (Supplementary Fig. 8c–e).
Myosin1G promotes Nodal signaling
In mice and zebrafish, Nodal expression at the LRO is initially induced by Notch signaling45, and then further upregulated through the capacity of Nodal ligands to induce their own expression46. While the analysis of the Notch target genes her4.1 and her15.1 suggests that MZ myo1g mutants present normal Notch signaling levels (Supplementary Fig. 8f, g), the reduced expression of spaw at the LRO (Fig. 4a) is similar to the one reported in animals mutant for spaw itself46.
Following the initiation of spaw expression, first at the level of the LRO and subsequently in the LPM, spaw propagates anteriorly through autoinduction. This posterior-to-anterior propagation is accompanied by the progressive induction of the nodal feedback-antagonist lft1 at the notochordal midline barrier32,33,46. Our data show that in addition to the delayed expression of spaw at the LRO and in the LLPM (Fig. 4a), MZ myo1g mutants present a nearly two-fold reduction in the rate of anterior propagation of notochordal lft1 expression (Fig. 4c and Supplementary Fig. 9a).
As Nodal ligands amplify their own expression by autoinduction as soon as they start to be expressed23, it is not possible to determine if the delays that we observe in spaw and lft1 expression are due to a defect in the initial induction of spaw, to a subsequent defect in spaw autoinduction or to a combination of the two. To circumvent this problem, we took advantage of germ ring stage embryos, which lack endogenous spaw, to introduce defined amounts of spaw RNA by microinjection and compare nodal target gene induction in WT control and MZ myo1g mutant animals. While high doses (20 pg) of Spaw readily induce ectopic lft1 expression in both WT and MZ myo1g mutants (Fig. 4d and Supplementary Fig. 9b), mutant animals present a reduced response to moderate (10 pg) doses of Spaw RNA (Fig. 4d and Supplementary Fig. 9b). Analysis of lft1 expression by quantitative RT-PCR reveals that while Spaw is still able to significantly induce lft1 in MZ myo1g mutants, the observed effect is weaker than in homozygous WT sibling controls (Fig. 4e, Cohen’s d effect size = 1.27 for MZ myo1g mutants versus 4.48 for WT controls).
Taken together, our observations suggest that Myo1G, while not strictly required for Spaw signal transduction, is essential to promote full-strength Nodal signaling. Injecting wild-type Myo1G RNA into MZ myo1g mutants significantly rescues the capacity of Spaw to induce lft1 expression, demonstrating the specificity of the observed effect (Supplementary Fig. 9c). To confirm that Myo1G promotes Spaw signaling, a lower amount of Spaw RNA (5 pg), that is on its own barely capable of inducing ectopic lft1 expression, was co-injected with wild-type Myo1G RNA into WT animals. qRT-PCR analysis shows that Myo1G overexpression promotes the capacity of this subliminal amount of Spaw to induce lft1 expression (Fig. 4f).
Myosin1G function in Southpaw-independent Nodal signaling
The observation that germ ring stage myo1g-deficient animals display a reduced induction of the nodal target gene lft1 in response to ectopic Spaw (Fig. 4d, e) raises the question whether Myo1G might also contribute to signaling mediated by cyclops (cyc) and squint (sqt), the two zebrafish nodal ligands that are responsible for the endogenous expression of lft1 in blastula/gastrula stage embryos.
The same experimental setup that has allowed us to show that Myo1G enhances Spaw activity (Fig. 4d and Supplementary Fig. 9b) was used to test if Myo1G also enhances Cyc / Sqt signaling. Our experiments show that while the injection of high doses of cyc or sqt RNA is sufficient to induce ectopic lft1 expression in WT as well as MZ myo1g mutant animals, mutant animals present a reduced response to moderate doses of cyc/sqt RNA (Fig. 5a, b). These observations indicate that, as already observed for Spaw, Myo1G is not strictly required for signaling by Cyc and Sqt but enhances the response to the ectopic expression of these Nodal ligands.

a, b MZ myo1g mutants display reduced lft1 induction in response to ectopic expression of the Nodal ligands cyclops (cyc, a) and squint (sqt, b). Animal pole views of germ ring stage embryos, arrows indicate patches of ectopic lft1 expression. c qPCR indicates that MZ myo1g mutants present a mild decrease in the endogenous expression levels of the Nodal target gene lft1. Box plots in c indicate mean values ± SD. d, e MZ myo1g mutants present a reduced immunoreactivity for activated Phospho-SMAD2/3 (n = 25 WT and 31 MZ myo1g mutant embryos from 2 independent experiments). Animal pole views of germ ring stage embryos. Scale bar: 100 µm. f Morpholino knockdown of cyc and sqt (MO cyc + MO sqt) elicits stronger Nodal loss of function phenotypes in MZ myo1g mutants than in WT controls. Nodal loss of function phenotypes at 32 hpf were categorized into five classes: class I (partial cyclopia), class II (complete cyclopia), class III (partial loss of the notochord), class IV (complete loss of the notochord), and class V (loss of posterior neural structures). All p values were obtained using non-directional statistical tests. Complete numerical and statistical information are provided in the Source Data files.
In the context of LR asymmetry, MZ myo1g mutants present a number of molecular and morphological phenotypes that are compatible with a reduction in Spaw-mediated Nodal signaling (Figs. 1, 2). In contrast, examination of MZ myo1g mutants using molecular markers (e.g., gastrulation stage endodermal sox17 expression, Supplementary Fig. 8a) or morphological examination failed to reveal any of the diagnostic phenotypes that are caused by defects in cyc/sqt-mediated germ layer specification (cyclopia or loss of axial structures, Fig. 5f)40,47,48. In spite of this lack of morphological phenotypes, germ ring stage MZ myo1g mutants present a partial reduction of lft1 expression that is detectable by qPCR (Fig. 5c) and a diminished immunoreactivity for activated Phospho-SMAD2/3 (Fig. 5d, e). These observations suggest that Myo1G does contribute to endogenous blastula stage cyc/sqt signaling, but that it’s importance for this process is only minor and myo1g loss of function therefore not associated with visible defects in Nodal-dependent germ layer specification. In accordance with a minor contribution of Myo1G to early cyc/sqt signaling, we observe a weak enhancement of the Nodal loss of function phenotypes that are observed upon partial cyc/sqt morpholino knock-down in MZ myo1g mutants compared to WT controls (Fig. 5f).
Taken together, our experiments indicate that in spite of its ability to promote cyc/sqt signaling (Fig. 5a, b) Myo1G is only of minor importance for Nodal signaling during the early blastula/gastrula period. In contrast, our analysis shows that by the end of gastrulation (bud stage), MZ myo1g mutants present a clear reduction of lft1 which is at this stage expressed together with cyc at the level of future LRO (Fig. 6a). This phenotype is observed more than 2 h before the onset of the expression of spaw itself, suggesting thereby that MZ myo1g mutants present a defect in cyc signaling/autoinduction that becomes detectable by the end of gastrulation.

a MZ myo1g mutants present a reduced expression of the Nodal ligand cyc and the Nodal feedback antagonist lft1 in the LRO/tail bud region of bud stage embryos. Mutant embryos also present a reduced expression of the Nodal ligand gdf3 in the eight-somites stage LRO. b MZ myo1g mutants display a reduced expression of the Nodal signaling antagonist dand5 from the bud stage onwards. a, b show vegetal views of the LRO/tail bud region, anterior up. c Eight-somites stage MZ myo1g mutants present a reduction in the antero-posterior extension of lft1 expression in the anterior brain. Dorsal views of the brain, anterior up. Box plots in c indicate mean values ± SD. Scale bars: a 50 µm, b, c 100 µm. All p values were obtained using non-directional statistical tests. Complete numerical and statistical information are provided in the Source Data files.
In addition to the effect observed on lft1 and cyc, MZ myo1g mutants present a reduced expression of the Cerberbus/Dan family member dand5 that antagonizes spaw signaling at the LRO24 (Fig. 6b) as well as the TGFβ superfamily member gdf3 that represents an obligate heterodimerization partner of different Nodal ligands, including Spaw49 (Fig. 6a). It is important to note that markers of LRO specification (sox17, sox32, foxj1a), LRO differentiation (dnah9, odad1), or other LRO signaling pathways (her4.1, her15.1) remain expressed normally (Supplementary Fig. 8), arguing that the observed defects in nodal pathway gene expression reflect a specific impairment in Nodal signaling and autoinduction rather than a general problem of LRO morphogenesis. In accordance with this hypothesis, morpholino knock-down of cyc or sqt or the genetic inactivation of spaw itself similarly cause a partial loss of gdf3 expression at the LRO (Supplementary Fig. 10).
Importantly our experiments indicate that Myo1G regulates Nodal signaling not only in the context of LR asymmetry, as eight-somites stage MZ myo1g mutants also present a reduced expression of lft1 in the anterior brain, which lacks spaw but expresses cyc39,40 (Fig. 6c). Taken together, our findings suggest that while Myo1G is essential for the Spaw-mediated establishment of zebrafish LR asymmetry, it also contributes to signaling by other Nodal ligands in specific biological contexts.
Myosin1G regulates activin receptor trafficking
How does Myo1G promote Spaw signaling? Proteomic studies identified Myo1G on exosomes, suggesting that this factor may be implicated in exovesicular secretion50. To determine if Myo1G is required for Spaw ligand secretion, we took advantage of a functional GFP-Spaw fusion construct that has been previously used to visualize Spaw secretion43. GFP-Spaw RNA injection into wild-type or MZ myo1g mutant animals results in similar labeling of the extracellular space (Fig. 7a, b). For quantitative assessment of Spaw-GFP levels, Spaw-GFP RNA was was co-injected with a Histone2B-RFP RNA that enabled us to estimate the amount of injected material received by individual embryos. Quantification of Spaw levels relative to the Histone2B tracer revealed that while MZ myo1g mutants present a minor reduction of normalized Spaw-GFP expression levels, this reduction is not statistically significant, suggesting that Myo1G has no major effect on Spaw ligand production and secretion (Supplementary Fig. 11a).

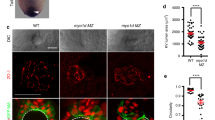
a, b Spaw-GFP localization is similar in WT (n = 20) and MZ myo1g mutants (n = 17). H2B-RFP was injected as a tracer to ascertain that embryos that had received equal amounts of RNA (see Supplementary Fig. 11a for quantification). c A constitutively activated form of the Nodal signal transducer SMAD2 (CA-SMAD2) elicits similar responses in WT and MZ myo1g mutants. Box plots in c indicate mean values ± SD. d Myo1G-GFP is detected at the cell cortex and in intracellular compartments (n = 10). e, e’, e” Myo1G-GFP is present on endosomes positive for the TGFβ signaling adapter SARA (n = 24, see also Supplementary Fig. 12a, b). f–j MZ myo1g mutants present a reduced number of endosomes positive for the Nodal receptors Acvr2Aa-GFP (f, g, j, n = 13 WT and 13 MZ myo1g mutant embryos) and Acvr2Ba-GFP (h–j, n = 14 WT and 13 MZ myo1g mutant embryos). In j data points represent the mean number of endosomes per cell for a particular embryo and lines indicate the overall mean ± SEM. k, l MZ myo1g mutants and WT siblings present a similar number of CD44a-positive endosomes (n = 7 WT and n = 7 MZ myo1g mutant embryos, see Supplementary Fig. 11b for quantification). a, b, d–i, k, l, animal pole views, germ ring stage. Scale bars: 10 µm. All p values were obtained using non-directional statistical tests. Complete numerical and statistical information are provided in the Source Data files.
While cytoplasmic Myosin1 proteins exert important roles in membrane trafficking51, nuclear isoforms of mammalian Myo1C can regulate TGFβ-responsive gene expression52. To determine if Myo1G controls the SMAD-mediated transcriptional downstream response to Spaw signaling, we injected RNA encoding a Constitutively Activated variant of SMAD2 (CA-SMAD2) into wild-type and MZ myo1g mutant animals and analyzed the effect on lft1 target gene induction by qRT-PCR. In contrast to the reduced induction of lft1 that is observed upon Spaw overexpression in MZ myo1g mutants (Fig. 4e), CA-SMAD2 elicited a similar induction of lft1 expression in myo1g-deficient animals (Fig. 7c, Cohen’s d effect size = 3.39 for MZ myo1g mutants versus 3.62 for WT controls).
The pharmacological Myosin antagonist Pentachloropseudilin (PCIP) inhibits TGFβ signaling by regulating the membrane trafficking of TGFβ type II receptors53. In accordance with a potential function in membrane trafficking, Myo1G-GFP localizes to both the cell cortex and to intracellular, potentially endosomal, compartments (Fig. 7d). The endosomal protein SMAD Anchor for Receptor Activation (SARA) has been shown to promote the activation of the Nodal signal transducers SMAD2 and 329. We, therefore, investigated if Myo1G-GFP positive intracellular compartments correspond to SARA endosomes. Strikingly, the use of an established mRFP-SARA construct54 revealed that in 24/24 embryos, SARA-positive compartments were always associated with Myo1G-GFP. Both standard laser scanning microscopy (Fig. 7e) and Airyscan super-resolution microscopy (Supplementary Fig. 12a, b) revealed that SARA-positive compartments are often part of larger, Myo1G-positive structures, in accordance with a potential role of Myo1G in the biology of TGFβ signaling endosomes.
In murine lymphocytes, Myo1G regulates the endocytic trafficking of the adhesion protein CD4455, a molecule that has, in other biological contexts, been shown to regulate TGFβ signaling by acting as Hyaluronan receptor56. In contrast to the situation described in the mouse immune system, our observations indicate that MZ myo1g mutants and WT controls have a similar number of CD44 endosomes (Fig. 7k, l and Supplementary Fig. 11b), suggesting thereby that Myo1G only regulates CD44 trafficking in specific biological contexts.
In the light of previous work linking Myosin1 activity to TGFβ type II receptor trafficking53, we next investigated the importance of Myo1G for the trafficking of Activin type II receptors. The zebrafish genome harbors four AcvrII receptor genes (acvrIIaa, acvrIIab, acvrIIba, acvrIIbb). Our analysis revealed that AcvrIIAa-GFP, AcvrIIBa-GFP, and AcvrIIBb-HA fusions all localize to the cell cortex and to intracellular compartments, similar to the localization pattern observed for Myo1G itself (Fig. 7d–i and Supplementary Fig. 11c). A completely different localization pattern was observed for AcvrIIAb, with similar behavior observed using a GFP-fusion generated in the course of the present work or a previously reported AcvrIIAb-HA57 construct (Supplementary Fig. 11d, e).
To study the potential impact of Myo1G on Activin receptor trafficking, we took advantage of our AcvrIIAa-GFP and AcvrIIBa-GFP constructs to quantify the number of AcvrII endosomes in MZ myo1g mutant and WT sibling embryos. Co-injection of a Histone2B-RFP construct was used to ensure that AcvrII endosomes were counted in animals that received comparable amounts of injected material. A first set of quantifications was carried out in blastoderm cells at the germ ring stage (onset of gastrulation, 5.5 h post fertilization). In accordance with a potential role of Myo1G in regulating AcvrII trafficking, our experiments reveal that MZ myo1g mutants present a significantly reduced number of AcvrIIAa and AcvrIIBa endosomes (Fig. 7f–j).
The observations that MZ myo1g mutants present a reduced number of AcvrII endosomes (Fig. 7f–j) and that Myo1G is found on SARA-positive compartments (Fig. 7e and Supplementary Fig. 12a, b) raise the question of whether Myo1G may be required for the formation of AcvrII/SARA-positive endosomes. Accordingly, MZ myo1g mutants present a reduction in the absolute number of SARA/AcvrIIAa-positive compartments and a higher fraction of AcvrIIAa compartments that lack the signaling endosome marker SARA (Fig. 8a–d). While the absolute numbers of endosomes observed at the germ ring and 12 somites stage cannot be compared due to technical reasons (see methods), a significant reduction of AcvrIIAa + SARA double-positive endosomes is observed in MZ myo1g mutants compared to WT controls both at the germ ring stage when early signaling by the Nodal ligands Cyclops and Squint regulates germ layer specification (Fig. 8a–c) and at the 12 somites stage (15 hpf) in the lateral plate mesoderm, when Spaw-mediated Nodal signaling controls LR asymmetry (Fig. 8d and Supplementary Fig. 12c, d).

a, b MZ myo1g mutants present an increased number of SARA-negative AcvrIIAa endosomes (white arrowheads). Animal pole views of germ ring stage blastoderm cells. Scale bars: 10 µm. c, d Quantifications of AcvrIIAa/SARA-positive endosomes in WT control and MZ myo1g mutant cells in the germ ring stage blastoderm (c) and the 12 somites stage Lateral Plate Mesoderm (d, see also Supplementary Fig. 12c, d). Left panels: Mean number of AcvrIIAa-GFP and mRFP-SARA double-positive endosomes per cell. Middle panels: Percentage of the total number of AcvrIIAa endosomes per cell that are SARA-negative. Right panels: Number of SARA endosomes per cell. The absolute number of AcvrIIAa+SARA double-positive endosomes decreases and the fraction of SARA-negative AcvrIIAa endosomes increases both in the early blastoderm (c) and later lateral plate mesoderm (d). In contrast, the mean number of SARA endosomes per cell remains unchanged in MZ myo1g mutants at the early germ ring stage while being significantly decreased at 12 somites. In c, d data points represent the mean number of endosomes per cell for a particular embryo and lines indicate the overall mean ± SEM. e Eight-somites stage SARA morphants (MO sara) present a reduction in the antero-posterior extension of forebrain lft1 expression. Dorsal views of the brain, anterior up. Scale bar: 100 µm. Box plots in e indicate mean values ± SD. All p values were obtained using non-directional statistical tests. Complete numerical and statistical information for all experiments are provided in the Source Data files.
Interestingly, our analysis suggests that MZ myo1g mutants present stage-specific defects in SARA endosome dynamics: While myo1g loss of function has no effect on the absolute number of SARA-positive endosomes in blastoderm cells at the germ ring stage (Fig. 8c), MZ myo1g mutants present a significant reduction in the number of SARA endosomes that are present in the 12 somites stage LPM (Fig. 8d). The observation that the specific decrease of SARA endosomes at later developmental timepoints is also observed if mRFP-SARA is injected alone without AcvrIIAa-GFP (Supplementary Fig. 12e) suggests that this phenotype is directly caused by the loss of function of Myo1G rather than being an indirect consequence of impaired AcvrIIAa-GFP trafficking.
SARA contributes to context-dependent Nodal signaling
The observations that Myo1G is dispensable for early Nodal signaling during germ layer specification but required for later Nodal signaling in LR asymmetry and that myo1g loss of function causes a specific reduction in the number of SARA endosomes at later developmental stages raises the question of whether SARA may itself contribute to the regulation of Nodal signaling in specific developmental contexts. To address this question, we took advantage of a knock-down strategy that has been validated in previous work54 in which both maternal and zygotic SARA functions are inhibited through the co-injection of one translation-blocking and one splice-blocking antisense morpholino oligonucleotide.
In accordance with previously reported phenotypes54 and similar to MZ myo1g mutants, SARA morphant embryos failed to display morphological defects indicative of an impairment in Nodal-dependent germ layer specification. Specific examination of LR asymmetry revealed, however, partial but significant defects in cardiac jogging (Supplementary Fig. 13a). While cardiac jogging is impaired in MZ myo1g mutants and SARA morphants, it is important to note that the jogging defects that are observed in the two types of animals are distinct: SARA morphants present a predominant occurrence of right-ward cardiac jogging (Supplementary Fig. 13a) while the defect most frequently observed in MZ myo1g mutants is a lack of jogging that leaves the heart in the middle (Fig. 1a). The occurrence of distinct laterality phenotypes was confirmed through the examination of spaw expression in the lateral plate mesoderm, with a reduction in left-sided spaw expression predominating in MZ myo1g mutants (Supplementary Fig. 3a) while SARA morphants frequently display bilateral spaw expression (Supplementary Fig. 13b).
Bilateral spaw expression can be indicative of defects in the formation of a functional midline barrier that restrains Spaw propagation32,33. To investigate the status of midline structures in SARA morphants, we performed in situ hybridizations against foxa1 which is normally expressed in midline cells of the floorplate and hypochord (located respectively above and below the notochord). SARA morphants present discontinuities in the hypochord (red arrowheads in Supplementary Fig. 13d) as well ectopic foxa1 expression in the notochord (green arrowheads in Supplementary Fig. 13d) that is normally devoid of staining (Supplementary Fig. 13c), indicating a defect in midline cell fate specification.
Although SARA was initially identified as a TGFβ signaling adapter29, subsequent studies revealed an additional function of SARA in promoting Notch signaling during spinal cord development54. As Notch signaling is also required for the specification of midline cell fates (including hypochord development)58,59 we investigated the status of Notch signaling in SARA morphants and observed a marked downregulation of the Notch target genes her4.1 and her15.1 (Supplementary Fig. 13e–h).
Our observations suggest, therefore, that, through its function in Notch signaling54, SARA contributes to the establishment of a functional midline barrier. As the midline barrier acts as a natural brake for Nodal signaling, the midline function of SARA prevented us from addressing its specific contribution to Myo1G-dependent Spaw signaling. While the observations that SARA morpholino injection enhances the defects of MZ myo1g mutants that are observed with respect to cardiac jogging (Supplementary Fig. 13i) and spaw expression (Supplementary Fig. 13j) are in potential accordance with a contribution of SARA to Myo1G/Spaw signaling, establishing such a function unambiguously would require to specifically inhibit only Notch-independent SARA activities.
To circumvent this problem, we analyzed the effect of SARA loss of function on the forebrain expression of lft1, which occurs in response to Myo1G-dependent Nodal signaling (Fig. 6c) and is independent of the more posteriorly located midline barrier. Similarly to MZ myo1g mutants, SARA morphants present a significantly reduced antero-posterior extension of the lft1 expression domain (Fig. 8e). Taken together, our findings suggest that SARA acts, like Myo1G, to promote Nodal signaling in specific biological contexts.
Discussion
A striking feature of LR asymmetry is that different species use seemingly distinct mechanisms for the determination of this body axis3. Only recently has the unconventional type I Myosin Myo1D, which was initially identified as a regulator of Drosophila laterality34,35, been identified as an evolutionarily conserved regulator of animal LR asymmetry4,5,6,7. While studies in fish and frogs uncovered an essential role of Myo1D in LRO morphogenesis4,5,6, several observations suggest that additional functions of Myosin1 proteins in LR asymmetry remain to be uncovered. First, previous studies had identified a function of Myo1D in the central LRO of fish and frogs, a biological structure that has no equivalent in Drosophila, where Myo1D ensures a local, organ-specific control of chiral morphogenesis. Second, in contrast to the unique myo1d gene present in flies, vertebrate genomes harbor not only myo1d, but also the closely related myo1g. We present an analysis of the function of this second myosin1 homolog in zebrafish and uncover an essential function of this gene in chiral morphogenesis that is distinct from the one exerted by myo1d4,5,6.
Myo1D controls the symmetry-breaking ciliary fluid flow in the central LRO4,5,6. Accordingly, myo1d loss of function causes defects in all lateralized organs4,5,6. In contrast, myo1g is required for the chiral morphogenesis of the heart and brain, but dispensable for visceral laterality (Fig. 1 and Supplementary Fig. 2). To specifically determine if Myo1G exerts an LRO flow-independent function, we inactivated myo1g in the context of animals that lack the ciliary motility gene dnaaf1 and therefore have no LRO flow. Lack of a LRO flow in dnaaf1 mutants causes a distinctive randomization of LR asymmetry, in which the heart, brain, and viscera develop either normally or as their mirror image. In contrast, a different phenotype is observed in dnaaf1 MZ myo1g double (or dnaaf1 MZ myo1d MZ myo1g triple) mutants where cardiac and brain laterality are altogether lost (Fig. 1). These findings establish an essential role of Myo1G in the flow-independent, tissue-specific control of LR asymmetry.
How does Myo1G exert this tissue-specific control? Myo1G could be involved in the local, organ-specific execution of chiral morphogenesis, like Drosophila myo1d18,35. Alternatively, Myo1G could be involved in the transmission of laterality information from the central LRO to different target tissues. While our experiments do not rule out the first possibility, MZ myo1g mutants present a reduced propagation of the Nodal ligand spaw and a reduction in the expression of different Nodal target genes (Fig. 2 and Supplementary Fig. 3). Our observations that residual spaw expression still reaches the posterior visceral primordium in MZ myo1g mutants (Supplementary Fig. 6), but fails to reach the more anterior cardiac territory (Fig. 3c, c’ and Supplementary Fig. 5) provides a potential explanation for the tissue-specificity of the observed laterality phenotypes. It is noteworthy that similar organ-specific laterality phenotypes are observed in zebrafish furin mutants that present a reduced Nodal signaling range due to impaired Nodal proprotein maturation43.
The conclusion that Myo1G is important for the Spaw-mediated control of LR asymmetry is further supported by the observation of similar cardiac laterality defects in MZ spaw single and MZ spaw MZ myo1g double mutants (Supplementary Fig. 4c). The observation that MZ myo1g mutants can be rescued through Spaw or FurinA overexpression (Fig. 2e, f) shows that Nodal signaling is reduced but not abolished in these animals. Quantitative analysis of Spaw-dependent gene induction indeed reveals that the responsiveness to Nodal ligands is reduced in MZ myo1g mutant embryos (Fig. 4).
In addition to the specific function of Spaw in LR asymmetry, two other zebrafish Nodal ligands, Cyc and Sqt play major roles in germ layer specification and patterning40,47,48. Although MZ myo1g mutants show a reduced response to ectopic Cyc/Sqt expression (Fig. 5a, b), they do not display phenotypes indicative of defects in Cyc/Sqt-dependent germ layer specification (Fig. 5f). MZ myo1g mutants do however display a reduced expression of Nodal target genes in different biological contexts at later developmental stages. Importantly a number of defects are observed well before the onset of spaw expression, e.g. in the tail bud region by the end of gastrulation (Fig. 6a, b), or in tissues that do not express spaw altogether (8 somites stage forebrain, Fig. 6c) demonstrating thereby that Myo1G is important for the context-dependent regulation of signaling by different Nodal ligands.
How does Myo1G control Nodal signaling? Myo1G localizes to the cellular cortex and to intracellular compartments that our experiments show to be positive for the endosomal TGFβ signaling adapter SARA29 (Fig. 7d, e), suggesting thereby that Myo1G may regulate the endosomal trafficking of Nodal pathway components. In accordance with this hypothesis, MZ myo1g mutants present a reduction in the number of endosomes that are positive for both SARA and Activin type II receptor (AcvrII) molecules (Figs. 7, 8).
How could the interplay between Myo1G, SARA, and AcvrII exert a context-specific regulation of Nodal signaling? While we cannot provide a definitive answer to this question at the present moment, we envisage two major hypotheses: On one hand, early and late nodal signaling may require different receptor complexes. Genetic studies of zebrafish AcvrII function have indeed revealed that AcvrII receptors are dispensable for early Nodal signaling in germ layer specification60, a finding that is in perfect accordance with our observation that Myo1G, which controls AcvrII trafficking, is itself not important for early Cyc/Sqt signaling. An alternative explanation for the occurrence of late but not early Nodal signaling phenotypes in MZ myo1g mutants could be that Nodal-related membrane trafficking and signaling are regulated differently according to the biological context. Our observation that MZ myo1g mutants present a reduction in the number of SARA endosomes in the 12 somites stage LPM but not in the germ ring stage blastoderm (Fig. 8c, dand Supplementary Fig. 12e) does indeed point to this direction. Interestingly, SARA and Myo1G are both dispensable for Nodal-dependent early germ layer specification (ref. 54 and Fig. 5f) but required to promote the expression of the nodal target gene lft1 during later forebrain development (Figs. 6c, 8e).
Taken together, our findings identify Myo1G as a context-dependent regulator of Nodal signaling whose function is specifically required for LR asymmetry. Investigating the molecular mechanism that underlies this context-dependent regulation will be an interesting aim for future studies. Finally, our work establishes a link between unconventional type 1 Myosins that are emerging as major regulators of animal laterality, and Nodal signaling, which has long been known to be the key pathway regulating vertebrate LR asymmetry.
Methods
Use of research animals
Zebrafish experiments have been performed in accordance with animal welfare guidelines in the iBV zebrafish facility (authorization #C06-088-17) in the context of the authorized animal experimentation projects APAFIS#5521-201605111041958v6 and APAFIS#15157-201805012112438v2, approved by the animal experimentation ethical committee Ciepal Azur.
Zebrafish strains and embryo maintenance
Embryos were raised in 0.3X Danieau medium (17.4 mM NaCl, 0.21 mM KCl, 0.12 mM MgSO4, 0.18 mM Ca (NO3)2, 1.5 mM Hepes, pH 7.6) at 28.5 °C and staged according to standard criteria61. If necessary, 1-phenyl-2-thiourea (Sigma) was added at 30 mg/l to prevent embryonic pigmentation.
myo1d inactivations were performed using the previously reported myo1dtj16b or myo1dtj16c alleles5. With the exception of Fig. 2b, myo1g inactivations were performed using the previously reported myo1gtj18b allele5. The analysis of spaw expression in the LLPM (Fig. 2b) additionally includes animals carrying the myo1gtj18c and myo1gtj18e alleles described below. Details concerning the mutant alleles used in different experiments are in the Source data files. All presented data were obtained using Maternal Zygotic (MZ) MZ myo1d or MZ myo1g single or MZ myo1d MZ myo1g double mutants.
Allele-specific PCR was used to identify the WT myo1d allele (forward primer 5′-AGAGTGGAGCTGGAAAAACAGA-3′, reverse primer 5′-CCCATCCCTCGTGTGAAACTAAATCAC-3′, 339 bp amplicon) as well as the mutant alleles tj16b (forward primer 5′-TGGAGCTGGAAAAAGGCTCGT-3′, reverse primer 5′-CCATCACTGCAGCAGAAATGAGAG-3′, 133 bp amplicon) and tj16c (forward primer 5′-GTGGAGCTGGAAAAAGGCTATAC-3′, reverse primer 5′-CCATCACTGCAGCAGAAATGAGAG-3′, 145 bp amplicon).
The allele-specific reverse primers 5′-TCTCATACAGTTCTCTTCCCCTAG-3′ (tj18b, 115 bp amplicon), 5′-GAGGTGGATTCTCATACAGTTCTCTTCCTCAA-3′ (tj18c, 120 bp amplicon), 5′-TCTTCCCTCGGATGTCTTCC-3′ (tj18e, 115 bp amplicon) and 5′- CTCATACAGTTCTCTTCCCCTGTAG-3′ (WT, 120 bp amplicon) were used with the generic forward primer 5′-GAGAAGAGTCGTATCTACACCTTC-3′ to genotype myo1g mutant fish. The myo1gtj18c and myo1gtj18e alleles both cause frame shift mutations after amino acid 64 due to a 10 bp deletion (CTACAGGGGA, myo1gtj18c) or a 6 bp deletion/13 bp insertion (CTACAG = > GGAAGACATCCGA, myo1gtj18e) in the myo1g open reading frame.
myo1Cb inactivation was performed using the myo1Cbsa16637 allele from the Zebrafish Mutation Project (http://www.sanger.ac.uk/resources/zebrafish/zmp/) obtained from the Zebrafish International Resource Center. The sa16637 allele introduces a premature stop codon at the 228th amino acid position. A generic forward primer 5′-GTCACATCCTGAACTACCTGCTAG-3′ was used along with a mutant-specific reverse primer 5′-TATTACCAGTATCTGGTCAAG-3′ (164 bp amplicon) to identify the mutant allele. The WT allele was identified using the generic forward primer with the WT-specific reverse primer 5′- CAGTACCAGTATCTGGTCAAG-3′ (164 bp amplicon).
dnaaf1 was inactivated using the dnaaf1tm317b mutant allele41. The forward primer 5′- GCAAGCTTTGCACGCTTAATGTCTC −3′ and reverse primer 5′ - AACACTGGAGAATGTTTGTGAC − 3′ were used to amplify the tm317b mutant allele (199 bp amplicon). The dnaaf1 WT allele was identified using the forward primer 5′-GCAAGCTTTGCACGCTTAATGTCTC-3′ and reverse primer 5′-CACACTGGAGAATGTTTGTGAC-3′ (199 bp amplicon). Beyond 24 h of development, dnaaf1 mutants can be identified through the oval phenotype that is diagnostic for ciliary mutations.
spaw was inactivated using the spaws457 allele62. The alternative forward primers 5′-AACCTCTCCATTCGCAAA-3′ (recognizing the s457 allele) 5′-AACCTCTCCATTCGCAAT-3′ (recognizing the WT allele) were used together with the generic reverse primer 5′-GGATAAAACTGGCTGGAG-3′ to yield 156 bp amplicons.
Plasmid generation
The myo1g ORF was amplified from a mixed stage pool of cDNAs using primers 5′-GATCCCATCGATTCGATGGCGGAGCTGGAGGGCTTG-3′ and 5′-AGGCTCGAGAGGCCTTACTGGGGCAGGAGTAAGG-3′ and cloned into the pCS2+ vector using Gibson assembly mix (NEB). Bold letters in the primer sequences indicate Gibson overhangs that are also present in the pCS2+ sequence. For generating the myo1g-GFP construct, the myo1g ORF was amplified from the myo1g-pCS2 construct using the primer pair
5′-GCAGGATCCCATCGATTCGACAGTAAACATGGCGGAGCTGGAGGGCTTG-3′ and
5′-ACCATGGACCCTCCGCTGGTGCCCTGGGGCAGGAGTAAGGTAAATC-3′, and was ligated onto GFP-pCS2 + . CD44a was amplified using the primers 5′-ATCCCATCGATTCGACAGTAAACATGTGGACTTTGTTATTTGTAGTGTT-3′ and 5′- ACCATGGACCCTCCGCTGGTGCCCATTAAATATTCTTTTTCGTGTTCA-3′ and ligated into GFP-pCS2 + . For the cloning of different AcvrII-GFP constructs, the following primers were used: Acvr2IIAa 5′-GGATCCCATCGATTCGACAGTAAACATGGGACCTGCAACAAAGCTGGC-3′ (forward) and
5′-ACCATGGACCCTCCGCTGGTGCCTAGACTAGACTCCTTTGGGGGATA-3′ (reverse), AcvrIIAb 5′-GGATCCCATCGATTCGACAGTAAACATGGTCAAGCAGGGCTGCTGGCT-3′ (forward) and 5′-TCACCATGGACCCTCCGCTGGTGCCTAGGCTGGACTCTTTAGGCGGGA-3′ (reverse), AcvrIIBa 5′- GATCCCATCGATTCGACAGTAAACATGTTCGCTTCTCTGCTCACTTTGG-3′ (forward) and 5′-ACCATGGACCCTCCGCTGGTGCCGATGCTGGACTCTTTGGGCGG-3′ (reverse) to amplify the ORFs, which were ligated into GFP-pCS2+. The her4.1-pBSK construct used to generate an in situ probe was cloned using the primer pair 5′- GTCGACGGTATCGATAAGCCACACAGCAATGACTCCTAC-3′ and
5′-CTAGAACTAGTGGATCCCCCTTAAGTCTACCAGGGTCTCC-3′. her15.1 was amplified using the forward primer 5′-GTCGACGGTATCGATAAGCGCTCAGAGAAACAGCATCTCTCC-3′ and reverse primer
5′-CTAGAACTAGTGGATCCCCCCTCCACAGGAGTTCAACATTGAC-3′ and cloned into pBSK.
RNA and morpholino injections
mRNAs were synthetized using the SP6 mMessage mMachine kit (Ambion). RNAs were diluted in 0.1 M KCl 0.2% Phenol Red. The following constructs and quantities were used: AcvrIIAa-GFP-pCS2+ (12.5/25 pg, this study), AcvrIIAb-GFP-pCS2+ (25 pg, this study), AcvrIIAb-HA-pCS2+ (25 pg57), AcvrIIBa-GFP-pCS2+ (12.5/25 pg, this study), AcvrIIBb-HA-pCS2+ (25 pg57), CA-SMAD2-pCS2+ (20 pg63), CD44a-GFP-pCS2+ (50 pg, this study), GFP-Spaw-pCS2+ (20 pg43), FurinA-pCS2+ (2.5 pg43), Histone2B-mRFP-pCS2+ (12.5 pg64), mRFP-SARA-pCS2+ (12.5/25 pg54), Myo1G-pCS2+ (50 pg, this study), Myo1G-GFP-pCS2+ (50 pg, this study). For Spaw65, Cyclops66, and Squint39, different concentrations used in individual experiments are indicated in the figures. For technical reasons, AcvrIIAa-GFP and mRFP-SARA were injected at 25 pg each for the analysis of Acvr/SARA endosomes at germ ring stages (Fig. 8c), while injection was cried out at 12.5 pg each for analysis in the 12 somites stage lateral plate mesoderm (Fig. 8d). Different concentrations were used at the two developmental stages to warrant sufficient labeling for imaging while at the same time preventing the occurrence of overexpression phenotypes. No attention should, therefore, be paid to the difference in the absolute number of endosomes that are observed at the two developmental stages.
The previously reported dnaaf1 Morpholino 5′-ATGCACTGTAATTTACCAAGTCAGG-3′36 was injected at a concentration of 500 µM. SARA knock-down was accomplished using a previously established combination54 of a translation-blocking morpholino (5′-CATGAAACTCCACCCTGCCAAGCGT-3′) and a splice-blocking morpholino (5′-TGAACTAGAGACTTTACCTTGCCAC-3′) injected at 150 µM each. cyclops (5′-GCGACTCCGAGCGTGTGCATGATG-3′) and squint (5′-ATGTCAAATCAAGGTAATAATCCAC-3′) were inhibited using previously validated morpholinos67. cyc and sqt morpholinos were injected either separately (cyc 64 µM, sqt 250 µM) or in combination (cyc 8 µM, sqt 31 µM). All morpholinos were diluted in 1x Danieau 0.2% Phenol Red.
For rescuing the cardiac jogging defects of myo1g mutant by Spaw mRNA injection, a mix of untagged Spaw RNA and GFP RNA was co-injected into one blastomere of two-cell stage embryos. At bud stage, embryos with a unilateral segregation of GFP expressing cells were selected using a fluorescent dissection scope (Leica M205FA), and grown further to score for cardiac jogging and looping phenotypes.
RNA in situ hybridization
Whole mount RNA in situ hybridizations were performed as previously described68. For the following genes probes were transcribed from previously reported plasmids: flh-pBSK69, spaw-pGEMT65, lefty1-pBSK31, lefty2-pBSK33, otx5-pBSK70, pitx2c-pBSK71, foxa1-pBSK72, cyclops-pBSK39, dand5-pBSK24, sox32-pBSK73, sox17-pBSK74, odad1-pME18S-FL368, dnah9-pCRII75, foxj1a-pBSK76, cmlc2/myl7-pCS277, gdf3-pBSK68, Her4.1-pBSK (this study), Her15.1-pBSK (this study). The elovl6 probe was transcribed from a PCR product containing a T7 promoter sequence at the 3′ end. elovl6 was amplified from genomic DNA using the forward primer 5′–CCCGTCCCATGTGCAGAACATTG–3′ and the reverse primer 5′–GGTGTCCATTGTGCTCGTGTGTCTCCCTATAGTGAGTCGTATTACGC– 3′.
qPCR analysis
qPCR was performed using PowerUP SYBR Green Master Mix (Applied Biosystems) in an Applied Biosystems Step-One PCR system. Individual reactions were performed in triplicates to account for pipetting errors. For sample preparation, whole cell mRNA was isolated from 50 embryos using TRI-Reagent (Sigma). Reverse transcription was performed on 2.5 µg of RNA using Superscript III (Invitrogen) to generate cDNA. Fold changes in gene expression were normalized to the internal control gene 36b4. The primers used for the amplification reactions are as follows: lefty1: forward 5′- AGAGGAGTTTGGGTCTAGTGG-3′, reverse 5′-TACGGAGAGAGGAAATGCG-3′. Spaw: forward 5′- TGACTTCGTCCTGAGCTTGA-3′, reverse 5′- TCAAGCTCAGGACGAAGTCA − 3′. 36b4: forward 5- ACGTGGAAGTCCAACTACT-3′, reverse: 5′- GTCAGATCCTCCTTGGTGA-3′. For estimating relative gene expression, the Ct values at 40 cycles of qPCR amplification were used according to the ΔΔCT method78. Individual data points in figures documenting qPCR experiments correspond to technical replicates. Complete statistical information, including numbers of biological and technical replicates, is provided in the Source Data files.
Immunocytochemistry
For AcvrII-HA antibody stainings, dechorionated embryos were fixed for 1.5 h at Room temperature in PEM (80 mM Sodium-Pipes, 5 mM EGTA, 1 mM MgCl2) - 4% PFA - 0.04% TritonX100 and then washed 2 × 5 min in PEMT (PEM - 0.2% TritonX100), 10 min in PEM 50 mM NH4Cl, 2 × 5 min in PEMT. Before incubation with primary antibody (Rat@HA, Roche 11867423001, used at 1:1000), embryos were blocked for a minimum of 2 h through preincubation in PEMT + 5% normal goat serum. Following primary antibody incubation, embryos were washed 5 – 10 – 4 × 15 min in PEMT before blocking again for a minimum of 2 h through preincubation in PEMT + 5% NGS, before incubation with the secondary antibody (Goat@Rat-Alexa488, Invitrogen A11006, used at 1:500). Following secondary antibody incubation, embryos were washed 5 – 10 – 4 × 15 min in PEMT.
For Phospho-SMAD2/3 antibody stainings, dechorionated embryos were fixed overnight at 4 °C in PBS − 4% PFA and then dehydrated through 5 min washes in PBS-25/50/75% Methanol before being stored in Methanol at −20 °C for at least 12 h. For immunostaining, embryos were rehydrated through 5 min washes in 75/50/25% Methanol-PBS. Embryos were then washed 4 × 5 min in PBS-1% Triton X-100, incubated 20 min in Acetone at −20 °C, and washed again 4 × 5 min in PBS-1% Triton. Before incubation with primary antibody (Rabbit@Phospho-SMAD2/3, Cell Signaling 8828 S, used at 1:2000) embryos were blocked for a minimum of 2 h through preincubation in PBS-Triton + 10% fetal bovine serum. Following primary antibody incubation, embryos were washed 8 × 15 min in PBS-Triton before blocking again for a minimum of 2 h through preincubation in PBS-Triton + 10% FBS, before incubation with the secondary antibody (Goat@Rabbit-AlexaFluorPlus488, Invitrogen A32731, used at 1:500). Following secondary antibody incubation, embryos were washed 8 × 15 min in PBS-Triton.
Microscopy and image analysis
Imaging was performed using Laser scanning confocal microscopes (Zeiss LSM710, 780 and 880). Airyscan super-resolution imaging was performed on a Zeiss LSM 880 system. For confocal imaging of Phospho-SMAD2/3 antibody stainings, embryos were incubated in Mowiol, the yolk removed manually, and the embryos flat-mounted between slide and coverslip for confocal imaging using a 10x dry objective.
For all other experiments, embryos were mounted in 0.75% low melting agarose (Sigma) in glass bottom dishes (Mattek) for confocal imaging using a 40x NA 1.1 water immersion objective.
In situ hybridizations were documented on a Leica M205 microscope with a Lumenera Infinity camera.
Image analysis was performed using ImageJ (http://rbs.info.nih.gov/ij/).
For Spaw-GFP intensity measurements (Supplementary Fig. 11a), Spaw-GFP signals of individual embryos were measured relative to the intensity of the co-injected Histone2B-RFP construct to correct for differences in the amount of injected material received by each embryo. To allow comparison between WT and MZ myo1g mutants, the mean Spaw-GFP intensity signal of the WT control population was then set to 1 and data from all individual embryos normalized with respect to this value.
For gdf3 LRO intensity measurements (Supplementary Fig. 10), RGB images were transformed to Luminance representations. Following the detection of the gdf3 signal through automated thresholding, integrated density signals were measured in ROIs that comprise the LRO but exclude the non-LRO gdf3 signal from the lateral mesoderm.
Statistical analyses
Appropriate statistical tests were selected for each experiment based on the nature of the comparison (bi- or multifactorial, ordinal or categorical data), data distribution, and variance. Statistical analysis and representations were performed using R/R-Studio. Complete information regarding the applied statistical tests, test statistics, sample sizes and displayed error bars for all experiments are provided in the Source Data files.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Source data are provided as Source Data files. Datasets and materials that were generated and analyzed during the current study are available from the corresponding author on request. Source data are provided with this paper.
References
Hamada, H. Molecular and cellular basis of left-right asymmetry in vertebrates. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 96, 273–296 (2020).
Schweickert, A. et al. Vertebrate left-right asymmetry: what can nodal cascade gene expression patterns tell us? J. Cardiovasc. Dev. Dis. 5, 1 (2017).
Hamada, H. & Tam, P. Diversity of left-right symmetry breaking strategy in animals. F1000Res 9, F1000 (2020).
Tingler, M. et al. A conserved role of the unconventional myosin 1d in laterality determination. Curr. Biol. 28, 810–816.e813 (2018).
Juan, T. et al. Myosin1D is an evolutionarily conserved regulator of animal left-right asymmetry. Nat. Commun. 9, 1942 (2018).
Saydmohammed, M. et al. Vertebrate myosin 1d regulates left-right organizer morphogenesis and laterality. Nat. Commun. 9, 3381 (2018).
Alsafwani, R. S. et al. Novel. Front. Med. 8, 724826 (2021).
Nonaka, S. et al. Randomization of left-right asymmetry due to loss of nodal cilia generating leftward flow of extraembryonic fluid in mice lacking KIF3B motor protein. Cell 95, 829–837 (1998).
Hashimoto, M. et al. Planar polarization of node cells determines the rotational axis of node cilia. Nat. Cell Biol. 12, 170–176 (2010).
Nonaka, S., Shiratori, H., Saijoh, Y. & Hamada, H. Determination of left-right patterning of the mouse embryo by artificial nodal flow. Nature 418, 96–99 (2002).
Kramer-Zucker, A. G. et al. Cilia-driven fluid flow in the zebrafish pronephros, brain and Kupffer’s vesicle is required for normal organogenesis. Development 132, 1907–1921 (2005).
Essner, J. J. et al. Conserved function for embryonic nodal cilia. Nature 418, 37–38 (2002).
Gros, J., Feistel, K., Viebahn, C., Blum, M. & Tabin, C. J. Cell movements at Hensen’s node establish left/right asymmetric gene expression in the chick. Science 324, 941–944 (2009).
Kajikawa, E. et al. Nodal paralogues underlie distinct mechanisms for visceral left-right asymmetry in reptiles and mammals. Nat. Ecol. Evol. 4, 261–269 (2020).
Levin, M., Thorlin, T., Robinson, K. R., Nogi, T. & Mercola, M. Asymmetries in H+/K+-ATPase and cell membrane potentials comprise a very early step in left-right patterning. Cell 111, 77–89 (2002).
Lebreton, G. et al. Molecular to organismal chirality is induced by the conserved myosin 1D. Science 362, 949–952 (2018).
Taniguchi, K. et al. Chirality in planar cell shape contributes to left-right asymmetric epithelial morphogenesis. Science 333, 339–341 (2011).
González-Morales, N. et al. The atypical cadherin dachsous controls left-right asymmetry in Drosophila. Dev. Cell 33, 675–689 (2015).
Naganathan, S. R., Fürthauer, S., Nishikawa, M., Jülicher, F. & Grill, S. W. Active torque generation by the actomyosin cell cortex drives left-right symmetry breaking. Elife 3, e04165 (2014).
Kuroda, R., Endo, B., Abe, M. & Shimizu, M. Chiral blastomere arrangement dictates zygotic left-right asymmetry pathway in snails. Nature 462, 790–794 (2009).
Minegishi, K. et al. A Wnt5 activity asymmetry and intercellular signaling via PCP proteins polarize node cells for left-right symmetry breaking. Dev. Cell 40, 439–452.e434 (2017).
Borovina, A., Superina, S., Voskas, D. & Ciruna, B. Vangl2 directs the posterior tilting and asymmetric localization of motile primary cilia. Nat. Cell Biol. 12, 407–412 (2010).
Hill, C. S. Establishment and interpretation of NODAL and BMP signaling gradients in early vertebrate development. Curr. Top. Dev. Biol. 149, 311–340 (2022).
Hashimoto, H. et al. The Cerberus/Dan-family protein Charon is a negative regulator of Nodal signaling during left-right patterning in zebrafish. Development 131, 1741–1753 (2004).
Sampaio, P. et al. Left-right organizer flow dynamics: how much cilia activity reliably yields laterality? Dev. Cell 29, 716–728 (2014).
Maerker, M. et al. Bicc1 and Dicer regulate left-right patterning through post-transcriptional control of the Nodal inhibitor Dand5. Nat. Commun. 12, 5482 (2021).
Minegishi, K. et al. Fluid flow-induced left-right asymmetric decay of Dand5 mRNA in the mouse embryo requires a Bicc1-Ccr4 RNA degradation complex. Nat. Commun. 12, 4071 (2021).
Gritsman, K. et al. The EGF-CFC protein one-eyed pinhead is essential for nodal signaling. Cell 97, 121–132 (1999).
Tsukazaki, T., Chiang, T. A., Davison, A. F., Attisano, L. & Wrana, J. L. SARA, a FYVE domain protein that recruits Smad2 to the TGFbeta receptor. Cell 95, 779–791 (1998).
Bodenstine, T. M., Chandler, G. S., Seftor, R. E., Seftor, E. A. & Hendrix, M. J. Plasticity underlies tumor progression: role of Nodal signaling. Cancer Metastasis Rev. 35, 21–39 (2016).
Thisse, C. & Thisse, B. Antivin, a novel and divergent member of the TGFbeta superfamily, negatively regulates mesoderm induction. Development 126, 229–240 (1999).
Lenhart, K. F., Lin, S. Y., Titus, T. A., Postlethwait, J. H. & Burdine, R. D. Two additional midline barriers function with midline lefty1 expression to maintain asymmetric Nodal signaling during left-right axis specification in zebrafish. Development 138, 4405–4410 (2011).
Bisgrove, B. W., Essner, J. J. & Yost, H. J. Regulation of midline development by antagonism of lefty and nodal signaling. Development 126, 3253–3262 (1999).
Hozumi, S. et al. An unconventional myosin in Drosophila reverses the default handedness in visceral organs. Nature 440, 798–802 (2006).
Spéder, P., Adám, G. & Noselli, S. Type ID unconventional myosin controls left-right asymmetry in Drosophila. Nature 440, 803–807 (2006).
Baker, K., Holtzman, N. G. & Burdine, R. D. Direct and indirect roles for Nodal signaling in two axis conversions during asymmetric morphogenesis of the zebrafish heart. Proc. Natl Acad. Sci. USA 105, 13924–13929 (2008).
Smith, K. A. et al. Rotation and asymmetric development of the zebrafish heart requires directed migration of cardiac progenitor cells. Dev. Cell 14, 287–297 (2008).
Rohr, S., Otten, C. & Abdelilah-Seyfried, S. Asymmetric involution of the myocardial field drives heart tube formation in zebrafish. Circ. Res. 102, e12–e19 (2008).
Rebagliati, M. R., Toyama, R., Fricke, C., Haffter, P. & Dawid, I. B. Zebrafish nodal-related genes are implicated in axial patterning and establishing left-right asymmetry. Dev. Biol. 199, 261–272 (1998).
Sampath, K. et al. Induction of the zebrafish ventral brain and floorplate requires cyclops/nodal signalling. Nature 395, 185–189 (1998).
Sullivan-Brown, J. et al. Zebrafish mutations affecting cilia motility share similar cystic phenotypes and suggest a mechanism of cyst formation that differs from pkd2 morphants. Dev. Biol. 314, 261–275 (2008).
Noël, E. S. et al. A Nodal-independent and tissue-intrinsic mechanism controls heart-looping chirality. Nat. Commun. 4, 2754 (2013).
Tessadori, F. et al. Nodal signaling range is regulated by proprotein convertase-mediated maturation. Dev. Cell 32, 631–639 (2015).
Petzoldt, A. G. et al. DE-cadherin regulates unconventional myosin ID and myosin IC in Drosophila left-right asymmetry establishment. Development 139, 1874–1884 (2012).
Raya, A. et al. Notch activity induces Nodal expression and mediates the establishment of left-right asymmetry in vertebrate embryos. Genes Dev. 17, 1213–1218 (2003).
Montague, T. G., Gagnon, J. A. & Schier, A. F. Conserved regulation of Nodal-mediated left-right patterning in zebrafish and mouse. Development 145, dev171090 (2018).
Feldman, B. et al. Zebrafish organizer development and germ-layer formation require nodal-related signals. Nature 395, 181–185 (1998).
Thisse, B., Wright, C. V. & Thisse, C. Activin- and Nodal-related factors control antero-posterior patterning of the zebrafish embryo. Nature 403, 425–428 (2000).
Pelliccia, J. L., Jindal, G. A. & Burdine, R. D. Gdf3 is required for robust Nodal signaling during germ layer formation and left-right patterning. Elife 6, e28635 (2017).
Perez-Hernandez, D. et al. The intracellular interactome of tetraspanin-enriched microdomains reveals their function as sorting machineries toward exosomes. J. Biol. Chem. 288, 11649–11661 (2013).
McIntosh, B. B. & Ostap, E. M. Myosin-I molecular motors at a glance. J. Cell Sci. 129, 2689–2695 (2016).
Arif, E. et al. The motor protein Myo1c regulates transforming growth factor-β-signaling and fibrosis in podocytes. Kidney Int. 96, 139–158 (2019).
Chung, C. L. et al. Pentachloropseudilin inhibits transforming growth factor-β (TGF-β) activity by accelerating cell-surface type II TGF-β receptor turnover in target cells. Chembiochem 19, 851–864 (2018).
Kressmann, S., Campos, C., Castanon, I., Fürthauer, M. & González-Gaitán, M. Directional Notch trafficking in Sara endosomes during asymmetric cell division in the spinal cord. Nat. Cell Biol. 17, 333–339 (2015).
López-Ortega, O. & Santos-Argumedo, L. Myosin 1g contributes to CD44 adhesion protein and lipid rafts recycling and controls CD44 capping and cell migration in B lymphocytes. Front. Immunol. 8, 1731 (2017).
Ito, T., Williams, J. D., Fraser, D. J. & Phillips, A. O. Hyaluronan regulates transforming growth factor-beta1 receptor compartmentalization. J. Biol. Chem. 279, 25326–25332 (2004).
Fu, X. X. et al. A spatiotemporal barrier formed by Follistatin is required for left-right patterning. Proc. Natl Acad. Sci. USA 120, e2219649120 (2023).
Appel, B. et al. Delta-mediated specification of midline cell fates in zebrafish embryos. Curr. Biol. 9, 247–256 (1999).
Latimer, A. J., Dong, X., Markov, Y. & Appel, B. Delta-Notch signaling induces hypochord development in zebrafish. Development 129, 2555–2563 (2002).
Preiß, H. et al. Regulation of Nodal signaling propagation by receptor interactions and positive feedback. Elife 11, e66397 (2022).
Kimmel, C. B., Ballard, W. W., Kimmel, S. R., Ullmann, B. & Schilling, T. F. Stages of embryonic development of the zebrafish. Dev. Dyn. 203, 253–310 (1995).
Kalogirou, S. et al. Intracardiac flow dynamics regulate atrioventricular valve morphogenesis. Cardiovasc. Res. 104, 49–60 (2014).
Dick, A., Mayr, T., Bauer, H., Meier, A. & Hammerschmidt, M. Cloning and characterization of zebrafish smad2, smad3 and smad4. Gene 246, 69–80 (2000).
Gong, Y., Mo, C. & Fraser, S. E. Planar cell polarity signalling controls cell division orientation during zebrafish gastrulation. Nature 430, 689–693 (2004).
Long, S., Ahmad, N. & Rebagliati, M. The zebrafish nodal-related gene southpaw is required for visceral and diencephalic left-right asymmetry. Development 130, 2303–2316 (2003).
Rebagliati, M. R., Toyama, R., Haffter, P. & Dawid, I. B. cyclops encodes a nodal-related factor involved in midline signaling. Proc. Natl Acad. Sci. USA 95, 9932–9937 (1998).
Dubrulle, J. et al. Response to Nodal morphogen gradient is determined by the kinetics of target gene induction. Elife 4, e05042 (2015).
Thisse, B. et al. Spatial and temporal expression of the zebrafish genome by large-scale in situ hybridization screening. Methods Cell Biol. 77, 505–519 (2004).
Talbot, W. S. et al. A homeobox gene essential for zebrafish notochord development. Nature 378, 150–157 (1995).
Gamse, J. T. et al. Otx5 regulates genes that show circadian expression in the zebrafish pineal complex. Nat. Genet. 30, 117–121 (2002).
Faucourt, M., Houliston, E., Besnardeau, L., Kimelman, D. & Lepage, T. The pitx2 homeobox protein is required early for endoderm formation and nodal signaling. Dev. Biol. 229, 287–306 (2001).
Odenthal, J. & Nüsslein-Volhard, C. fork head domain genes in zebrafish. Dev. Genes Evol. 208, 245–258 (1998).
Alexander, J., Rothenberg, M., Henry, G. L. & Stainier, D. Y. casanova plays an early and essential role in endoderm formation in zebrafish. Dev. Biol. 215, 343–357 (1999).
Alexander, J. & Stainier, D. Y. A molecular pathway leading to endoderm formation in zebrafish. Curr. Biol. 9, 1147–1157 (1999).
Essner, J. J., Amack, J. D., Nyholm, M. K., Harris, E. B. & Yost, H. J. Kupffer’s vesicle is a ciliated organ of asymmetry in the zebrafish embryo that initiates left-right development of the brain, heart and gut. Development 132, 1247–1260 (2005).
Yu, X., Ng, C. P., Habacher, H. & Roy, S. Foxj1 transcription factors are master regulators of the motile ciliogenic program. Nat. Genet. 40, 1445–1453 (2008).
Rottbauer, W. et al. Cardiac myosin light chain-2: a novel essential component of thick-myofilament assembly and contractility of the heart. Circ. Res. 99, 323–331 (2006).
Livak, K. J. & Schmittgen, T. D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-delta delta C(T)) method. Methods 25, 402–408 (2001).
Acknowledgements
This study was supported by the ARC project grants PJA20181208167 (M.F.), PJA2023090007156 (M.F.), the ANR DroZeMyo (ANR-17-CE13-0024-02) (M.F.), and a CSI grant from Université Côte d’Azur (M.F.). A.J.K. benefited from a fourth-year PhD fellowship from La Ligue Contre le Cancer. Confocal microscopy was performed with the help of the iBV PRISM imaging platform. We thank J.Bakkers, J.Batut, E.Cau, P.Dufourcq, M.Gonzalez-Gaitan, C.P.Heisenberg, T.Lepage, S.Lopes, M.Roussigné, C. & B.Thisse, J.Vermot & PF Xu for the sharing of reagents. We are grateful to S.Polès, R.Rebillard, G.Dupuy, C.Girardin, and F.Salmy for technical assistance and excellent fish care.
Author information
Authors and Affiliations
Contributions
The genetic analysis of myosin1 function in zebrafish Nodal signaling was performed by A.J.K. F.B. generated reagents used in the present study. M.F. designed the study, performed experiments, analyzed the data, and wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Kurup, A.J., Bailet, F. & Fürthauer, M. Myosin1G promotes Nodal signaling to control zebrafish left-right asymmetry. Nat Commun 15, 6547 (2024). https://doi.org/10.1038/s41467-024-50868-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-50868-y
- Springer Nature Limited