

Abstract
Saponins are plant secondary metabolites comprising glycosylated triterpenoids, steroids or steroidal alkaloids with a broad spectrum of toxicity to microbial pathogens and pest organisms that contribute to basal plant defense to biotic attack. Secretion of glycosyl hydrolases that enzymatically convert saponins into less toxic products was thus far the only mechanism reported to enable fungal pathogens to colonize their saponin-containing host plant(s). We studied the mechanisms that the fungus Botrytis cinerea utilizes to be tolerant to well-characterized, structurally related saponins from tomato and Digitalis purpurea. By gene expression studies, comparative genomics, enzyme assays and testing a large panel of fungal (knockout and complemented) mutants, we unraveled four distinct cellular mechanisms that participate in the mitigation of the toxic activity of these saponins and in virulence on saponin-producing host plants. The enzymatic deglycosylation that we identified is novel and unique to this fungus-saponin combination. The other three tolerance mechanisms operate in the fungal membrane and are mediated by protein families that are widely distributed in the fungal kingdom. We present a spatial and temporal model on how these mechanisms jointly confer tolerance to saponins and discuss the repercussions of these findings for other plant pathogenic fungi, as well as human pathogens.
Similar content being viewed by others


Introduction
Plant defense reactions against microbial pathogens commonly involve the production of chemical defense compounds. This chemical armory involves constitutively produced phytoanticipins and pathogen-induced phytoalexins. A common cellular target of various defense substances from both groups is the pathogen plasma membrane, such that the presence of the plant defense compound destroys the integrity of this cellular barrier, resulting in rapid cell death. As part of the arms race between plants and pathogens, microorganisms have evolved resistance mechanisms against these plant defense compounds, which in some cases also confer resistance against (multiple) synthetic antimicrobials, such as fungicides or clinical drugs. An important category of membrane-targeting phytoanticipins are the saponins, a large group of secondary metabolites comprising glycosylated triterpenoids, steroids or steroidal alkaloids that are widely distributed in the plant kingdom1,2,3. They are considered basal defense compounds in plants because of their toxicity to a wide range of plant pathogens and pests, including fungi, bacteria, oomycetes, nematodes and herbivorous insects1,4,5,6,7,8. The importance of saponins in plant defense was illustrated by the observation that saponin-deficient (sad) mutants of the wild oat species Avena strigosa were significantly compromised in resistance against a variety of fungal pathogens9. The growth inhibition of fungi by saponins is achieved through membrane disruption following their binding to sterols10. This mode of action was demonstrated in the interactions between different saponins with natural and artificial membrane preparations11,12,13. So far, the only known fungal resistance mechanism against saponins is their enzymatic detoxification. To detoxify saponins, fungi can secrete diverse glycosyl hydrolases (GHs) that convert saponins into less toxic products via hydrolytic deglycosylation1,2,14. The detoxification of saponins mediated by GHs is important for the capacity of fungal pathogens to infect host plants containing saponins. The ability of different isolates of Septoria avenae to degrade the oat leaf saponins 26-desglucoavenacosides A and B positively correlated with their pathogenicity15. The avenacinase activity in the fungus Gaeumannomyces graminis var. avenae that mediates hydrolysis of the oat root saponin avenacin A-1 is a ß-glucosidase16 that was later assigned to the CAZyme family GH3. Avenacinase-deficient mutants of G. graminis var. avenae exhibited increased in vitro sensitivity to avenacin A-1 and lost the ability to infect oat while retaining pathogenicity on wheat which does not accumulate avenacin A-116. A survey of 161 fungal isolates sampled from roots of field-grown oat and wheat plants for their sensitivity to avenacin A-1 indicated that nearly all isolates sampled from oat were resistant to avenacin, while the isolates sampled from wheat comprised both avenacin-sensitive and avenacin-resistant fungi17.
The importance of saponin degradation is also evident for α-tomatine, a steroidal glycoalkaloid (SGA) consisting of the steroidal aglycon tomatidine and a tetrasaccharide sugar chain (β-lycotetraose). It is the most abundant saponin in tomato vegetative tissues, green fruit as well as roots, with the highest fresh weight concentrations reported to exceed 1 mM18. Deglycosylation of α-tomatine by several tomato pathogens has been reported and is mediated by tomatinases from different glycosyl hydrolase (GH) families with distinct modes of action18. Hydrolysis of the intact tetrasaccharide and the concomitant release of the aglycon tomatidine is catalyzed by GH10 family tomatinases in fungi, including Cladosporium fulvum, Fusarium oxysporum f. sp. lycopersici, F. graminearum and F. solani19,20,21. Second, cleavage of the terminal glucose resulting in the generation of β2-tomatine is mediated by tomatinases belonging to the GH3 family in Septoria lycopersici, Verticillium albo-atrum and Colletotrichum coccodes22,23,24. Third, the generation of β1-tomatine by hydrolytic removal of the terminal xyloside was reported to occur exclusively in Botrytis cinerea25 though the β-xylosidase protein was not identified. The GH10 tomatinases in C. fulvum and F. oxysporum are not essential for pathogenicity but rather contribute to full virulence on tomato, as manifested by less severe symptoms caused by the tomatinase-deficient mutants21,26. Remarkably, the contribution of saponin-degrading enzymes to plant infection is not only by their role in saponin detoxification but was also reported to be related to the suppression of plant defense responses mediated by the enzymatic breakdown products, indicating another step in the arms race between pathogen and host27,28.
It is important to note that all saponin tolerance mechanisms that have been reported in fungal pathogens are limited to enzymatic degradation. Studies on fungal mutants unable to degrade saponins suggest, however, that so far unknown non-hydrolytic mechanisms exist that contribute to tolerance to saponins and virulence on saponin-containing plants. For instance, S. lycopersici tomatinase mutants were more sensitive to α-tomatine but their growth was not fully inhibited even at 1 mM, and the virulence on tomato was not reduced29. Given the fact that saponins can disrupt fungal membrane integrity via binding to 3β-hydroxy sterols (free sterols), modification of sterols or a reduction in sterol content might lead to increased tolerance to saponins30. This can be inferred from the relatively high tolerance to α-tomatine in Phytophthora species, which lack the biosynthetic capacity to synthesize sterols31. Moreover, it was proposed that tomato and potato cells withstand high levels of their own saponins (α-tomatine and solanine, respectively) due to their capacity to glycosylate plasma membrane sterols31,32. In addition to target modifications, active membrane repair could also counteract the membrane-disruptive action of saponins. Schumann et al.33 recently reported that in the saprotrophic fungus Neurospora crassa, a mutant lacking the PEF1 protein shows increased sensitivity to α-tomatine. PEF1 likely mediates a membrane repair mechanism and readily accumulates at the plasma membrane in response to α-tomatine.
Botrytis cinerea, the causal agent of gray mold disease, is a broad host range fungal pathogen affecting the global production of food and ornamental plants34. It produces a unique type of tomatinase activity that could be induced in liquid cultures by α-tomatine but not by other SGAs25. However, the B. cinerea gene encoding the tomatinase remains to be identified. The availability of B. cinerea genome information combined with a recently established CRISPR/Cas9 mutagenesis system render this fungus an ideal model to study tolerance mechanisms to saponins and their contribution to virulence35,36.
Based on existing findings on fungal saponin tolerance, we investigated whether B. cinerea employs hydrolysis combined with other (so far unknown) resistance mechanisms to gain tolerance to α-tomatine. In order to obtain a comprehensive view of fungal cellular responses to saponins, we employed genomics, transcriptomics and functional genetic approaches. We identified a gene encoding a novel, secreted saponin hydrolase and present evidence for at least three additional, intracellular mechanisms contributing to saponin tolerance. These mechanisms are activated on transcriptional or post-translational levels, probably representing pre-formed and induced systems. The respective genes are widespread in the fungal kingdom suggesting broadly conserved functions in pathogenic but also saprophytic species.
Results
Isolate B05.10 converts α-tomatine into β1-tomatine, while isolate M3a lacks tomatinase activity
Previous studies by Quidde et al.25 identified a B. cinerea isolate, designated M3a, which was unable to hydrolyze α-tomatine, in stark contrast to many other isolates including B05.10. To confirm these early observations and verify the phenotype of the M3a culture, we compared the formation of hydrolytic degradation products in liquid cultures of B. cinerea B05.10 and M3a amended with α-tomatine by liquid chromatography–mass spectrometry (LC-MS). After 9 h of incubation with α-tomatine, the supernatant of the B05.10 liquid culture contained β1-tomatine as the only α-tomatine breakdown product (Fig. 1A), indicating that tomatinase activity is indeed conferred by a β-xlyosidase, as proposed by Quidde et al.25. No hydrolytic conversion product of α-tomatine was identified in supernatant of the culture of M3a (Fig. 1B).

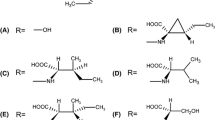
A LC-MS profiles of liquid cultures of wild type isolate B05.10 (highlighted in gray) and various knockout mutants upon incubation with 200 µM α-tomatine. B LC-MS profiles of liquid cultures of wild type isolate M3a (highlighted in gray) and various complemented mutants upon incubation with 200 µM α-tomatine. C LC-MS profile of α-tomatine upon incubation with purified BcTOM1 protein for 2 h. D Structures of the metabolites 1–6. Note that the commercial α-tomatine used as substrate contained traces of dehydrotomatine (peak 2), which differs in the aglycone moiety, but is also a substrate for tomatinases.
M3a displays increased sensitivity to saponins and compromised virulence on tomato
To test whether the ability or inability to degrade α-tomatine correlates with differences in saponin sensitivity and virulence, tolerance and infection assays were conducted with isolates B05.10 and M3a. The sensitivity to saponins of B05.10 and M3a was tested on agar containing different concentrations of α-tomatine or digitonin, a saponin from Digitalis purpurea that is structurally related to α-tomatine. It contains a pentasaccharide that has one extra galactose moiety at the β2-position but shares with α-tomatine the terminal xylose at the β1-position. M3a was more sensitive to α-tomatine and digitonin than B05.10 (Fig. 2A). Virulence of M3a on tomato leaves was severely compromised (Fig. 2B). Under conditions where B05.10 produced expanding lesions on tomato leaves, M3a displayed a disease incidence of ~20%, meaning that 80% of the inoculation droplets resulted in infection that was confined to necrotic spots that did not expand, even upon longer incubation (Fig. 2B). Inoculation of M3a on D. purpurea was performed with agar plugs as its leaf surface contains many trichomes that interfere with inoculations using spore suspensions. On D. purpurea, lesions of M3a were 20% smaller than those of B05.10 (Fig. 2C). On Nicotiana benthamiana, which is not known to produce saponins, M3a displayed a disease incidence of >90% though it produced lesions that were ~20% smaller than B05.10 (Fig. 2D).

A Mycelium growth on plates containing 400 µM α-tomatine and 30 µM digitonin, colony diameters were measured after 3 days. The mock treatment for experiments with α-tomatine was methanol, while for experiments with digitonin, it was water. Data are presented as mean values ± standard deviation (SD) of all biological replicates. B–D Infection of B05.10 and M3a on tomato (B), Digitalis purpurea (C) or Nicotiana benthamiana (D). Disease incidence represents the proportion of inoculation droplets that resulted in expanding lesions. Open dots represent individual values of disease incidence or lesion diameter, based on three-to-four independent inoculation assays. Data are presented as mean values ± SD of all biological replicates. P-values of two-tailed Student’s t-tests are provided above the bars. Source Data are provided as a Source Data file. E Illustration of the absence of genes Bcin08g00060 and Bcin08g00070 in isolate M3a. Red and blue boxes represent gene models in B05.10 and M3a, respectively, with the gene IDs plotted above or below the box. Dotted lines join orthologous genes. TE, transposable element in M3a. Start and end coordinates are plotted at the extremities of the contigs. Annotations of the genes in panel (E) are provided in Supplementary Table S1A.
Genome comparison of M3a with B05.10 reveals candidate genes for α-tomatine tolerance
We reasoned that the ability and inability of B05.10 and M3a to tolerate α-tomatine should be reflected in genomic differences between the strains and that comparison of their genome sequences might reveal the molecular basis of α-tomatine tolerance in B05.10. The genome sequence of the tomatinase-producing isolate B05.10 was previously published and serves as a reference for the species B. cinerea36. The genome of isolate M3a was sequenced using a combination of Illumina and Nanopore technology, resulting in an assembly of 23 contigs, of which four contain telomeric repeats on both ends and 12 contain telomeric repeats on one end. The annotated M3a genome was examined for polymorphisms with isolate B05.10. Specifically, the M3a genome was inspected for the absence of genes or for polymorphisms that would render a B05.10 gene dysfunctional. The most striking difference was the absence in M3a of the entire minichromosome 18, and of a region of ~12 kb from B05.10 Chromosome 8 (Fig. 2E, Supplementary Data S1A), which contains two genes: Bcin08g00060 and Bcin08g00070, encoding a secreted glycosyl hydrolase GH43 and a cytoplasmic glycosyl transferase GT28, respectively. The M3a genome contains in this region two different genes and a transposon (Fig. 2E, Supplementary Data S1A). M3a displayed across the genome ~92500 single nucleotide polymorphisms, as well as ~65 major polymorphisms (multiple SNPs, indels, rearrangement, translocation), as compared with B05.10. These major polymorphisms affect 166 proteins in M3a, of which 96 have no functional description (Supplementary Data S1B).
Identification of α-tomatine-responsive genes in B. cinerea
After identifying two candidate genes in isolate B05.10 that could either perform enzymatic glycoside hydrolysis or glycosylation, we analyzed the transcriptional response to α-tomatine in the assumption that exposure to a sublethal dosage of α-tomatine would result in the transcriptional upregulation of genes contributing to tolerance to the metabolite. An overnight culture of isolate B05.10 was divided in two parts. One half was supplemented with medium containing 200 μM α-tomatine, while the other half only received fresh medium. Samples were taken at 3 and 6 h, and RNA was extracted for sequencing. RNA-seq analysis revealed that α-tomatine treatment in B05.10 strongly induced the transcript levels of multiple genes. Only 21 genes displayed >10-fold increased transcript levels at 3 h after α-tomatine addition to the culture (Supplementary Table 1), among which are the Bctom1 (Bcin08g00060) and Bcgt28a (Bcin08g00070) genes which are absent in M3a (Fig. 2E), three additional genes from the GT28 family, four genes encoding RTA1-like proteins, three dioxygenase genes and a gene encoding an ABC transporter designated BcAtrT. In order to obtain a detailed view of the temporal dynamics of transcript induction after α-tomatine addition, we used reverse transcription quantitative polymerase chain reaction (RT-qPCR) to study the expression profiles of a subset of α-tomatine-responsive genes as well as Bcpef1 (Supplementary Fig. 1). In accordance with RNA-seq results, all tested genes showed low, stable transcript levels in the absence of α-tomatine (Supplementary Fig. 2) and were induced in response to α-tomatine treatment (Supplementary Fig. 1A) with comparable kinetics and induction levels (fold-changes), except for Bcpef1, whose mRNA level remained stable throughout all time points. Induction of α-tomatine-responsive genes was observed at 30 min and peaked between 3 and 9 h (Supplementary Fig. 1A). Eventually, the transcript levels decreased at 24 h after supplementing the culture with α-tomatine. Transcript levels of the same genes were also analyzed in M3a. Remarkably, induction of α-tomatine-responsive genes was less pronounced in M3a, except for RTA1a which still displayed high induction (Supplementary Fig. 1D), suggesting that the absence of Bctom1 and Bcgt28a genes in M3a might not be the sole reason for its sensitivity to α-tomatine.
To test whether increased transcript levels of the above genes in B05.10 were specific to α-tomatine or resulted from a general response to membrane damage, we analyzed their expression upon treatment with the structurally related saponin digitonin or treatment with nystatin, a chemically distinct polyene antibiotic known to disrupt fungal membranes. Transcript levels of α-tomatine-responsive genes, both in B05.10 and M3a, strongly increased upon digitonin treatment but not upon nystatin treatment (Supplementary Fig. 1B, C), suggesting that transcript upregulation was in response to a saponin, rather than a response to membrane damage.
Expression levels of α-tomatine-responsive genes during plant infection
To test if α-tomatine-responsive genes are also induced during infection on the host plant tomato, we analyzed their expression during infection on tomato leaves by RT-qPCR (Supplementary Fig. 3A). Most of the genes, with exception of Bcrta1b, Bcrta1c and Bcgt28b, displayed low transcript levels in early infection stages that strongly increased at later time points, with the exception of Bcrta1c. The increase was prominent at 24 h post-inoculation (hpi) and coincided with the initiation of lesion expansion. Bcpef1 transcript levels did not increase during tomato infection. The α-tomatine-responsive genes were also induced during infection on D. purpurea leaves from 12 hpi onward (Supplementary Fig. 3B), but not induced during infection on N. benthamiana or the common French bean Phaseolus vulgaris, which do not produce α-tomatine or related compounds (Supplementary Fig. 3C, D).
Enzymatic detoxification of α-tomatine in B. cinerea is catalyzed by a GH43 protein
The hydrolytic conversion of α-tomatine by B. cinerea into an inactive compound was reported to be mediated by a β-xylosidase25,37. According to the carbohydrate-active enzymes (CAZymes) database38, proteins from the GH43 family have potential β-xylosidase activity and we thus presumed that the α-tomatine-responsive gene Bcin08g00060 encodes an enzyme with tomatinase activity. To test this hypothesis, we produced a recombinant protein in Escherichia coli and tested it for α-tomatine-degrading activity. After 2 h of incubation with α-tomatine, LC-MS analysis confirmed the formation of β1-tomatine (Fig. 1C), the product obtained by hydrolytic removal of xylose from α-tomatine. To confirm its function, the Bcin08g00060 gene was deleted in B05.10 and was overexpressed in M3a through CRISPR/Cas9-mediated transformation. Indeed, the B05.10 Bcin08g00060 knockout mutant lost the ability to degrade α-tomatine (Fig. 1A), whereas its overexpression in M3a conferred tomatinase activity, as manifested by detection of β1-tomatine by LC-MS (Fig. 1B). These results confirm the exclusive role of Bcin08g00060 in converting α-tomatine into β1-tomatine in B. cinerea. The gene was therefore designated as Bctom1.
Phylogenetic analysis of BcTom1
Phylogenetic analysis of the BcTOM1 protein (Fig. 3) indicated that within Sclerotiniaceae, this gene is only present in three species in the genus Botrytis. Specifically, we identified orthologs in B. aclada (BACL_015g04100) and B. calthae (BCAL_0134g00010)39,40, pathogens that infect onion (Allium cepa) and marsh-marigold (Caltha pallustris), respectively. Orthologs of BcTOM1 were also identified in distantly related fungi that interact with tomato, including Dothideomycete pathogens Stemphylium lycopersici and Macrophomina phaseolina, the Sordariomycete endophyte Colletotrichum tofieldiae, as well as in 13 Eurotiomycetes from the genera Aspergillus and Penicillium that are not known to interact with plants.

Fungal taxa are color-coded based on the classes to which they belong.
Hydrolytic enzymes contribute to tolerance to plant saponins and virulence
After demonstrating the enzymatic activity of BcTOM1, we tested its role in tolerance to α-tomatine and in virulence. Bctom1-KO mutants displayed increased sensitivity to α-tomatine on agar plates, manifested as slower radial growth than the wild type (WT) B05.10, as well as reduced virulence on tomato (Fig. 4A, E). To test whether heterologous tomatinase activity can also enhance tolerance to α-tomatine and virulence of B. cinerea on tomato, we overexpressed tomatinase genes from a different CAZyme family, that possess a different mode of action (Supplementary Fig. 4). To this end, we transformed Cftom1 (GH10) from C. fulvum and Sltom1 (GH3) from S. lycopersici in B. cinerea isolate M3a, which lacks the Bctom1 gene. The overexpression of heterologous tomatinase genes conferred on M3a the corresponding ability to degrade α-tomatine, manifested by the detection of the appropriate breakdown products deduced from their catalytic activity: tomatidine and β2-tomatine, respectively (Fig. 1). Expression of these heterologous tomatinases in M3a resulted in an increased tolerance to α-tomatine (Fig. 4B–D) and a higher proportion of expanding lesions (disease incidence) upon inoculation on tomato (Fig. 4F–H).

A Comparison of wild type B05.10 and ΔBcTom1 knockout mutant. B–D Comparison of M3a with transformants expressing C. fulvum Cftom1 (B), S. lycopersici Sltom1 (C), or Bctom1 (D), respectively. For each type of mutant strain, experiments were performed with two independent transformants. Mycelium growth was on plates containing 800 µM (panel A) or 400 µM (panels B–D) α-tomatine or lacking α-tomatine (mock), and colony diameters were measured at 3 dpi. Open dots represent individual colony diameters. Data are presented as mean values ± standard deviation (SD) of five biological replicates. P-values of two-tailed Student’s t-tests are provided above the bars. Source Data are provided as a Source Data file. E–H Infection of B. cinerea transformants on tomato leaves. Wild type recipient was inoculated on the left side of the central vein (dashed line), transformants on the right side. Scale bars indicate 1 cm. For B05.10 knockout mutants, the lesion diameter is presented. For overexpression transformants of M3a, the disease incidence is shown. Open dots represent individual values of disease incidence or lesion diameter, based on three-to-four independent inoculation assays. Data are presented as mean values ± SD of all biological replicates. P-values of two-tailed Student’s t-tests are provided above the bars. Source Data are provided as a Source Data file.
These same transformants were tested for tolerance to digitonin in plate assays and for virulence on D. purpurea. The Bctom1-KO mutants displayed increased sensitivity to digitonin on agar plates, while M3a transformants expressing CfTOM1 or BcTOM1 displayed increased tolerance and the M3a transformants expressing SlTOM1 grew equal to the recipient (Fig. 5). Inoculation of these transformants on D. purpurea showed a reduced virulence for the B05.10 BcTom1-KO mutants, while M3a transformants expressing SlTOM1 or BcTOM1 displayed increased virulence and M3a transformants expressing CfTOM1 were equally virulent as the recipient (Fig. 5). When inoculated on N. benthamiana leaves, M3a transformants expressing different tomatinase genes or B05.10 Bctom1-KO mutants did not exhibit differences in lesion sizes as compared with their recipients (Supplementary Fig. 5), except for M3a expressing SlTOM1, which formed slightly smaller lesions (Supplementary Fig. 5K).

A–D Sensitivity to digitonin in mycelial growth assays. A Comparison of B05.10 with ΔBctom1 mutant. B–D Comparison of M3a with transformants expressing C. fulvum CfTOM1 (B), S. lycopersici SlTOM1 (C), or BcTOM1 (D), respectively. For each type of mutant strain, experiments were performed with two independent transformants. Mycelium growth was on plates containing 30 µM digitonin (panel A) or 20 µM digitonin (panels B–D) or lacking digitonin (mock). Open dots represent individual colony diameters, measured at 3 dpi. Data are presented as mean values ± standard deviation (SD) of five biological replicates. P-values of two-tailed Student’s t-tests are provided above the bars. Source Data are provided as a Source Data file. E–H Infection on D. purpurea leaves. E Comparison of B05.10 and ΔBctom1 mutant. F–H Comparison of M3a with transformants expressing C. fulvum CfTOM1 (F), S. lycopersici SlTOM1 (G), or BcTOM1 (H), respectively. Wild type recipient was inoculated on the left side of the central vein (depicted by a dashed line), while transformants were inoculated on the right side of the vein. Scale bar, 2 cm. For each type of mutant strain, experiments were performed with two independent transformants. Open dots represent individual values of disease incidence or lesion diameter, based on two-to-three independent inoculation assays. Data are presented as mean values ± SD of all biological replicates. P-values of two-tailed Student’s t-tests are provided above the bars. Source Data are provided as a Source Data file.
Contribution of non-hydrolytic mechanisms for tolerance to saponins
Based on the transcriptome data, which revealed significant upregulation of several genes in the presence of α-tomatine, we considered that besides enzymatic degradation by BcTOM1, additional cellular mechanisms might contribute to saponin tolerance. To test this hypothesis, we generated a large set of single and multiple gene knockout mutants for the α-tomatine-responsive genes of isolate B05.10. Specifically, the attention was focused on the GT28 glycosyl transferase family and the RTA1 gene family, for which multiple members of each family were strongly upregulated by α-tomatine. Also, the PEF1 gene was studied that was earlier reported to contribute to membrane damage mitigation in response to α-tomatine in N. crassa33. Single knockout mutants in the Bcpef1 gene, as well as triple mutants in three Bcrta1 genes, or in three BcGT28 genes all retained the capacity to hydrolyze α-tomatine (Fig. 1A) as well as the capacity to induce expression of the Bctom1 gene upon addition of α-tomatine (Supplementary Fig. 1F). The ABC transporter gene BcatrT was not included in further studies, as it appeared to be a pseudogene with a single nucleotide insertion that causes a frameshift, resulting in a protein that only contains six (instead of 12) transmembrane domains. Combinations of mutants in distinct types of genes were also generated. The mutants were tested in growth assays for sensitivity to α-tomatine and digitonin, using both germination assays with dilution series of spores (Source Data) and radial growth assays with mycelium on agar plugs to distinguish the effects on colony establishment and the growth of mature hyphae, respectively. Furthermore, mutants were tested for virulence on tomato, D. purpurea and N. benthamiana (Table 1). We here only discuss the results for a subset of mutants that showed an altered phenotype (in saponin sensitivity and/or virulence). The characterization of these mutants in radial growth assays and infection assays is presented in Fig. 6 and Supplementary Fig. 7. B05.10 knockout mutants in the Bcgt28a gene displayed increased sensitivity to α-tomatine and digitonin in a spore germination assay, but not in a mycelial growth assay. The mutant showed a reduced virulence on tomato but not on D. purpurea. Complementing the knockout mutant with the Bcgt28a gene restored the phenotype to that of the wild type. The increased sensitivity was not exacerbated by deleting additional GT28 genes; however, the ΔBctom1ΔBcgt28a double mutant was significantly more sensitive to α-tomatine and digitonin than B05.10ΔBctom1 and B05.10ΔBcgt28a single mutants, separately. These observations suggest that BcTOM1 and BcGT28a function in independent pathways. B05.10 triple knockout mutants in three Bcrta1 genes displayed an increased sensitivity only to digitonin, and this was associated with a reduced virulence on D. purpurea, but not on tomato or N. benthamiana. Analysis of Bcrta1 single and double mutants revealed that the phenotype was predominantly resulting from the deletion of the Bcrta1b gene (Table 1).

A–E Sensitivity to α-tomatine in mycelial growth assays. A Comparison of M3a with BcGT28a-OE transformant. B, C Comparison of B05.10 with mutants deleted in three Bcgt28 genes (B) or three Bcrta1 genes (C). Comparison of M3a (D) or B05.10 (E) with their respective mutants in the Bcpef1 gene. For each type of mutant strain, experiments were performed with two independent transformants. Mycelium growth was on plates containing 400 µM (panels A and D) or 800 µM (panels B, C and E) α-tomatine or lacking α-tomatine (mock). Open dots represent the individual colony diameters measured at 3 dpi. Data are presented as mean values ± standard deviation (SD) of all biological replicates from at least three independent inoculations. P-values of two-tailed Student’s t-tests are provided above the bars. Source Data are provided as a Source Data file. F–J Infection on tomato leaves. WT strains were inoculated on the left side of the vein (as depicted by the dashed line). Transformant strains were inoculated on the right side of the vein. F Comparison of M3a with BcGT28a-OE transformant. G–I Comparison of B05.10 with mutants deleted in three Bcgt28 genes (G), three Bcrta1 genes (H), or in the Bcpef1 gene (J), or comparison of M3a deleted in the Bcpef1 gene (I), respectively. Experiments for panels (F–H) and (J) were performed with inoculation medium containing 15 mM sucrose, while for panel (I), the sucrose was increased to 50 mM to obtain a high incidence of expanding lesions of strain M3a. Scale bar, 1 cm. For each type of mutant strain, experiments were performed with two independent transformants. Open dots represent individual lesion diameters at 3 dpi, in mm. Data are presented as mean values ± SD of at least two independent inoculation assays. P-values of two-tailed Student’s t-tests are provided above the bars. Source Data are provided as a Source Data file.
α-Tomatine induces membrane disruption and recruits BcPEF1 and BcGT28 to ergosterol-rich domains
As α-tomatine causes membrane disruption by complexing with sterols, we examined the distribution of sterols in B. cinerea membranes by filipin staining and observed that ergosterol is enriched at hyphal tips and in septa (Supplementary Fig. 9). Studies in N. crassa have demonstrated that the NcPEF1 protein is recruited to damaged sites at the membrane in response to Ca2+ influx and thereby acts as a marker for membrane damage33. We studied the pore-forming activity of α-tomatine on B. cinerea using transformants of B05.10 that overexpress BcPEF1-GFP. Comparable to observations in N. crassa, the BcPEF1-GFP signal was mainly cytoplasmic with some accumulation at the endoplasmic reticulum around nuclei in the absence of α-tomatine (Fig. 7A). Addition of α-tomatine resulted in rapid cell lysis indicated by the vacuolized appearance of the fungal cells. BcPEF1-GFP accumulated in punctate structures at the cell periphery, mostly co-localizing with ergosterol-enriched sites as determined by filipin stain (Fig. 7A). More than 55% of the hyphae treated with α-tomatine showed recruitment of BcPEF1-GFP to the tips (Fig. 7C). These observations confirm the membrane-targeting action of α-tomatine and reveal that BcPEF1 was efficiently recruited to this damaged site, as was also reported in N. crassa33. To determine the subcellular localization of BcGT28a, a GT28a-GFP fusion protein was expressed in B05.10 under control of a constitutive promoter. In medium without α-tomatine, fluorescent signal was homogenously distributed in the hyphae, indicating BcGT28a is predominantly a cytoplasmic protein. After addition of α-tomatine or the chemically unrelated antibiotic nystatin to the culture, the fluorescent signal upon both treatments accumulated at hyphal tips (Fig. 7B, D) and the recruitment of BcGT28a-GFP to hyphal tips was even higher than for BcPEF1-GFP (Fig. 7C). Since BcPEF1 and BcGT28a showed similar localization patterns, we tested if BcPEF1 influences the dynamics of BcGT28a. A Bcpef1 deletion mutant was constructed in the background of B05.10 expressing BcGT28a-GFP and the dynamics of GT28 was tested. The mobilization of BcGT28a-GFP to the membrane of hyphal tips was still observed with high incidence in the absence of BcPEF1 (Fig. 7C, D), indicating that BcGT28a does not require BcPEF1 for translocation to the damaged sites in the membrane. Germlings of B05.10 that express a cytoplasmic GFP did not display an accumulation of fluorescence at hyphal tips upon α-tomatine application (Fig. 7F), while germlings of recipient strain B05.10 that lacks a GFP gene did not display any fluorescence upon α-tomatine application (Fig. 7G).

A BcPEF1-GFP signal observed at 10 min after application of 190 µM α-tomatine. Then the sample was stained with filipin at 20 min to detect ergosterol in the membrane. Signals are indicated by a white arrow (GFP) or a star (*) (filipin staining). B BcGT28a-GFP signal at 10 min after application of 190 µM α-tomatine. C Quantification of recruitment of BcPEF1-GFP and BcGT28a-GFP to the tips of the germlings. Untreated samples are indicated in blue bars, α-tomatine treated samples in red bars and nystatin-treated samples in green bars. Quantification of recruitment of BcPEF1-GFP and BcGT28a-GFP to the tips of the germlings. For each experiment, 100 germlings were counted (total n = 300). Data are presented as percentage mean recruitment ± SD from three independent experiments. P-values obtained from chi-square tests are provided above the bars. D BcGT28a-GFP signal in Bcpef1 KO mutant at 10 min after application of 190 µM α-tomatine; E BcGT28a-GFP signal at 10 min after treatment with or 0.05 mg/mL nystatin; F absence of membrane targeting in B05.10 strain expressing cytoplasmic GFP; G absence of fluorescence in non-GFP recipient strain B05. 10 after application of 190 µM α-tomatine. Experiments in panels (D–H) were performed three times with similar results. Scale bars in panels (A), (B), (D–G), 5 µm.
Discussion
B. cinerea employs a novel tomatinase for saponin degradation
Saponins are a common class of plant defense compounds found in many different plant genera. Fungal pathogens of these host plants must therefore have evolved efficient resistance mechanisms to cope with these membrane toxic compounds. For the last ca. 30 years, enzymatic deglycosylation and absence of the saponin target ergosterol have been the only known resistance mechanism against saponins in fungi14,30. Consistent with earlier studies, we identified the α-tomatine hydrolyzing enzyme BcTOM1 as a major factor in α-tomatine resistance in B. cinerea. BcTOM1 is the first α-tomatine-degrading β-xlyosidase, thus representing a previously unknown class of α-tomatine-degrading enzymes. The comparison of the host range of isolates M3a (lacking Bctom1) and B05.10 (containing Bctom1) confirmed the earlier observed correlation of the host range and the ability to chemically degrade the host defense compounds. A study by Mercier et al.41 compared various field isolates of B. cinerea sampled from tomato or grapevine. Their findings revealed that strains isolated from tomato consistently exhibit three copies of a 25 kbp genomic region, which includes the Bctom1 and Bcgt28a genes. By contrast, four strains obtained from grape lack the Bctom1 gene. The annotation employed in that study suggested that the Bctom1 gene participates in hemicellulose degradation, but did not allude to its role in α-tomatine detoxification41. The presence of multiple copies of two genes that confer tolerance to α-tomatine provides clear evidence for evolutionary adaptation in B. cinerea to host species. It may be interesting to explore if gene duplication events in B. cinerea isolates of other host plant species may also be indicative of the involvement of such duplicated genes in host preference.
Our study demonstrated that introducing tomatinases from unrelated fungi in B. cinerea isolate M3a enhances its tolerance to this saponin and increases disease incidence in tomatoes. This suggests that the overall degradation ability of the enzymes is more significant for the plant-fungus interaction outcome than their specific enzymatic activity in breaking down the saponin. The fact that different α-tomatine-degrading enzymes are found within the fungal kingdom supports the notion that the ability to degrade α-tomatine has independently evolved in different lineages from distinct ancestral glycosyl hydrolases of the GH3, GH10 and GH43 families.
Bctom1 orthologs were not detected in any other Sclerotiniaceae except for two Botrytis species that are not reported to be pathogenic on tomato. Orthologs of Bctom1 were, however, identified in distantly related fungi that interact with tomato, either as a pathogen or an endophyte, as well as in saprophytic Aspergillus and Penicillium species. The patchy distribution over fungal taxa that interact with various plant hosts or act as saprotrophs is intriguing yet provides insufficient evidence for proposing horizontal transfer as a means for broadening the host range of B. cinerea. In fact, it would have been more probable that B. cinerea would have acquired a tomatinase gene from the GH3 or GH10 family present in a broad spectrum of microbes that are pathogenic on tomato18.
Additional mechanisms contribute to α-tomatine resistance
Previous studies on gene knockout mutants have hinted at the existence of additional saponin resistance mechanisms in fungi. These mutants displayed a reduced yet persistent ability to infect host plants and demonstrated higher tolerance to saponin concentrations compared to non-pathogenic isolates or species, that are non-pathogenic on the respective hosts21,26,29. Consistent with this notion, the B. cinerea Bctom1 mutant tolerates higher α-tomatine concentrations than the saprotrophic fungus N. crassa33. Our comprehensive study of the response of B. cinerea to α-tomatine unveils previously undescribed molecular factors representing additional resistance mechanisms beyond enzymatic detoxification or the absence of sterol. Our genetic analysis indicated that BcGT28a, BcRTA1b and BcPEF1 all contribute independently from each other to α-tomatine resistance. While BcTOM1, BcGT28a and BcRTA1 are transcriptionally upregulated in the presence of α-tomatine, BcPEF1 represents a post-translationally activated response mechanism.
The B. cinerea GT28 enzymes show sequence and structural similarity to ergosterol glycosyl transferases of baker’s yeast and plants (Supplementary Fig. 10). In the saponin-producing plant species potato and tomato, membrane sterols are glycosylated by enzymes of the GT28 family to protect them from toxic interaction with their own defense compound42. The discovery of the conserved steroid binding domain in BcGT28 family members strongly supports the hypothesis that also in B. cinerea, these enzymes function in modifying the α-tomatine target ergosterol in the plasma membrane, thereby providing resistance against this saponin. Upon exposure to α-tomatine or other membrane-disturbing drugs, the BcGT28a protein is rapidly translocated from the cytoplasm to distinct regions in the plasma membrane. These dynamics suggest that BcGT28a undergoes post-translational regulation in response to membrane damage. The protein predominantly accumulates at ergosterol-rich cell tips, corresponding to the cellular region where most saponin damage is expected to occur. The mechanisms controlling BcGT28a recruitment to the membrane remain to be resolved but do not include BcPEF1, which exhibits similar subcellular dynamics. BcGT28 homologs are widely found in filamentous fungi, suggesting a broader role than just conferring protection against a single specific plant defense compound. Given that membrane sterols serve as targets for numerous plant defense compounds, fungicides and clinical drugs, further investigation into the role of GT28 enzymes in drug resistance holds potential.
RTA1 proteins are broadly conserved and were first identified in S. cerevisiae, where they confer resistance to the membrane-disrupting compound 7-aminocholesterol43. RTA1 is proposed to operate as a fungal lipid-translocating exporter that participates in restoring membrane integrity upon exposure to membrane-disrupting compounds. It has thus far only been studied in other yeasts, such as Candida and Cryptococcus44, and functional studies on RTA1 genes in filamentous fungi are lacking. Based on the available information from yeasts, we postulate that RTA1 proteins in B. cinerea contribute to mitigating membrane damage caused by saponins, rendering them potential virulence factors. Investigating their role in other phytopathogenic fungi and studying their ability to confer tolerance to various membrane-targeting antifungal compounds, such as agricultural fungicides and clinical drugs, is of broad interest.
The calcium-binding protein PEF1 also contributes to saponin resistance and is part of a conserved membrane repair mechanism in filamentous fungi. Both in B. cinerea and N. crassa, the protein rapidly translocates to the plasma membrane in response to drug-induced membrane damage. A study in N. crassa indicated that it also contributes to membrane repair in response to mechanical membrane rupture, for example, during aberrant plasma membrane fusion, suggesting a general function in securing membrane integrity33. Activation of the PEF1 response does not include gene activation but occurs post-translationally in a calcium-dependent manner, suggesting a role in an immediate cellular emergency response. The study in N. crassa indicated that at least one additional PEF1-independent repair mechanism exists, which might also participate in this response in B. cinerea. The future identification and comprehensive description of membrane repair mechanisms in filamentous fungi will therefore significantly improve our understanding of the global cellular response to membrane-targeting antifungal compounds.
Based on our observations, we propose a multifactorial model for saponin resistance, which likely also applies to fungal responses to other membrane-targeting compounds, including fungicides and clinical drugs (Fig. 8). In this model, post-translational and transcriptional activation of synergistic response mechanisms occur in a stepwise and spatially distinct fashion, allowing initial survival and subsequent adaptation of the fungus upon exposure to the membrane-perforating compounds. Activation of the transcriptional response requires physical contact of the cell with the membrane-perforating compound. In the natural infection process of necrotrophic plant pathogenic fungi, this contact occurs when the fungus destroys the integrity of host plant cells and the saponins are released in the damaged tissue. Disintegration of the plant cells likely occurs already at a distance from the fungal mycelium via secreted fungal pathogenicity factors, such as the cytotoxic sesquiterpene botrydial45, proteins with cytolytic activity on plant cells such as NLPs46,47 or other cell death-inducing compounds48,49. The extending fungal hyphae will come in direct contact with the plant defense compound, while the fungus colonizes the tissue. This contact induces the rapid transcriptional activation of inducible fungal resistance mechanisms but will inevitably cause immediate membrane damage. Disintegration of the membrane during this early phase would, however, result in instantaneous collapse of ion gradients across the plasma membrane and subsequent cell death. Such a life-threatening situation is counteracted by the instant activation of pre-formed membrane repair mechanisms, including PEF1 (Fig. 8). Since such repair does not depend on gene activation, it can respond within seconds to enable fungal survival until inducible systems are activated. A common signal for membrane disintegration and the activation of adequate repair mechanisms in eukaryotic cells is a rapid increase in calcium ions in the cytoplasm. We hypothesize that saponin-induced pore formation results in such an increase and activation of PEF1. After the short-term survival of the fungus is ensured, the inducible systems will implement its long-term adaptation. As described above, these mechanisms likely involve alteration of the cellular target (GT28a) and additional reinforcement of the membrane (RTA1). Secretion of BcTOM1 will then reduce the saponin levels in surrounding plant tissue, clearing the path for hyphal extension (Fig. 8).

The responses start from the moment that α-tomatine is released from the plant vacuoles upon plant cell damage or death inflicted by fungal invasion. Details are discussed in the text (Created with BioRender.com released under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International license).
For many antifungal plant compounds, this model also includes the rapid induction of efflux pumps of the MDR-MFS and MDR-ABC transporter type by their substrates. Efflux is a common cellular protection mechanism against toxic compounds and likely functions between the rapid pre-formed survival and long-term adaptation mechanisms. The combined action of efflux and enzymatic degradation in the resistance against plant defense compounds has also been reported in B. cinerea, including during its interaction with tomato50,51,52,53. So far, efflux has not been described as a saponin resistance mechanism. However, the protective role of MDR transporters has been demonstrated for other compounds, which also target the plasma membrane and disrupt its integrity, such as eugenol or benzylisothiocyanate52,54,55,56. Interestingly, our transcriptome analysis revealed induction of an MDR-ABC transporter gene upon exposure to α-tomatine, which we designated BcatrT. In B05.10, however, this gene contains an early stop codon and therefore does not encode a functional transporter protein. By contrast, the BcatrT gene appeared to be intact in M3a. Following up on the potential contribution of efflux to saponin resistance might further extend the model presented in Fig. 8. While induced drug resistance mechanisms are a known and common theme in fungal biology, the cellular mechanisms mediating transcriptional induction of drug-responsive genes remain cryptic. Interestingly, the observed gene induction by α-tomatine in the tolerant B. cinerea isolate B05.10 was largely absent in the sensitive isolate M3a, while the gene induction by digitonin was similar in both isolates. Future comparison of the two respective genomes therefore holds much potential for further resolving this important question.
The interaction between a host plant and a pathogenic fungus is intricate and encompasses numerous molecular interactions. These interactions arise from an evolutionary arms race between both partners, and the combinatorial defense response against saponins likely reflects the result of such a stepwise evolution. While obtaining an initial, single defense mechanism might not provide full resistance, it could allow limited survival in the presence of the plant defense compound. This critical scenario, on the verge of death, would impose intense selection pressure for adapting to such a toxic environment. As a result, acquiring a single initial defense mechanism could rapidly lead to adaptation and an expansion of the host range of the fungus. This adaptation may also involve general non-specific defense mechanisms, such as the broad and conserved PEF1-mediated membrane repair mechanisms. While not individually capable of providing full resistance, these mechanisms can make important contributions when combined with specific factors. The evolution of broad combinatorial pathogenicity mechanisms remains mysterious, yet it presents significant potential for comprehending the emergence of new pathogens and developing effective control measures.
Methods
Fungal growth and transformation
B. cinerea culturing and spore production were performed as described57. CRISPR/Cas9-mediated transformation of B. cinerea was performed according to Leisen et al.35. Single Guide RNAs (sgRNAs) targeting the gene of interest were synthesized prior to the transformation using T7 RNA polymerase. The template for sgRNA synthesis was made from two long primers (Supplementary Table 2). For complementation of knockout mutants or overexpression, nitrate reductase (BcniaD; Bcin07g01270) and nitrite reductase (BcniiA; Bcin01g05790) gene loci were used for targeted integration of an overexpression cassette generated in pNDH-OGG and pNAN-OGG vectors58. The donor DNA comprising selection marker templates were amplified by PCR using primers containing 60 bp homologous recombination regions. PCR reactions using flanking primers spanning target genes were used for genotyping (Supplementary Table 2). Whether transformants were homokaryotic was checked by PCR for the absence of the target locus. For each combination of mutations, at least two independent homokaryotic transformants were used for characterization.
Plant growth and infection assays
Tomato S. lycopersicum cv. Moneymaker, D. purpurea, N. benthamiana and P. vulgaris were grown in a greenhouse at 21/19 °C (day/night) temperatures. Detached leaves (S. lycopersicum) or whole plants (N. benthamiana, P. vulgaris) were used for inoculation under lab conditions, either in Gamborg’s B5 (Duchefa) minimal medium (GB5) supplemented with 15 mM sucrose and 10 mM potassium phosphate (pH 6.0) or in potato dextrose broth (PDB) (12 g/L) at a concentration of 1 × 106/mL. Detached leaves of D. purpurea were inoculated with agar plugs (5 mm) containing mycelium. Comparisons of mutants with their respective recipient wild type were always performed with two independently obtained mutants. Infection assays were performed in at least three biological repetitions with different batches of plants. Leaves were photographed and lesion sizes were measured with a digital caliper at 3 dpi (tomato, N. benthamiana and P. vulgaris) or 4 dpi (D. purpurea). Statistical analysis of comparisons of lesion sizes was performed with a two-tailed Student’s t-test.
Gene expression analysis of α-tomatine-responsive genes in B. cinerea
B. cinerea spores were inoculated in a liquid medium containing 3 g/L GB5 salts, 100 mM fructose, 10 mM potassium phosphate, 0.5% yeast extract and adjusted to pH 5.5 at a final concentration of 1 × 106/mL. After overnight incubation at 20 °C 120 RPM, half of the culture was supplemented with α-tomatine (TCI Europe), and the other half was supplemented with solvent control (methanol + 0.5% formic acid). Mycelium was sampled at 0, 3 and 6 h after α-tomatine/mock treatment and used for total RNA isolation using a Maxwell 16 LEV Plant RNA Kit (Promega). RNA-seq was carried out at Beijing Genomics Institute (BGI), Shenzhen, China. The reads were mapped to the reference B05.10 genome36 and expression levels were quantified as transcripts per million reads (TPM).
Induction of α-tomatine-responsive genes was further validated by RT-qPCR using samples generated as described above, with more time points (B05.10: 0.5, 1, 2, 3, 6, 9 and 24 h; M3a: 1, 3 and 9 h). Treatments with nystatin (Sigma Aldrich) or digitonin (Carl Roth) were carried out on B05.10 in a similar way and sampled at 1, 3 and 9 h after treatment. Expression of α-tomatine-responsive genes was investigated during tomato and N. benthamiana infection at different time points (tomato: 0, 6, 12, 18, 24, 30, 36 and 43 h post-inoculation; N. benthamiana: 0, 12, 24, 36 and 48 h post-inoculation). RT-qPCR was performed as described59 using primers listed in Supplementary Table 2.
Sensitivity test to membrane-disrupting compounds using plate assays
Mycelium was plated on GB5 agar plates containing 10 mM sucrose, 10 mM phosphate buffer (pH 6.5) and varying amounts of α-tomatine (B05.10 800 µM; M3A 400 µM), digitonin (B05.10 30 µM; M3A 20 µM). Agar plugs of 5 mm were taken from the growing edge of B. cinerea colonies on MEA plates and placed on the saponin-containing plates with mycelium facing the agar. Plates were incubated at 20 °C and colony diameters were measured at 3 dpi.
The sensitivity assays using spot inoculation were performed as described for N. crassa33. Serial dilutions of conidia (105–102 per droplet) of wild type B05.10 and various mutant strains were plated on BDES medium and on BDES medium containing a concentration range of α-tomatine (100–400 μg/mL). Growth was documented after 3 dpi.
Comparisons of mutants with their respective recipient wild type were always performed with two independently obtained mutants, in at least two biological repetitions with different batches of plates.
M3a genome sequencing, assembly and comparative genomics analysis with B05.10
Genomic DNA of B. cinerea strain M3a was extracted and sequenced by Illumina and Nanopore sequencing. The genome of M3a was assembled using Oxford Nanopore reads. Adapters of the reads were trimmed with porechop, version 0.2.4 (https://github.com/rrwick/Porechop), and the assembly was carried out using NECAT60. An assembly of 43.8 Mb was obtained with 23 contigs, of which four contained telomeric repeats on both ends and 12 contained telomeric repeats on one end. After assembly, we polished the genome using Illumina short reads using Pilon61. Gene annotation was performed using FUNANNOTATE v.1.8.962 with B. cinerea strain B05.10 proteins36 as external evidence.
The genomes of B05.10 and M3a were compared using four different methods. First, a global alignment between the two genomes was produced using the NUCmer algorithm63. Second, an alignment-free comparison was performed using the shortest unique substrings (shustring) algorithm64. Third, short reads obtained from Illumina sequencing on the M3a genome were aligned to the B05.10 genome using the BWA-MEM algorithm. Fourth, long reads obtained from Nanopore sequencing on M3a were aligned to the B05.10 genome using Minimap265. Based on the Illumina reads alignment, Single Nucleotide Polymorphisms (SNPs) and small insertions and deletions (indels) were mapped using a FreeBayes algorithm66. The snpEff algorithm was used to predict the effect of SNPs on proteins67.
Phylogenetic analysis
The NCBI web-based BLAST tool was used to identify homologs of BcTOM1 with NCBI non-redundant databases. Homologs were selected based on BLAST criteria that at least 80% of query length is covered by the hit sequence with at least 70% of sequence identity. The homologs were aligned using MAFFT with default settings68 and alignment was curated using Gblocks69 with non-stringent parameters. The phylogenetic tree of the curated alignment was constructed using RAxML70 with 100 as bootstrap value.
LC-MS analysis of steroidal glycoalkaloids
Flash-frozen material was powdered using a mortar and pestle cooled with liquid nitrogen. An aliquot of 50 (±2) mg fresh weight from each ground sample was extracted with 99% methanol containing 0.133% formic acid (FA) in a 3:1 ratio (μL methanol-FA: mg sample), followed by sonication and centrifugation for 15 min each. Chromatographic separation was performed using an Acquity UPLC module (Waters) on a reversed Luna C18 column (2.0 × 150 mm and 3 µm (Phenomenex, the Netherlands)) at 40 °C, using a linear gradient from 5 to 75% acetonitrile (0.1% FA) at a flow rate of 0.19 mL/min in 45 min. The injection volume was 5 μL. Detection of compounds eluting from the column was performed with a Q‐Exactive Plus Orbitrap FTMS mass spectrometer (Thermo Scientific). Full scan MS data were generated with electrospray in switching positive/negative ionization mode at a mass resolution of 35.000 in a range of m/z 95–1350. Subsequent MS/MS experiments for the identification of selected metabolites were performed with positive electrospray ionization at a normalized collision energy of 27 and a mass resolution of 17.500. The ionization voltage was optimized at 3.5 kV; the capillary temperature was set at 250 °C; the auxiliary gas heater temperature was set to 220 °C; sheath gas, auxiliary gas and the sweep gas flow were optimized at 36, 10 and 1 arbitrary units, respectively. Automatic gain control was set at 3 e6 and the injection time at 100 ms. MS/MS fragmentation patterns of steroidal glycoalkaloids were manually compared to those in the literature. Having analyzed in positive ESI mode, the main detected ions were [M+H]+ and its formic acid adduct [M+FA+H]+. The in-source and MS/MS fragments for most compounds comprised a loss of hexose (neutral loss of 162.05282), deoxyhexose (neutral loss of 146.05791), pentose (neutral loss of 150.052823), a methyl group (neutral loss of 14), CO2 (neutral loss of 44), and the aglycone of the related metabolite. All these indicative masses were used, when possible, to verify the annotations of the identified compounds (Supplementary Table 3).
For quantifications of α-tomatine and tomatidine, samples were diluted ten times by pipetting 30 µL of α-tomatine extract in 270 µL 50% v/v methanol prior to analysis on an Acquity Ultra Performance LC (Waters, Milford, USA) combined with a Xevo TripleQuad MS (Waters). Samples were placed in an autosampler kept at 10 °C and steroidal glycolkaloids were separated chromatographically on a Waters Acquity UPLC® BEH C18 1.7 µm 2.1 × 100 mm column (Waters) at 40 °C using 0.1% v/v formic acid in water (A) and methanol (B). The eluent gradient started with 0.25 min 95% A, followed by 1 min 80% A, 2.5 min 75% A, 1.7 min 68% A, 4.6 min 15% A, 2.95 min 95% A. The flow rate was 0.4 mL/min. The column was washed in between samples with strong and weak wash, consisting of respectively 90% and 10% v/v acetonitrile. The mass spectrometer had ESI fitted as source combined with the following parameters: capillary voltage of 3 kV, cone voltage of 70 V, source temperature of 150 °C, desolvation temperature of 600 °C, desolvation gas flow of 1000 L/h, cone gas flow of 50 L/h and LM resolution 1 at 2.80. Multiple reaction monitoring (MRM) parameters for α-tomatine were: parent mass charge ratio of 1034.6 m/z, daughter mass charge ratio of 145.2 m/z, dwell time of 0.036 s, cone voltage of 70 V and collision voltage of 60 V. For tomatidine the MRM parameters were: parent mass charge ratio of 416.6 m/z, daughter mass charge ratio of 161.2 m/z, dwell time of 0.036 s, cone voltage of 45 V and collision voltage of 38 V. Relative quantification of α-tomatine and tomatidine was done by integration of the area under the curve using Masslynx version 4.1 (Waters).
α-Tomatine and digitonin degradation assay
Recombinant BcTOM1 protein was produced in E. coli strain BL21 using a pET-15b expression vector71. Then, 12.5 μg pure BcTOM1 protein was incubated with 15 µM of α-tomatine for 2 h. Isolates B05.10 and M3a and their transformants were grown in liquid as described above and incubated with 200 µM α-tomatine or 100 µM digitonin for 9 h.
General microscopy and analysis
Fungal cells were observed on a Zeiss Observer 2.1 microscope using Nomarski optics with a Plan-Neofluar 100×/1.30 oil immersion objective (420493-9900) with CoolLED pE4000 as a light source for fluorescence microscopy. Images were captured with a PCO Edge 5.5 Gold (16 bit) camera and analyzed using ImageJ.
Sample preparation for microscopy and quantitative recruitment assay
To analyze BcGT28a-GFP recruitment in response to nystatin and α-tomatine, 5 µL of 2 × 107 spores were incubated in 150 µL liquid mineral medium in Ibidi eight-well μ-slides (Sigma Aldrich) for 5–6 h at 20 °C. After imaging the untreated cells, 100 µL of 193 µM α-tomatine solution was added. Similarly, for nystatin, 10 µL of 0.05 mg/mL was added and mixed by pipetting and incubated for 5–10 min before analysis by Nomarski and fluorescence microscopy. For quantitative assays, 100 germlings were tested for BcGT28a-GFP recruitment. Each test was performed three times with multiple technical replicates.
Sterol staining
To stain membrane sterols in germlings, 100 μL of 100 μg/mL of filipin III solution in 1% (v/v) DMSO was added to the germlings grown in Ibidi eight-well μ-slides as mentioned above. Cells were incubated for 20 min and analyzed by fluorescence microscopy using a DAPI filter setup.
Software used
The following software packages were used during the research: porechop, version 0.2.4 (https://github.com/rrwick/Porechop); NECAT60; Pilon61; FUNANNOTATE (v.1.8.9)62; NUCmer algorithm63; shortest unique substrings (shustring) algorithm64; BWA-MEM algorithm; Minimap265; FreeBayes algorithm66; snpEff algorithm67; MAFFT with default settings68; Gblocks69 with non-stringent parameters; RAxML70 with 100 bootstrap value; Image Lab: 6.0.1; GraphPad Prism: 9.3.1.
Statistics and reproducibility
For plant infection experiments, leaves of appropriate size were excised from the plants grown in the greenhouse and transported to the laboratory. Leaves were randomly distributed over plastic trays, and inserted into floral foam. We systematically inoculated one leaf half with one fungal genotype (usually a wild type isolate) and the other half with a different fungal genotype (either a mutant of the same isolate or a different wild type isolate). The tray was closed with a transparent lid, and trays were incubated in random stacks at ambient temperature. Following 3–4 days of incubation, all lesions were measured using a digital caliper connected to a USB port. Every inoculation was done with two leaves, and every inoculation was repeated at least three times with different batches of plants at different times. For each type of mutant (~25 different in total), two independently obtained transformants were used in each experiment. In general, one infection experiment would yield ~30 datapoints per plant/fungal genotype combination.
No statistical method was used to predetermine the sample size. Statistical analyses were conducted with Student’s t-tests for the vast majority of experiments, specifically the fungal radial growth tests and plant infection assays. A chi-square test was used for the protein recruitment analysis shown in Fig. 7. All experiments with fungi in vitro were highly reproducible, while the plant infection assays could show some variation, either because of seasonal influences, the quality of the plant material or issues with humidity or temperature during incubation of inoculated plant material. If infection experiments showed deviating outcome, such data were excluded from analysis and the experiment was repeated with a new batch of plants. The experiments were not randomized and investigators were not blinded to allocation during experiments and outcome assessment, as this would interfere with the scoring.
Data availability
The sequence data in this manuscript are deposited in NCBI. Sequence reads for the RNAseq experiment on α-tomatine-induced gene expression (BioProject number PRJNA955032) were deposited under SRA accession numbers SRR24174271–SRR24174280. The genome assembly and annotation of B. cinerea isolate M3a are deposited under accession number JARWBL000000000, version JARWBL020000000. LC-MS raw data are deposited in the MassIVE database of CCMS (massive.ucsd.edu) with the identifier MSV000094370. Source data are provided with this paper.
References
Osbourn, A. E. Preformed antimicrobial compounds and plant defense against fungal attack. Plant Cell 8, 1821–1831 (1996).
Osbourn, A. E. Saponins and plant defence—a soap story. Trends Plant Sci. 1, 4–9 (1996).
Piasecka, A., Jedrzejczak-Rey, N. & Bednarek, P. Secondary metabolites in plant innate immunity: conserved function of divergent chemicals. New Phytol. 206, 948–964 (2015).
Akinpelu, D. A., Aiyegoro, O. A., Akinpelu, O. F. & Okoh, A. I. Stem bark extract and fraction of Persea americana (Mill.) exhibits bactericidal activities against strains of Bacillus cereus associated with food poisoning. Molecules 20, 416–429 (2014).
Augustin, J. M. et al. UDP-glycosyltransferases from the UGT73C subfamily in Barbarea vulgaris catalyze sapogenin 3-O-glucosylation in saponin-mediated insect resistance. Plant Physiol. 160, 1881–1895 (2012).
D’Addabbo, T. et al. Control of plant parasitic nematodes with active saponins and biomass from Medicago sativa. Phytochem. Rev. 10, 503–519 (2010).
Hussain, M. et al. Role of saponins in plant defense against specialist herbivores. Molecules 24, 2067 (2019).
Potter, D. A. & Kimmerer, T. W. Inhibition of herbivory on young holly leaves: evidence for the defensive role of saponins. Oecologia 78, 322–329 (1989).
Papadopoulou, K., Melton, R. E., Leggett, M., Daniels, M. J. & Osbourn, A. E. Compromised disease resistance in saponin-deficient plants. Proc. Natl Acad. Sci. USA 96, 12923–12928 (1999).
Sandrock, R. W. & VanEtten, H. D. Fungal sensitivity to and enzymatic degradation of the phytoanticipin α-tomatine. Phytopathology 88, 137–143 (1998).
Armah, C. N. et al. The membrane-permeabilizing effect of avenacin A-1 involves the reorganization of bilayer cholesterol. Biophys. J. 76, 281–290 (1999).
Claereboudt, E. J. S., Eeckhaut, I., Lins, L. & Deleu, M. How different sterols contribute to saponin tolerant plasma membranes in sea cucumbers. Sci. Rep. 8, 10845 (2018).
Sudji, I. R., Subburaj, Y., Frenkel, N., García-Sáez, A. J. & Wink, M. Membrane disintegration caused by the steroid saponin digitonin is related to the presence of cholesterol. Molecules 20, 20146–20160 (2015).
Westrick, N. M., Smith, D. L. & Kabbage, M. Disarming the host: detoxification of plant defense compounds during fungal necrotrophy. Front. Plant Sci. 12, 684 (2021).
Wubben, J. P., Price, K. R., Daniels, M. J. & Osbourn, A. E. Detoxification of oat leaf saponins by Septoria avenae. Phytopathology 86, 986–992 (1996).
Bowyer, P., Clarke, B. R., Lunness, P., Daniels, M. J. & Osbourn, A. E. Host range of a plant pathogenic fungus determined by a saponin detoxifying enzyme. Science 267, 371–374 (1995).
Carter, J. P., Spink, J., Cannon, P. F., Daniels, M. J. & Osbourn, A. E. Isolation, characterization, and avenacin sensitivity of a diverse collection of cereal-root-colonizing fungi. Appl. Environ. Microbiol. 65, 3364–3372 (1999).
You, Y. & van Kan, J. A. L. Bitter and sweet make tomato hard to (b)eat. New Phytol. 230, 90–100 (2021).
Carere, J. et al. A tomatinase-like enzyme acts as a virulence factor in the wheat pathogen Fusarium graminearum. Fungal Genet. Biol. 100, 33–41 (2017).
Lairini, K. & Ruiz-Rubio, M. Detoxification of α-tomatine by Fusarium solani. Mycol. Res. 102, 1375–1380 (1998).
Ökmen, B. et al. Detoxification of α-tomatine by Cladosporium fulvum is required for full virulence on tomato. New Phytol. 198, 1203–1214 (2013).
Sandrock, R. W., DellaPenna, D. & VanEtten, H. D. Purification and characterization of beta 2-tomatinase, an enzyme involved in the degradation of alpha-tomatine and isolation of the gene encoding beta 2-tomatinase from Septoria lycopersici. Mol. Plant Microbe Interact. 8, 960–970 (1995).
Sandrock, R. W. & VanEtten, H. D. The relevance of tomatinase activity in pathogens of tomato: disruption of the β2-tomatinase gene in Colletotrichum coccodes and Septoria lycopersici and heterologous expression of the Septoria lycopersici β2-tomatinase in Nectria haematococca, a pathogen of tomato fruit. Physiol. Mol. Plant Pathol. 58, 159–171 (2001).
Woodward, S. & Pegg, G. F. Synthesis and metabolism of α-tomatine in tomato isolines in relation to resistance to Verticillium albo-atrum. Physiol. Mol. Plant Pathol. 28, 187–201 (1986).
Quidde, T., Buttner, P. & Tudzynski, P. Evidence for three different specific saponin-detoxifying activities in Botrytis cinerea and cloning and functional analysis of a gene coding for a putative avenacinase. Eur. J. Plant Pathol. 105, 273–283 (1999).
Pareja-Jaime, Y., Isabel, M., Roncero, M. I. G. & Ruiz-Roldán, M. C. Tomatinase from Fusarium oxysporum f. sp. lycopersici is required for full virulence on tomato plants. Mol. Plant Microbe Interact. 21, 728–736 (2008).
Bouarab, K., Melton, R., Peart, J., Baulcombe, D. & Osbourn, A. A saponin-detoxifying enzyme mediates suppression of plant defences. Nature 418, 889–892 (2002).
Ito, S. et al. Tomatidine and lycotetraose, hydrolysis products of alpha-tomatine by Fusarium oxysporum tomatinase, suppress induced defense responses in tomato cells. FEBS Lett. 571, 31–34 (2004).
Martin-Hernandez, A. M., Dufresne, M., Hugouvieux, V., Melton, R. & Osbourn, A. E. Effects of targeted replacement of the tomatinase gene on the interaction of Septoria lycopersici with tomato plants. Mol. Plant Microbe Interact. 13, 1301–1311 (2000).
Défago, G. & Kern, H. Induction of Fusarium solani mutants insensitive to tomatine, their pathogenicity and aggressiveness to tomato fruits and pea plants. Physiol. Mol. Plant Pathol. 22, 29–37 (1983).
Steel, C. C. & Drysdale, R. B. Electrolyte leakage from plant and fungal tissues and disruption of liposomemembranes by α-tomatine. Phytochemistry 27, 1025–1030 (1988).
Walker, B. W., Manhanke, N. & Stine, N. J. Comparison of the interaction of tomatine with mixed monolayers containing phospholipid, egg sphingomyelin, and sterols. Biochim. Biophys. Acta 1778, 2244–2257 (2008).
Schumann, M. R., Brandt, U., Adis, C., Hartung, L. & Fleißner, A. Plasma membrane integrity during cell-cell fusion and in response to pore-forming drugs is promoted by the Penta-EF-Hand protein PEF1 in Neurospora crassa. Genetics 213, 195–211 (2019).
Williamson, B., Tudzynski, B., Tudzynski, P. & van Kan, J. A. L. Botrytis cinerea: the cause of grey mould disease. Mol. Plant Pathol. 8, 561–580 (2007).
Leisen, T. et al. CRISPR/Cas with ribonucleoprotein complexes and transiently selected telomere vectors allows highly efficient marker-free and multiple genome editing in Botrytis cinerea. PLoS Pathog. 16, e1008326 (2020).
van Kan, J. A. L. et al. A gapless genome sequence of the fungus Botrytis cinerea. Mol. Plant Pathol. 18, 75–89 (2017).
Lombard, V., Golaconda Ramulu, H., Drula, E., Coutinho, P. M. & Henrissat, B. The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 42, D490–D495 (2014).
Drula, E. et al. The carbohydrate-active enzyme database: functions and literature. Nucleic Acids Res. 50, D571–D577 (2022).
Valero Jiménez, C. A., Veloso, J., Staats, M. & van Kan, J. A. L. Comparative genomics of plant pathogenic Botrytis species with distinct host specificity. BMC Genomics 20, 203 (2019).
Valero-Jiménez, C. A. et al. Dynamics in secondary metabolite gene clusters in otherwise highly syntenic and stable genomes in the fungal genus Botrytis. Genome Biol. Evol. 12, 2491–2507 (2020).
Mercier, A. et al. Population genomics reveals molecular determinants of specialization to tomato in the polyphagous fungal pathogen Botrytis cinerea in France. Phytopathology 111, 2355–2366 (2021).
Ramirez-Estrada, K. et al. Tomato UDP-glucose sterol glycosyltransferases: a family of developmental and stress regulated genes that encode cytosolic and membrane-associated forms of the enzyme. Front. Plant Sci. 8, 984 (2017).
Soustre, I., Letourneux, Y. & Karst, F. Characterization of the Saccharomyces cerevisiae RTA1 gene involved in 7-aminocholesterol resistance. Curr. Genet. 30, 121–125 (1996).
Smith-Peavler, E. S. et al. RTA1 is involved in resistance to 7-aminocholesterol and secretion of fungal proteins in Cryptococcus neoformans. Pathogens 11, 1239 (2022).
Colmenares, A. J., Aleu, J., Durán-Patrón, R., Collado, I. G. & Hernández-Galán, R. The putative role of botrydial and related metabolites in the infection mechanism of Botrytis cinerea. J. Chem. Ecol. 28, 997–1005 (2002).
Schouten, A., van Baarlen, P. & van Kan, J. A. L. Phytotoxic NLPs from the necrotrophic fungus Botrytis cinerea associate with membranes and the nucleus of plant cells. New Phytol. 177, 493–505 (2008).
Cuesta Arenas, Y. et al. Functional analysis and mode of action of phytotoxic Nep1-Like Proteins of Botrytis cinerea. Physiol. Mol. Plant Pathol. 74, 376–386 (2010).
Leisen, T. et al. Multiple knockout mutants reveal a high redundancy of phytotoxic compounds that determine necrotrophic pathogenesis of Botrytis cinerea. PLoS Pathog. 18, e1010367 (2022).
Bi, K., Liang, Y., Mengiste, T. & Sharon, A. Killing softly: a roadmap of Botrytis cinerea pathogenicity. Trends Plant Sci. 28, 211–222 (2022).
Bulasag, A. S. et al. Botrytis cinerea tolerates phytoalexins produced by Solanaceae and Fabaceae plants through an efflux transporter BcatrB and metabolizing enzymes. Front. Plant Sci. 14, 1177060 (2023).
Schoonbeek, H., del Sorbo, G. & de Waard, M. A. The ABC transporter BcatrB affects the sensitivity of Botrytis cinerea to the phytoalexin resveratrol and the fungicide fenpiclonil. Mol. Plant Microbe Interact. 14, 562–571 (2001).
Schoonbeek, H., van Nistelrooij, J. G. M. & De Waard, M. A. Functional analysis of ABC transporter genes from Botrytis cinerea identifies BcatrB as a transporter of eugenol. Eur. J. Plant Pathol. 109, 1003–1011 (2003).
Stefanato, F. L. et al. The ABC transporter BcatrB from Botrytis cinerea exports camalexin and is a virulence factor on Arabidopsis thaliana. Plant J. 58, 499–510 (2009).
Wang, C., Zhang, J., Chen, H., Fan, Y. & Shi, Z. Antifungal activity of eugenol against Botrytis cinerea. Trop. Plant Pathol. 35, 137–143 (2010).
Vela-Corcia, D. et al. MFS transporter from Botrytis cinerea provides tolerance to glucosinolate-breakdown products and is required for pathogenicity. Nat. Commun. 10, 2886 (2019).
Zhang, M. et al. 2-Phenylethyl isothiocyanate exerts antifungal activity against Alternaria alternata by affecting membrane integrity and mycotoxin production. Toxins 12, 124 (2020).
Zhang, L. et al. A novel Zn2Cys6 transcription factor BcGaaR regulates D-galacturonic acid utilization in Botrytis cinerea. Mol. Microbiol. 100, 247–262 (2016).
Schumacher, J. Tools for Botrytis cinerea: new expression vectors make the gray mold fungus more accessible to cell biology approaches. Fungal Genet. Biol. 49, 483–497 (2012).
Qin, S. et al. Molecular characterization reveals no functional evidence for naturally occurring cross-kingdom RNA interference in the early stages of Botrytis cinerea-tomato interaction. Mol. Plant Pathol. 24, 3–15 (2023).
Chen, Y. et al. Efficient assembly of nanopore reads via highly accurate and intact error correction. Nat. Commun. 12, 60 (2021).
Walker, B. J. et al. Pilon: integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE 9, e112963 (2014).
Palmer, J. & Stajich, J. nextgenusfs/funannotate: funannotate v1.5.3. Zenodo https://doi.org/10.5281/zenodo.2604804 (2019).
Kurtz, S. et al. Versatile and open software for comparing large genomes. Genome Biol. 5, R12 (2004).
Haubold, B., Pierstorff, N., Möller, F. & Wiehe, T. Genome comparison without alignment using shortest unique substrings. BMC Bioinformatics 6, 123 (2005).
Li, H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100 (2018).
Garrison, E. & Marth, G. Haplotype-based variant detection from short-read sequencing. Preprint at https://arxiv.org/abs/1207.3907 (2012).
Cingolani, P. et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 6, 80–92 (2012).
Katoh, K. & Standley, D. M. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780 (2013).
Castresana, J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol. Biol. Evol. 17, 540–552 (2000).
Stamatakis, A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313 (2014).
Abedi, D., Beheshti, M., Najafabadi, A. J., Sadeghi, H. M. & Akbari, V. Optimization of the expression of genes encoding poly(3-hydroxyalkanoate) synthase from Pseudomonas aeruginosa PTCC 1310 in Escherichia coli. Avicenna J. Med. Biotechnol. 4, 47–51 (2012).
Acknowledgements
The research of Y.Y. and S.Q. was funded by the China Scholarship Council, while the research of H.M.S. is supported by TKI grant LWV21.291. The authors are grateful to the Tudzynski lab (WWU Münster, Germany) for providing the B. cinerea M3a isolate, and to Rachel Melton and Prof. Anne Osbourne (John Innes Center, Norwich, UK) for providing a Septoria lycopersici tomatinase construct.
Author information
Authors and Affiliations
Contributions
Y.Y., J.A.L.v.K. and A.F. designed the study; Y.Y., H.M.S., L.M., A.L.H.V., P.R., A.O., H.G.B., S.Q. and F.V. performed the experiments; Y.Y., X.S.K., F.P., E.A.C.C., I.K., A.F. and J.A.L.v.K. analyzed the data; Y.Y., H.M.S., L.M. and X.S.K. made the figures and tables; Y.Y., A.F. and J.A.L.v.K. wrote the manuscript; all authors approved the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Nathaniel Westrick and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
You, Y., Suraj, H.M., Matz, L. et al. Botrytis cinerea combines four molecular strategies to tolerate membrane-permeating plant compounds and to increase virulence. Nat Commun 15, 6448 (2024). https://doi.org/10.1038/s41467-024-50748-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-50748-5
- Springer Nature Limited