

Abstract
Mogrosides constitute a series of natural sweeteners extracted from Siraitia grosvenorii fruits. These mogrosides are glucosylated to different degrees, with mogroside V (M5) and siamenoside I (SIA) being two mogrosides with high intensities of sweetness. SgUGT94-289-3 constitutes a uridine diphosphate (UDP)-dependent glycosyltransferase (UGT) responsible for the biosynthesis of M5 and SIA, by continuously catalyzing glucosylation on mogroside IIe (M2E) and on the subsequent intermediate mogroside products. However, the mechanism of its promiscuous substrate recognition and multiple catalytic modes remains unclear. Here, we report multiple complex structures and the enzymatic characterization of the glycosyltransferase SgUGT94-289-3. We show that SgUGT94-289-3 adopts a dual-pocket organization in its active site, which allows the two structurally distinct reactive ends of mogrosides to be presented from different pockets to the active site for glucosylation reaction, thus enabling both substrate promiscuity and catalytic regioselectivity. We further identified a structural motif that is essential to catalytic activity and regioselectivity, and generated SgUGT94-289-3 mutants with greatly improved M5/SIA production from M2E in an in vitro one-pot setup.
Similar content being viewed by others
Introduction
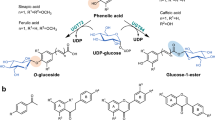
Mogrosides are a class of secondary metabolites found primarily in the fruit extract of Siraitia grosvenorii (Monk fruit or Luo Han Guo in Chinese), known for their high sweetness and low calorie content1,2. The Food and Drug Administration has approved S. grosvenorii fruit extract as a sugar substitute for food, with mogroside V (M5) being the major sweetness component3. Mogrosides comprise a family of triterpenoid saponins that are composed of a mogrol aglycone attached with multiple glucose groups4. These glucoses are linked to the hydroxyl groups on C24 and C3 positions (hereby termed R1 and R2 end) distributed on two ends of the mogrol aglycone, yielding a set of mogrosides with linear and branched sugar chains5 (Fig. 1a and Supplementary Table 1). The sweetness intensity of different mogrosides varies, and is determined by the number and more critically, the linkage pattern of glucose groups attached2,6,7. M5 contains five glucose groups and exhibits a relative sweetness intensity of approx. 400 times compared to aqueous sucrose of the same concentration8,9. A four glucose-containing mogroside isomer, siamenoside I (SIA), exhibits the highest sweetness intensity of over 500-fold compared to that of sucrose8,9.

a Chemical structures of various mogrosides. Glucose groups are colored blue for R1-G1 and R2-G1, red for R1-G2(1-6), green for R2-G2(1-6), and yellow for R1- G3(1-2). The atoms of O1, O2 and O6 of these glucose groups are labeled as 1, 2 and 6, respectively. b M5 biosynthesis pathway. Glucose groups are shown as spheres and colored the same as in (a). Reactions catalyzed by SgUGT94-289-3 are highlighted in the box. Reactions of β (1–6) glucosylation at R1 end, R2 end and β (1–2) glucosylation at R1 end are indicated by red, green and yellow arrows, respectively. The pathway from M3E to SIA is a minor pathway with low efficiency, thus the arrow is shown in 50% transparency. The UGT(s) responsible for producing M3E from M2E was not identified, thus this process is indicated by arrow with dashed line.
The production of M5 and SIA requires serial glucosylation events on mogrol, catalyzed by a set of uridine diphosphate (UDP)-dependent glycosyltransferases (UGTs). Recently, the precise steps involved in biosynthesis of mogrosides were identified10 (Fig. 1b). The first two steps of this pathway are catalyzed by SgUGT720-269-1, adding one glucose molecule each on aglycone at both R1 and R2 ends and yielding M2E, a bitter-tasting intermediate10. We termed the two glucoses as R1-G1 and R2-G1. SgUGT94-289-3 is responsible for the high sweetness of mogrosides by catalyzing the latter steps, transferring the glucose group from UDP-Glucose (UPG) to R1-G1 and R2-G1 of various mogrosides, thus generating M5 and several intermediate products. Previous reports showed that SgUGT94-289-3 catalyzes the transfer of the terminal glucose of UPG (UPG-Glc) onto mogrosides in at least three different modes10 (Fig. 1b). First, SgUGT94-289-3 catalyzes the conversion of M2E-to-M3 and M3E-to-SIA, adding the UPG-Glc on 6-OH of R1-G1 through β (1-6) glycosylation. The newly added glucose is termed R1-G2(1-6) hereafter. The second mode catalyzed by SgUGT94-289-3 involves the conversion of M3-to-M4A and SIA-to-M5, installing the UPG-Glc onto R2-G1 through β (1-6) linkage (the newly added glucose is termed R2-G2(1-6)). Moreover, SgUGT94-289-3 catalyzes the conversion of M3-to-SIA and M4A-to-M5, through β (1-2) linking of the UPG-Glc onto R1-G1 (the newly added glucose is termed R1-G3(1-2)) provided that the 6-OH of R1-G1 is already occupied.
SgUGT94-289-3 recognizes at least five different sugar acceptor substrates and adopts different catalytic modes, thus showing strong promiscuity (Fig. 1b)10. Substrate promiscuity is commonly found among plant UGTs11. Recently, the substrate-bound structures of Stevia rebaudiana UGT76G1 (SrUGT76G1) and Oryza sativa UGT91C1 (OsUGT91C1), two plant UGTs capable of adding glucose on two reactive ends of their sugar acceptors (steviol glucosides), were reported12,13, revealing that substrate promiscuity is a result of a large substrate binding pocket within the enzyme and non-specific enzyme-substrate interactions14. However, the steviol aglycone exhibits a pseudo-two-fold symmetry, whereas mogrosides are characterized by an asymmetric structure, with a longer and branched R1 end, as well as a shorter and linear R2 end (Supplementary Fig. 1). As a result, SgUGT94-289-3 may adopt a mechanism for promiscuous substrate recognition different from those of SrUGT76G1 and OsUGT91C1. Nevertheless, the structural basis of non-symmetric sugar acceptor recognition and catalysis of UGTs remain unclear.
Industrial production of mogrosides currently relies exclusively on extraction from mature S. grosvenorii fruits5,15,16. However, the low abundance and impurity limit the application of M5/SIA as sweeteners. Recently, an improved production of mogrol aglycone has been achieved using engineered biocatalysts17. Moreover, a recent study reported designed glycosylation routes by which three kinds of mogrosides (M5, M5A and M5X) were generated from the aglycone mogrol utilizing three engineered UGTs (SgUGT74AC1, OsUGT91C1 and MS1, a homologous protein of SgUGT94-289-3), with a total conversion rate of 99%18. Previous studies provided insights critical for improving mogrosides production through synthetic biology strategies4,17,18,19,20, however, efficient production of M5 and SIA, the mogrosides with high sweetness, remains challenging. Since SgUGT94-289-3 is responsible for most of the mogroside glucosylation steps for M5 and SIA biosynthesis, it is essential to understand the molecular mechanisms of sugar acceptor recognition and catalysis of SgUGT94-289-3.
Here, we set out to investigate the mechanisms of sugar acceptor recognition and catalysis of SgUGT94-289-3, and perform structure-based engineering of SgUGT94-289-3, resulting in mutated enzymes with greatly improved efficiency of M5/SIA production.
Results
Enzymatic characterization of SgUGT94-289-3
We expressed and purified the recombinant SgUGT94-289-3 protein (Supplementary Fig. 2), and measured its products specificity towards various sugar acceptors involved in M5 biosynthesis (M2E, M3, M3E, M4A, SIA). Albeit adopting different catalytic modes, we found that SgUGT94-289-3 largely maintained the dominance of only one specific product generated from each of the sugar acceptors, except for M4A (Fig. 2a, b). More specifically, SgUGT94-289-3 catalyzes the addition of R1-G2(1-6) (M2E-to-M3 and M3E-to-SIA conversion) with a product specificity of nearly 90%. Similarly, the product specificity of adding the R2-G2(1-6) (M3-to-M4A and SIA-to-M5 conversion) is approximately 70%. However, when we used M4A as sugar acceptor, we found that no single product was dominant, with M5 production (adding the R1-G3(1-2)) constituting less than 30% of all products. Moreover, our kinetic data (Supplementary Table 2) showed that SgUGT94-289-3 failed to reach a steady state when using M4A as sugar acceptor. These data suggested that the enzyme exhibits lower activity and/or less specificity for β (1-2) glucosylation than β (1-6) linkage. Furthermore, we also found that the enzyme exhibits higher Kcat/Km on the R1 end substrates (M2E and M3E) than on the R2 end substrates (M3 and SIA) (Supplementary Table 2), indicating a higher catalytic efficiency at the R1 end. Together, our results showed that while SgUGT94-289-3 exhibits promiscuous mogroside recognition and catalytic modes, it displays β (1-6) glucosylation preference and favors the R1 end than R2 end of mogrosides, thus exhibiting a relatively stringent regioselectivity on both the reactive end and the linkage of glycosidic bond.

a UPLC chromatographic curves of each single glucosylation step catalyzed by SgUGT94-289-3 towards different mogrosides, with the type of mogroside substrates and the reaction time labeled in the right. The peak of target product of each reaction is indicated by a black arrow. Mogrosides (peaks) containing the same number of glucoses are shown with the same background color. b Histogram of conversion efficiency of the target product using M2E, M3, M3E, M4A or SIA as substrates, calculated from the curves in (a). c Dynamics of products converted in an in vitro continuous glucosylation assay using M2E as the starting substrate. Time course: 720 min. SIA is not included in the calculation due to its very low production (yield of 2%). d Histogram of products’ conversion rate in different time points (0, 40, 300 and 720 min), calculated from the curves in (c). Conversion rate of each product (%) is represented by the percentage of corresponding product in total products. Data are presented as mean values ± SD (n = 3 biological replicates). The negative control group consisted of buffer without enzyme or substrates. Source data are provided as a Source Data file.
We further incubated purified SgUGT94-289-3 with M2E and UPG to verify its ability of in vitro M5 synthesis in a one-pot cascade. We measured the amounts of various mogrosides produced in a time course of 720 min, a time point at which steady-state was nearly reached (Fig. 2c, d and Supplementary Fig. 3). We showed that during the catalytic process, M2E was rapidly consumed after approx. 40 min reaction time, whereas M3 and M4A emerged almost immediately after the onset of the reaction. While M3 reached its maximal proportion in approx. 20 min before slowly decaying to undetectable levels, the amount of M4A exhibited its maximum levels at approx. 100 min. M5 appeared at approx. 60 min and slowly accumulated to the end of our measurements. Byproducts (other isomers that could not be identified due to lacking of standard mogrosides) became the most abundant type by approx. 180 min. Together, our results clearly showed that SgUGT94-289-3 catalyzes the sequential conversion from M2E to M3, then M4A and finally M5 in an in vitro one-pot setup. However, the M5 production is less than 10% in our in vitro assay, which is presumably a result of low β (1-2) glucosylation efficiency of SgUGT94-289-3 as revealed by our enzymatic assay (Fig. 2b).
To reconstitute the reaction of M5 production from M3E10, we incubated SgUGT94-289-3 with M3E and UPG. This resulted in sequential production of SIA and M5 with much lower byproducts accumulation (Supplementary Fig. 4). These results were consistent with our enzymatic data showing that SgUGT94-289-3 exhibits strong preference for β (1-6) linkage. However, the amount of M3E produced in S. grosvenorii fruits is negligible, suggesting that this pathway plays only a minor role in the in vivo biosynthesis of M5/SIA.
Overall structure of SgUGT94-289-3 and the bound UPG
To investigate the catalytic mechanisms of SgUGT94-289-3 and its binding mode with the sugar acceptor/donor, we solved ten crystal structures of SgUGT94-289-3 (Supplementary Tables 3, 4 and Supplementary Figs. 5, 6), namely the apo form (apo), the sugar donor UPG-bound (termed UPG) and UDP-bound forms (two structures termed UDP-1 and UDP-2), and six acceptor-bound forms. Two of the six acceptor-bound structures were obtained from wild type (WT) SgUGT94-289-3 bound with either M3 (termed M3) or M3E (termed M3E), while other structures were obtained using mutated SgUGT94-289-3 proteins generated in the present study (describe later), including V148G, G152S and V148W/G152S. These four structures were termed according to the sugar acceptor and the protein type, as SIAV148G, SIAV148W/G152S, M5V148G and M5G152A. All acceptor-bound structures contained one mogroside molecule accommodated in a same shallow groove on the surface of SgUGT94-289-3 (termed site 3). Moreover, we found a second mogroside bound inside the protein molecule in M3E, M3 and SIAV148G structures, but located at two different positions. While the apo, M3 and SIAV148G structures contain only one SgUGT94-289-3, other seven structures possess two SgUGT94-289-3 molecules in one asymmetric unit of the crystal (Supplementary Table 3). Both protein and ligands are well superposed between two molecules in each of these seven structures (Supplementary Fig. 7). Therefore, we only describe one molecule of these structures in the following text.
Analysis of our structures showed that SgUGT94-289-3 adopts a typical plant UGT fold21,22 (Fig. 3a), with its N- and C-terminal domain (NTD and CTD) responsible for binding sugar acceptor and donor, respectively23. The NTD consists of seven β-strands (Nβ1-Nβ7) surrounded by ten α-helices (Nα1-Nα10), whereas the CTD contains six β-strands and nine α-helices (Cβ1-6 and Cα1-9). All ten structures exhibit almost identical overall folding, especially their CTDs, but adopt slightly different conformations of the Nα6-Nα8 fragment (Fig. 3b). The Nα8 helix in apo, UDP-1 and UPG structures tilts ~30 degree compared with that in other structures. These conformational differences suggest that the Nα6-Nα8 region in SgUGT94-289-3 is inherently mobile, which may be related to its specific catalytic property.

a Cartoon and surface representation of UPG structure. NTD and CTD are colored marine and magenta, respectively, with secondary structures labeled. b Superposition of ten SgUGT94-289-3 structures. The zoom-in view of sugar donor and the Nα6-Nα8 region are shown in the left and the top, respectively. Conformational changes of Nα6-Nα8 region are indicated by red arrows. c The binding of UPG molecule. UPG structure is shown in surface mode (the same view as in a). UPG molecule is shown in stick-ball mode and residues involved in the UPG interactions are shown as sticks. The UPG-Glc is filled with brown color.
In all nine complex structures, the UPG/UDP molecules are closely superimposed (Fig. 3b), and accommodated within the CTD (Fig. 3c). The UPG-Glc is hydrogen-bonded with E273, D374 and Q375, and is located in proximity to conserved residues, H22 and D121, which correspond to the catalytic dyad in other UGT family members24,25 (Fig. 3c). Mutating any of these five residues resulted in complete loss of catalytic activity of SgUGT94-289-3 (Supplementary Fig. 8), highlighting their essential roles in binding the UPG molecule and/or catalyzing the glucose transfer reaction26. Alignment of our UPG structure with the previously reported structure of UPG-bound VvGT127 (PDB code: 2C1Z) showed that UPG molecules in the two structures are positioned at the same site, and adopt largely identical conformations (Supplementary Fig. 9). However, the UPG-Glc in VvGT1 structure is flipped perpendicularly to the UDP moiety, thus exposing the anomeric carbon atom (C1), representing a productive conformation of UPG. In contrast, in our structure, the UPG molecule adopts a relaxed conformation with the UPG-Glc extending towards catalytic center, thus shielding the catalytic C1 atom, suggesting a non-productive state of the UPG molecule in our structure. This conformational difference is presumably caused by the absence of sugar acceptor in our UPG structure, whereas a sugar acceptor (kaempferol) is present in VvGT1 structure and may induce the specific conformation of UPG.
The pocket 2 for sugar acceptor binding in SgUGT94-289-3
Two of our six acceptor-bound structures, namely M3 and SIAV148G, have the same acceptor binding mode, with one M3/SIA molecule binding to site 3, and the other M3/SIA located in a pocket shaped by Nα6-Nα8 region (Fig. 4a). The latter M3/SIA adopts a R2-in orientation, with the R2 end penetrating deeply inside the pocket and approaching to the catalytic center, thus we termed this site pocket 2. The M3 and SIA molecules are perfectly superposed in the two structures, especially their R2 ends (Fig. 4b), which are identical in the two acceptor molecules (Fig. 1a). Therefore, we only describe the M3 structure in the following text.

a Cartoon representation of M3 structure. Two M3 molecules are shown in stick-ball mode, and one of the two M3 molecules binds in pocket 2 and is further shown in surface mode, with its 6-OH (O6) of R2-G1 indicated by a red arrow. UDP and the catalytic residue H22 are shown as sticks. b Structure alignment of M3 and SIAV148G. NTD and CTD of M3 structure are shown in marine and magenta. SIAV148G structure is shown in gray. Substrates bound in M3 and SIAV148G structures are shown in purple and orange, respectively. c Superposition of M3 structure and the structure of OsUGT91C1 complexed with UDP and Reb E (PDB code 7ES0). Proteins are shown in cartoon mode, UDP, sugar acceptors and H22 are shown as sticks. Molecules M3 (green stick) and Reb E (white stick), including their oxygen atoms waiting to be glucosylated (colored red and indicated by arrows), are superposed well in pocket 2. d Binding of M3 molecule in pocket 2 and its interactions with SgUGT94-289-3. M3, UDP and adjacent residues are shown as stick-ball, stick and lines, respectively. Hydrogen-bond interactions are shown by black dashed lines. Hydrophobic residues are shown as dots.
Interestingly, the M3 structure aligned well with OsUGT91C1 structure (PDB code 7ES013 [https://doi.org/10.2210/pdb7es0/pdb]), which also contains a sugar acceptor (Rebaudioside E, Reb E) bound in the position corresponding to pocket 2 (Fig. 4c). The 6-OH of R2-G1 of the M3 molecule in pocket 2 is hydrogen-bonded with H22, and is located at the same position as 2-OH (the reactive group) of glucose 1-R2 (the glucose at the catalytic site) of Reb E in OsUGT91C1 structure, strongly indicating that M3 molecule in pocket 2 is in a productive state, and is ready to be glucosylated through β (1-6) linkage to produce M4A. Residues W17, H96 and E273 interact with other three hydroxyl groups of the R2-G1 of M3 molecule (Fig. 4d), suggesting that these residues play an important role in orientating the sugar acceptor in pocket 2 and determining the product specificity of SgUGT94-289-3. Moreover, the R2-G1 group of M3 molecule is sandwiched by L123 and V373 from both sides, and residues L197 and F377 stabilize the mogrol scaffold through hydrophobic interactions. V148, G152 and I156 on Nα6 also participate in the hydrophobic interaction with M3. These results strongly suggested that pocket 2 of SgUGT94-289-3 is able to accommodate sugar acceptors in R2-in orientation, and thus facilitating the β (1-6) glucosylation of their R2-G1 group.
The pocket 1 for sugar acceptor binding in SgUGT94-289-3
Next, we analyzed the sugar acceptor binding mode in our M3E structure, and found that in addition to one M3E molecule at site 3, another M3E occupies a pocket distinct from pocket 2, and formed by Nα4 and Nα8. The latter M3E molecule adopts a R1-in orientation, with its R1 end pointing towards the catalytic center (Fig. 5a). We thus named this binding site pocket 1. When we compared our M3E structure with other UGT structures, we found that the M3E molecule is located at a position similar to the sugar acceptor in the SrUGT76G1 structure (PDB code 6O8828) (Fig. 5b). However, the 6-OH group of M3E molecule is a little distant from the catalytic residue H22. The non-productive conformation of M3E in the structure is presumably due to a tris molecule in the catalytic site (Fig. 5c), which might be a result of the high concentrations of tris buffer (0.1 M) used in the crystallization solution. A stably bound tris molecule in the catalytic center was also observed in other UGT structures29,30, presumably because the hydroxyl groups of tris mimic those of the glucose moiety, thereby stabilizing its binding31.

a Cartoon representation of M3E structure. Two M3E molecules are shown in stick-ball mode, and one of the two M3E molecules binds in pocket 1 and is further shown in surface mode, with its 6-OH (O6) of R1-G1 indicated by a red arrow. UDP and a tris molecule are shown as sticks. b Superposition of M3E structure and the structure of SrUGT76G1 complexed with UDP and product Reb A (PDB code 6O88). Proteins are shown in cartoon mode, UDP, sugar acceptors and H22 are shown as sticks. Molecule M3E (yellow stick) and Reb A (white stick) can be aligned in pocket 1, and the oxygen atom waiting to be glucosylated in M3E and that already glucosylated in Reb A are shown as red spheres and indicated by arrows. c Binding of M3E and tris molecules in pocket 1 and their interactions with SgUGT94-289-3. M3E, tris and UDP are shown in ball-stick mode, residues nearby are shown as lines. Hydrogen-bond interactions are shown by black dashed lines. d Binding of M3E in pocket 1 after MD simulation. M3E and UPG are shown in ball-stick mode, residues nearby are shown as lines. The 6-OH (O6) of R1-G1 of M3E is indicated by a red arrow. Hydrogen-bond interactions are shown by black dashed lines. Hydrophobic residues are shown as dots.
To verify the binding mode of M3E in SgUGT94-289-3, we performed molecular dynamics (MD) simulation using a tris-free SgUGT94-289-3 model, in which a UDP molecule was replaced with a UPG. We found that the M3E molecule is stably bound within pocket 1 in the R1-in state, and moving closer to the UPG molecule at the end of simulation. The 6-OH of R1-G1 moves 4.3 Å towards the UPG-Glc and is positioned within a hydrogen bond distance (3.5 Å) to the catalytic residue H22 (Fig. 5d and Supplementary Fig. 10a–c). Moreover, we found that upon the M3E approaching, the UPG-Glc flips and switches to the active conformation, similar to that observed in VvGT1 structure (PDB code 2C1Z27) (Supplementary Fig. 10b). These findings suggest that the M3E molecule in the MD model represents its productive conformation. Based on our structural and simulation data, we propose that several residues, including W17, H96, L123 and V373 are presumably involved in the binding of the M3E molecule.
Although we failed to obtain the complex structures of SgUGT94-289-3 bound with M2E or M4A, we were able to dock the two sugar acceptors separately into the SgUGT94-289-3 structure and perform MD simulation. We found that both sugar acceptors adopt R1-in orientation and stably bind in pocket 1, similar to M3E (Supplementary Figs. 10d–i and 11). M2E exposes its 6-OH of R1-G1 closely to H22 and UPG-Glc (Supplementary Fig. 10d–f), supporting our enzymatic result that SgUGT94-289-3 prefers to catalyze the β (1-6) glucosylation of M2E. Moreover, the UPG molecule also adopts the active conformation in our simulation, with the flipped UPG-Glc (Supplementary Fig. 10e). For M4A, while its mogrol aglycone located in pocket 1, its R1-G2(1-6) group extends towards pocket 2. This conformation enables the exposure of the 2-OH (and other hydroxyl groups) of R1-G1 towards H22 (Supplementary Fig. 10g–i). Together, our results indicated that pocket 1 in SgUGT94-289-3 preferably binds sugar acceptors in R1-in orientation, which is available for further glucosylation on the R1-G1 group.
A dual-pocket mode for the catalysis of SgUGT94-289-3
Our M3E and M3 structures showed that although located in different pockets, M3E and M3 molecules interact with SgUGT94-289-3 in similar manners, namely through non-polar interactions surrounding the scaffold region, and additional hydrogen bond interactions with distinct reactive ends. We further superimposed the two structures and found that R1-G1 of M3E and R2-G1 of M3 are located in the same active site that is shaped by W17, H22, H96, L123 and V373 in SgUGT94-289-3 (Fig. 6a). While both substrates in M3E and M3 structures share similar residues for their stabilization, they also form distinct interactions with the enzyme.

a Superposition of M3 and M3E structures. Sugar acceptors are shown in lines. Residues crucial for recognition and binding of sugar acceptors in both pocket 1 and pocket 2 are shown in stick mode. R1-G1 in M3E (green) and R2-G1 in SIA (brown) are filled with green and brown color, respectively. b–e Catalytic activity and specificity of WT and mutated forms of SgUGT94-289-3 after 40 min reaction time. Conversion from M2E to M3 (b), from M3E to SIA (c), from M3 to M4A (d), and from SIA to M5 (e). The activity (%) was measured as substrate consumption (hollow) and the specificity (%) was measured as product yield (solid). b, c represent the case of R1-in substrates, while d, e represent that of R2-in substrates. Data are presented as mean values ± SD (n = 3 biological replicates). The WT enzyme-catalyzed reaction served as the positive control group, while the negative control group consisted of buffer without enzyme or substrates. Source data are provided as a Source Data file.
To verify the functional role of the dual-pocket organization in catalysis, we analyzed the enzymatic activities of single-site mutations targeting residues involved in binding M3E (pocket 1) and M3 (pocket 2). We found that these residues contribute differently to the catalytic activity and specificity on various mogroside substrates, in agreement with their specific locations on the enzyme (Fig. 6). Residue W17 is closely located to the catalytic residues H22 and D121 (Fig. 6a), and its bulky side chain may be critical for the stabilization and/or correct orientation of sugar acceptors. In line with this structural observation, we found that W17A mutation abolishes the activity of SgUGT94-289-3 (Fig. 6b–e). Residues H96 and V373 are located proximal to the G1 group of both reactive ends (Fig. 6a), accordingly, both H96A and V373A mutant forms exhibited decreased activity on all substrates. Moreover, H96A mutation greatly reduced the catalytic specificity on R2-in substrates (M3 and SIA) (Fig. 6d, e), confirming our suggestion that H96 is critical in orienting R2-G1 of M3/SIA for β (1-6) glucosylation (Fig. 4d). The L123A and L197G mutations reduced the catalytic efficiency of SgUGT94-289-3 on R1-in (M2E and M3E) and R2-in (M3 and SIA) substrates, respectively (Fig. 6b–e). These results are in line with our structures showing that L123 lies in pocket 1 whereas L197 in pocket 2.
In summary, our enzymatic assay of SgUGT94-289-3 mutants confirmed our structural and simulation findings, demonstrating that SgUGT94-289-3 utilizes a dual-pocket mode for mogroside binding and catalysis. The enzyme binds sugar acceptors with R1-in orientation (M2E, M3E, M4A) in pocket 1, whereas those in R2-in state (M3 and SIA) in pocket 2. The orientation-specific assignment of sugar acceptors in different pockets may be determined by the asymmetric structure of mogrosides and the different shapes of the two pockets. Our structural analysis showed that the opening of pocket 1 is wider and that of pocket 2 is narrower (Supplementary Fig. 12a), thus the branched and bulky R1 may enter the wider pocket 1, whereas the linear R2 end is able to enter the narrower pocket 2. Furthermore, sugar acceptors may enter the wider pocket 1 more easily, which may explain our enzymatic results that SgUGT94-289-3 favors the R1 end of mogrosides during catalysis (Fig. 2b).
The site 3 for sugar acceptor binding
Comparison of M3 and M3E structures also revealed conformational changes occurred in the Nα6-Nα8 region (Supplementary Fig. 12b). Nα8 is located more distally from Nα4 in the M3E structure, but away from Nα6 in the M3 structure, thus creating space for substrate binding in each pocket. These results confirmed our previous observation that the Nα6-Nα8 region is mobile, and highlighted the important role of this region in facilitating sugar acceptor binding and directing.
Interestingly, in all acceptor-bound structures, we observed one acceptor molecule bound at site 3, which is on the molecular surface and located close to Nα8 (Supplementary Fig. 13a). These acceptor molecules adopt an identical orientation and superimposed well, with the R1 end pointing towards pocket 1 and R2 end approaching pocket 2. We therefore hypothesized that site 3 may promote the shuttling of acceptors between pocket 1 and 2, thus facilitating the alternating glucosylation of different ends of sugar acceptors. In addition, mogrosides are non-polar chemicals with low solubility in aqueous solution, thus they may prefer to attach to the mostly hydrophobic groove on the protein surface. Site 3 may serve as a hub that recruits substrate and exchanges more hydrophilic products with more hydrophobic substrate. A similar case was found in the enzyme Diphosphoinositol pentakisphosphate kinase 232, which possesses a second substrate binding pocket at the protein surface adjacent to the primary catalytic pocket, serving to capture the substrates from the bulk phase. However, site 3 is distantly located from the catalytic center (Supplementary Fig. 13a), and our enzymatic assay showed that mutation of residues in site 3 has minor effect on the catalytic activity of SgUGT94-289-3 (Supplementary Fig. 13b), suggesting that this site might be an artifact not present under physiological conditions.
Continuous catalytic model of SgUGT94-289-3
Based on our structural and enzymatic analysis, we propose a model that describes the continuous conversion from M2E to M5 catalyzed by SgUGT94-289-3, and explains the promiscuity and stringent regioselectivity exhibited during SgUGT94-289-3’s catalysis on different mogrosides (Supplementary Fig. 14). Initially, the sugar acceptor M2E prefers to enter pocket 1 due to its wider opening, and adopts the R1-in orientation, thus exposing the 6-OH of R1-G1. Simultaneously, UPG binds to the active site where it is poised to donate the UPG-Glc to M2E. Through a series of potential conformational changes, M2E is glucosylated at the 6-OH of R1-G1, yielding the major product M3. Following the consumption of M2E and the accumulation of M3, SgUGT94-289-3 utilizes M3 as its substrate. At that time, SgUGT94-289-3 presumably exhibits a lower affinity for the R1 end of M3 because its sophorose moiety may introduce growing steric hindrance to pocket 1, while pocket 2 exhibits higher affinity for its R2 end. Thus SgUGT94-289-3 binds M3 with R2-in orientation in pocket 2, with the 6-OH of R2-G1 exposed to the catalytic site. The UPG-Glc is then transferred to R2-G1 of M3 through β (1-6) glycosidic bond formation, yielding M4A. M4A subsequently enters pocket 1, positioning its R1-G1 group at the catalytic site, and is converted into M5, accompanied by various additional byproducts.
Residue important for β (1-2) glucosylation activity
Although SgUGT94-289-3 is capable of conducting a continuous glucosylation reaction on M2E to produce M5, the conversion is inefficient due to the production of large quantities of unwanted byproducts. To identify the potential sites that may be responsible for increasing the regioselectivity and activity of SgUGT94-289-3, we constructed a series of single mutations targeting the key structural elements identified in our structures, especially the Nα6-Nα8 region, generating over 70 mutants (Fig. 7a). We then screened these mutants in a continuous glucosylation assay for increased accumulation of M4A production after 40 min, and found that three mutants, namely V148M, G152A and S185L, exhibited significantly increased M4A production when compared to production by WT forms (Fig. 7b). Notably, all three mutated positions are located within the Nα6-Nα8 region, with V148 and G152 on Nα6, and S185 on Nα7. By extending the reaction time to 720 min, the S185L mutant was excluded from further analysis because of more accumulation of byproduct and reduced M5 production (Supplementary Fig. 15a, b).

a Mapping of mutated residues (salmon surface) of SgUGT94-289-3. b M4A production of mutants compared to WT in the time course of 40 min and using M2E as the substrate. Positive hits are highlighted in red and labeled. Conversion rates of SIA (c), M5 (d) and total byproducts (e) by WT and mutated forms of SgUGT94-289-3 in a continuous glucosylation assay, using M2E as the only input sugar acceptor, and monitored at 40, 300 and 720 min. Better-performed mutants are highlighted with red star. f Conversion rate dynamics of various products during continuous glucosylation reactions, catalyzed by WT and four mutants with improved M5 production. Data are presented as mean values ± SD (n = 3 biological replicates). The WT enzyme-catalyzed reaction served as the positive control group, while the negative control group consisted of buffer without enzyme or substrates. Source data are provided as a Source Data file.
We next generated three additional mutant forms targeting the residue at position 148, namely V148G (no side chain), V148F and V148W (bulky side chain), and calculated the yield of all intermediate products of the four 148-mutants at three intervals of time (40, 300 and 720 min), using M2E as the sugar acceptor (Fig. 7c–e and Supplementary Fig. 15c, d). We found that at longer reaction times (300 and 720 min), V148G exhibited lower activity compared to WT enzyme. In comparison, V148M and V148F mutants exhibited greatly increased M5 production (Fig. 7d), and higher catalytic efficiency (Kcat/Km) of M4A-to-M5 conversion (Supplementary Table 2), with V148F being more efficient. These results suggested that these two mutants exhibit higher β (1-2) glucosylation activity. Structural comparison of WT (M3 structure) and 148 mutants (SIVV148G, M5V148G and SIAV148W/G152S structures) showed that these mutants exhibit identical conformation as WT (Supplementary Fig. 16a), thus the effect of V148 mutation on catalysis should be a result of the change of side chains. Analysis of our structures showed that V148 is located inside pocket 2, proximally to the catalytic site, thus mutation of V148 to residues with larger side chain narrows down the space of the active center at the side of pocket 2. Our MD simulation models of WT and V148F mutant bound with M4A suggested that the R1-G2(1-6) group of M4A extends into pocket 2, which may facilitate the exposure of the 2-OH group of R1-G1 to the catalytic residue H22 (Supplementary Fig. 16b). During the simulation, the 2-OH of R1-G1 in the V148F mutant is positioned closer to the C1 of UPG than that in WT (Supplementary Fig. 16c), presumably because the narrower pocket 2 restricts the mobility of R1-G2(1-6) and thus helps to orient the 2-OH of R1-G1 towards the catalytic residue. In comparison, V148G mutation creates a larger space of the catalytic center, hence reducing the conversion rate of M5 (Fig. 7d). Curiously, V148W possesses the largest side chain, but does not exhibit increased activity as V148F/V148M. Superposition of our M3 and SIAV148W/G152S structures showed that the side chain of W148 overlaps with the M3 molecule in pocket 2 (Supplementary Fig. 16d, e). As a consequence, slight conformational change of mogrosides and/or a rotamer switch of W148 side chain may be necessary for mogroside substrates to be fully accommodated in pocket 2. This might be one of the reasons why V148W mutation did not promote the enzymatic activity of SgUGT94-289-3. Together, our results indicated that a moderately large residue at the position 148 increases β (1-2) glucosylation activity and the regioselectivity on M4A.
Residue important for catalytic efficiency
Our enzyme kinetics analysis showed that the G152A mutant form exhibits significantly higher catalytic activity compared to WT (Supplementary Table 2), resulting in a greatly increased M4A-to-M5 conversion (Fig. 7d), but also considerable amounts of unwanted byproducts (Fig. 7e). These results indicated that the G152A mutation greatly stimulated the enzymatic activity of SgUGT94-289-3, but showed negligible effects on its catalytic specificity. Similar results were also obtained from G152S and G152T mutants (Fig. 7c–e). Our M5G152A and SIAV148W/G152S structures revealed that mutation of G152 to residues with small side chain failed to result in conformational changes compared to WT (Supplementary Fig. 16a). G152 is positioned in pocket 2, similar to V148, but is located more distant from the catalytic center than V148. Thus, replacing the glycine at the position 152 with small side chain residues (A/S/T) may affect the enzymatic activity, but not specificity of SgUGT94-289-3. Intriguingly, compared to WT, G152A exhibited comparable catalytic efficiency (1.33 and 2.42 times) on R1-in substrate (M2E and M3E), but drastically higher efficiency (11.57 and 368.22 times) on R2-in substrate (M3 and SIA) (Supplementary Table 2), suggesting that enzyme activity was stimulated, particularly for R2 end reactions. The underlying reason for this observation could not be explained solely from structural data, and the mechanism requires further investigation.
SgUGT94-289-3 mutants with highly efficient M5 production
We further generated double mutants (V148M/F/W together with G152A) based on our above findings, hoping to combine the effects of mutations at positions 148 and 152. As expected, the double mutants exhibit greatly increased M5 yields in the continuous glucosylation system as well as a decrease in accumulated byproducts (Fig. 7d, e). Particularly, V148M/G152A mutant exhibited the highest M5 yields (94%), while simultaneously we observed minimum amount of unwanted byproducts (3%). Interestingly, we noticed that at the 300-minutes time point, V148F and V148W mutations increased the M3-to-SIA conversion compared to WT. Similarly, double mutants V148F/G152A and V148W/G152A exhibit increased SIA production, accompanied with decreased M3-to-M4A conversion (Fig. 7c, f). These experimental data suggested that mutation of V148F/G152A and V148W/G152A of SgUGT94-289-3 change its end-selectivity from R2 end to R1 end, and enhance the β (1-2) glucosylation activity on R1 end of M3, thus producing higher amount of SIA. The increased M3-to-SIA conversion is also presumably a result of the narrower catalytic center at the side of pocket 2 in these mutant enzymes.
Together, our work presents a set of SgUGT94-289-3 derived catalysts, such as V148F, V148M/G152A and V148W/G152A, with high regioselectivity and catalytic efficiency on M5 and SIA production (Fig. 7f). Using the primary mogroside M2E as sole input, our established in vitro one-pot synthesis system with a single catalyst achieved production of up to 94% M5 (V148M/G152A) and 43% SIA (V148W/G152A), with minimal unwanted byproducts.
Discussion
UGTs constitute a large superfamily of enzymes and are usually able to catalyze the glycosylation of multiple sugar acceptor substrates or of the distinct reactive ends of one substrate33. The recognition and properly binding of various sugar acceptors are the prerequisite of catalysis of UGTs. Structures of two plant UGTs, SrUGT76G1 and OsUGT91C1, in complex with their sugar acceptors have been reported12,13,28,34, showing that both enzymes use one single pocket to bind and catalyze glycoside substrates with two distinct orientations. While the almost symmetric structure of steviol aglycone and non-specific hydrophobic enzyme-substrate interaction allow the one-pocket mode for SrUGT76G1 and OsUGT91C1, mogrol exhibit apparent difference of the two reactive ends in both structure and size (Supplementary Fig. 1). If SgUGT94-289-3 utilizes the one-pocket mechanism, it may face a tradeoff between large pocket size to match the larger reactive end and a reasonable pocket size to maintain regioselectivity on the smaller reactive end. We showed here that SgUGT94-289-3 adopts a hitherto unknown dual-pocket organization, accommodating the two structurally distinct reactive ends of mogrosides separately into the two pockets, thus achieving a broader substrate spectrum while stringent regioselectivity. Interestingly, the sugar acceptor binding pocket in SrUGT76G1 and OsUGT91C1 structures correspond to pocket 1 and pocket 2 in SgUGT94-289-3, respectively, implying that the dual-pocket organization of SgUGT94-289-3 is developed from the one-pocket mode, and may ubiquitously exist in other UGTs. Many triterpenoid saponins possess various glycosylation modifications and usually exhibit non-symmetric structure11. Glycosylation on their different reactive ends requires multiple UGTs acting synergistically35. However, the utilization of multiple-enzyme pathways may lead to increased complexity, making the industrial synthesis of target products less practical. Developing one enzyme capable of catalyzing several reaction steps and yielding the final product independently constitutes a promising approach for directed synthesis of target products36. The dual-pocket mode found in SgUGT94-289-3 could be applied to design and engineer other UGTs that catalyze sugar acceptors with distinct/non-symmetric reactive ends.
Furthermore, our structure-based engineering of SgUGT94-289-3 revealed that mutation of V148 to residues with moderately large side chains increases β (1-2) glucosylation activity on R1 end, whereas mutation of G152 to residues with small side chains greatly enhances the catalytic activity of SgUGT94-289-3 but has minor effects on regioselectivity. Furthermore, double mutants V148M/G152A, V148F/G152A and V148W/G152A exhibit greatly improved efficiency of M5 and SIA production when compared to the WT form (Fig. 7f). Both positions (148 and 152) are located at Nα6, suggesting that this structural motif may serve as a key element in both regioselectivity and catalytic efficiency of SgUGT94-289-3. In conclusion, our work showed it is possible to develop efficient biocatalysts for single-pot synthesis of M5/SIA products, and should provide a reference for future engineering of SgUGT94-289-3 and potentially other plant UGTs.
Methods
Molecular cloning
The gene encoding SgUGT94-289-3 was amplified from a cDNA library of S. grosvenorii and cloned into pET28a (+) plasmid (Merck Millipore, 69864) using Gibson assembly method (Transgen), with both N- and C-terminus linked with one hexa-His tag. Primer sequences to subclone SgUGT94-289-3 to pET28a are as follows: Forward primer 5′- ACTGGTGGACAGCAAATGGGTCGCGGAATGGATGCTGCCCAACAAG-3′ and reverse primer 5′- ATCTCAGTGGTGGTGGTGGTGGTGCTCTATTTTAAGCAAGAGAGAAATTTCAGCGAC-3′. All the point mutations were generated using Gibson assembly method. For the structural based manipulation of SgUGT94-289-3, a list of mutated positions is provided in Supplementary Table 5. These residues were mutated to alanine and/or other conserved residues among the homologous UGTs from Cucurbitaceae.
Protein expression and purification
The recombinant plasmid was transformed into E. coli Rosetta (DE3) competent cells. The transformed cells were cultured in Lysogeny Broth medium containing 50 µg/ml kanamycin and shaken under 220 r.p.m. at 37 °C until the O.D.600 reaching around 0.8. The protein expression was induced by adding 0.5 mM IPTG in the culture which was shaken under 180 r.p.m. at 18 °C for additional 20 h. The cells were harvested by centrifugation at 6000 × g for 10 min at 4 °C and resuspended in the Lysis Buffer (50 mM Tris-HCl, pH 8.0, 500 mM NaCl, 5% glycerol and 10 mM imidazole). The cells were then lysed by sonication, and centrifuged at 18,000 × g for 40 min at 4 °C. The supernatant was loaded onto Ni-NTA column (GE Life Sciences) pre-equilibrated with Lysis Buffer. The column was washed with 50 volumes of wash buffer (50 mM Tris 8.0, 500 mM NaCl, 100 mM Imidazole) and subsequently eluted with elution buffer (50 mM Tris 8.0, 500 mM NaCl, 300 mM Imidazole). The eluate was buffer-exchanged to buffer A (20 mM Tris-HCl pH 8.0, 0.1 mM EDTA pH 8.0, 5 mM β-ME) with a 30kD-cutoff centrifugal filter (Merck Millipore, C7719) and directly loaded onto RESOURCE Q anion exchange chromatography column (Cytiva). The column was equilibrated with buffer A and eluted with linear gradient of buffer B (buffer A supplemented with 500 mM NaCl), with the target protein eluted under the salt concentration of ~150 mM. Homogeneity of purified protein sample was confirmed by size exclusion chromatography using Superdex 200 (Cytiva). The protein sample was desalted and concentrated to 15 mg/ml, and stored at −80 °C for future use.
Crystallization
All the sugar acceptors used for soaking or co-crystallization with SgUGT94-289-3 were dissolved in 50% DMSO, resulting in 80 mM stock solutions of M2E, M3, M3E, SIA and M5. UDP and UPG were prepared as 40 mM stock solution in water (Supplementary Table 1). All the crystals were grown by the hanging-drop vapor diffusion method at 16 °C. Crystals of apo SgUGT94-289-3 (used to solve apo structure) were obtained from the reservoir solution containing 0.1 M Tris-HCl pH 8.0, 35% (w/v) polyethylene glycol 3350, 0.2 M NaCl and 4% (v/v) Polypropylene glycol P400. The crystals of SgUGT94-289-3 in complex with UPG (used to solve UPG structure) were obtained by soaking apo protein crystals overnight in the same reservoir solution supplemented with 4 mM UPG. The crystals of SgUGT94-289-3 in complex with UDP were obtained by incubating protein with either 4 mM UPG (for UDP-1 structure), or 4 mM UPG and 8 mM M2E (for UDP-2 structure) for 30 min, and crystallized under the same reservoir condition of apo protein. Crystals used to solve M3E, M3 and SIAV148G structures were obtained by incubating SgUGT94-289-3 proteins with 4 mM UDP and 8 mM of either M3E, M3 or SIA prior to crystallization. Crystals for M3E structure was grown in reservoir solution containing 0.1 M Tris-HCl pH 8.0, 35% (w/v) polyethylene glycol 3350, 0.2 M NaCl and 4% (v/v) Polypropylene glycol P400. Crystals for M3 structure were grown using 0.2 M sodium iodide and 20% (w/v) polyethylene glycol 3,350 pH 7.0. Crystals for SIAV148G structure were obtained in reservoir solution composed of 4% v/v TacsimateTM pH 7.0 and 12% w/v Polyethylene glycol 3350. The crystals used for solving other acceptor-bound complex structures were all obtained by incubating the proteins with 4 mM UDP and 8 mM of corresponding sugar acceptor prior to crystallization under the same reservoir conditions of the apo-protein. Crystals were directly flash-frozen in liquid nitrogen without additional cryoprotection. The use of UDP instead of UPG for incubation with the enzyme and various mogrosides is to avoid proceeding of the catalytic reaction during the long period time of crystallization, thereby enabling the obtaining of the acceptor-bound complex of SgUGT94-289-3.
Data collection and structure determination
The X-ray diffraction data sets were collected under cryogenic conditions at 100 K at beamline BL17U137, BL18U1, and BL19U138 of Shanghai Synchrotron Radiation Facility at a wavelength of 0.97918 Å. Diffraction data were processed and scaled with XDS package39,40. The UDP-1 structure was solved by molecular replacement method using the phaser software in PHENIX package41, and the structure of TcCGT1 (PDB code 6JTD42) was used as a search model. Other structures were solved by phaser using the refined UDP-1 structure as an initial search model. Structures were manually adjusted by Coot43, and refined by Phenix. refine41. Ramachandran plot statistics based on an analysis of each structure and a good quality model would be expected to have over 95% in the most favored regions. A summary of data collection and structure refinement statistics is provided in Supplementary Table 3. All the figures of the structures were prepared by PyMOL44.
Docking and molecular dynamics simulation
The M3E structure was used to generate a tris-free SgUGT94-289-3 structural model, by manually removing tris and M3E molecules. The V148F mutant model was generated through mutagenesis in PyMOL44. Missing residues in loop regions and mutated residue were refined by Rosetta45. M2E, M3E and M4A molecules were separately docked in SgUGT94-289-3 structural models (WT and V148F) using LeDock46. LeDock is based on a combination of simulated annealing and evolutionary optimization of the ligand pose (position and orientation) and its rotatable bonds, using a physics/ knowledge hybrid scoring scheme derived from prospective virtual screening campaigns. The hydrogen atoms were added to these molecules using Openbabel47. A root-mean-square deviation (RMSD) value and the number of binding poses were set to 1 and 1000, respectively. Models of SgUGT94-289-3 in complex with M2E, M3E or M4A with the lowest energy state were chosen as the initial model for MD simulation analysis.
For MD simulation, the UDP molecule in each model was replaced by UPG molecule. All MD simulations were performed by GROMACS version 2019.648,49, using the Amber14SB_OL15 force field50. Ligand (UPG and sugar acceptor) charges and atom types were assigned with the AM1-BCC model51 and the second-generation generalized AMBER force field (GAFF2)52,53, respectively. The system was solvated with a transferrable intermolecular potential with three points (TIP3P)54 explicit solvent model in a cubic box with a 10 Å buffer region and electrically neutralized with sodium ions. The number of each type of molecules involved in system are listed in Supplementary Table 6. Energy minimization was performed with the steepest descent algorithm. Equilibration was performed with harmonic restraints (1000 kJ/mol/nm2) on heavy atoms with a stepdown equilibration. The 100 ps NPT ensemble was used and the timestep was 2 fs. Temperature and pressure regulations were controlled by the V-rescale55 and Parrinello-Rahman56 methods, respectively. Short-range nonbonded interactions were calculated with a cutoff of 1.0 nm, and long-range electrostatic interactions were calculated with Particle-Mesh-Ewald57,58. Hydrogen bonds were constrained with the LINCS method59. The production simulations were conducted for 130 ns in the NPT ensemble. Each production MD simulation had been considered as independent action and repeated at three times with random velocity seed for generating initial speed. The related indices to evaluate the equilibration of production MD runs, include protein backbone RMSD, potential energy, temperature and pressure variations from simulation system, shown in Supplementary Fig. 17. All repeated MD simulations yielded similar results, and the data of only one replicate was provided as a representative.
In vitro glucosylation assay and products preparation
In vitro glucosylation assay was performed in the reaction buffer containing 50 mM PBS, pH 6.5 and 5 mM β-mercaptoethanol. Mogrosides (M2E, M3, M3E, M4A, SIA and M5) were dissolved in 50% DMSO to a concentration of 10 mM. UPG was dissolved in water to 80 mM concentration.
Following reaction setup, 0.5 mM mogrosides, 8 mM UPG and 10 µg purified enzymes were mixed into a reaction system of 100 μl. The reaction system was incubated at 45 °C for different time courses and was terminated by adding 50 μl of methanol into the system following 5 min incubation at 95 °C. The reaction system was centrifuged at 12,000 × g for 10 min. The supernatant containing the glucosylated products was collected, stored at 4 °C for further UPLC or UPLC-ESI-QTOF-MS/MS analysis.
Ultra performance liquid chromatography analyses
Glucosylated products were analyzed on a Waters ACQUITY UPLC I-Class system (Waters, Milford, MA, USA) with diode array detector (Waters), using an ACQUITY UPLC BEH C18 column (2.1 × 100 mm, 1.7 μm particle, Waters) with flow rate of 0.25 ml/min and the injection volume of 5 µl at 30 °C. The mobile phase is consisted of 0.1% formic acid in water (v/v, solvent A) and pure acetonitrile (solvent B) with a gradient elution: 0–10 min, 21–28% (solvent B). The detection wavelength was 203 nm. The target products were identified by comparing the elution time and Mass spectrometry with those of mogroside standards (Chengdu Must Bio-Technology Co., Ltd). The products were quantified by extra-standard method of chromatographic peak area. Conversion rate was calculated by dividing the quantity of certain products by that of total products.
Mass spectrometric analysis
The qualitative analysis of the glycosylated products was conducted using the Xevo G2-XS Q/TOF mass spectrometer (Waters) system in the negative mode of the electrospray ionization (ESI) interface. The desolvation gas flow rate was set to 600 L/h at 250 °C. The cone gas was set to 50 L/h and the source temperature was set at 100 °C. The capillary and cone voltages were set at 2.5 kV and 40 V, respectively. MS/MS fragment information was obtained using a collision energy ramp from 10 to 45 V. Mass spectrometry was performed in full scan mode from m/z 50 to 1500. Accurate mass and fragment ions were got using MassLynx™ software (version 4.1, Waters). One set of samples was analyzed.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The atomic coordinates and crystallographic structure factors generated in this study have been deposited into the protein data bank (PDB) database under accession codes: 8HJQ (apo), 8HJP (UDP-1), 8HJO (UDP-2), 8HJN (UPG), 8J66 (M3), 8HJL (M3E), 8HJK (SIAV148G), 8HJH (SIAV148W/G152S), 8HJG (M5V148G), 8HJF (M5G152A). The mass spectrometry data have been deposited into the Science Data Bank [https://doi.org/10.57760/sciencedb.09947]. The molecular dynamics trajectories and parameters files generated in this study are provided in Supplementary Data 1. PDB codes of previously published structures used in this study are 7ES0, 6O88, 6JTD and 2C1Z. Source data are provided with this paper.
References
Pawar, R. S., Krynitsky, A. J. & Rader, J. I. Sweeteners from plants-with emphasis on Stevia rebaudiana (Bertoni) and Siraitia grosvenorii (Swingle). Anal. Bioanal. Chem. 405, 4397–4407 (2013).
Chun, L. I. et al. Chemistry and pharmacology of Siraitia grosvenorii: a review. Chin. J. Nat. Med. 12, 89–102 (2014).
Soejarto, D. D., Addo, E. M. & Kinghorn, A. D. Highly sweet compounds of plant origin: from ethnobotanical observations to wide utilization. J. Ethnopharmacol. 243, 112056–112066 (2019).
Seki, H., Tamura, K. & Muranaka, T. Plant-derived isoprenoid sweeteners: recent progress in biosynthetic gene discovery and perspectives on microbial production. Biosci. Biotechnol. Biochem. 82, 927–934 (2018).
Gong, X. et al. The fruits of Siraitia grosvenorii: a review of a Chinese food-medicine. Front. Pharmacol. 10, 1400–1411 (2019).
Wang, L. et al. Cucurbitane glycosides derived from mogroside IIE: structure-taste relationships, antioxidant activity, and acute toxicity. Molecules 19, 12676–12689 (2014).
Kim, S.-K., Chen, Y., Abrol, R., Goddard, W. A. & Guthrie, B. Activation mechanism of the G protein-coupled sweet receptor heterodimer with sweeteners and allosteric agonists. Proc. Natl Acad. Sci. 114, 2568–2573 (2017).
Matsumoto, K., Kasai, R., Ohtani, K. & Tanaka, O. Minor cucurbitane-glycosides from fruits of Siraitia grosvenori (Cucurbitaceae). Chem. Pharm. Bull. 38, 2030–2032 (1990).
Murata, Y. et al. Sweetness characteristics of the triterpene glycosides in Siraitia grosvenori. J. Jpn. Soc. Food Sci. 53, 527–533 (2006).
Itkin, M. et al. The biosynthetic pathway of the nonsugar, high-intensity sweetener mogroside V from Siraitia grosvenorii. Proc. Natl Acad. Sci. 113, E7619–E7628 (2016).
Kurze, E. et al. Structure-function relationship of terpenoid glycosyltransferases from plants. Nat. Prod. Rep. 39, 389–409 (2022).
Yang, T. et al. Hydrophobic recognition allows the glycosyltransferase UGT76G1 to catalyze its substrate in two orientations. Nat. Commun. 10, 3214–3228 (2019).
Zhang, J. et al. Catalytic flexibility of rice glycosyltransferase OsUGT91C1 for the production of palatable steviol glycosides. Nat. Commun. 12, 7030–7042 (2021).
Hult, K. & Berglund, P. Enzyme promiscuity: mechanism and applications. Trends Biotechnol. 25, 231–238 (2007).
Fry, J. C. Natural low-calorie sweeteners. in Natural Food Additives, Ingredients and Flavourings (eds. Baines, D. & Seal, R.) 41–75 (Woodhead Publishing, 2012).
Shivani et al. Introduction, adaptation and characterization of monk fruit (Siraitia grosvenorii): a non-caloric new natural sweetener. Sci. Rep. 11, 6205–6018 (2021).
Wang, S. et al. Construction and optimization of the de novo biosynthesis pathway of mogrol in Saccharomyces cerevisiae. Front. Bioeng. Biotech. 10, 919526–919540 (2022).
Li, J. et al. Glycosyltransferase engineering and multi-glycosylation routes development facilitating synthesis of high-intensity sweetener mogrosides. iScience 25, 105222–105240 (2022).
Philippe, R. N., De Mey, M., Anderson, J. & Ajikumar, P. K. Biotechnological production of natural zero-calorie sweeteners. Curr. Opin. Biotechnol. 26, 155–161 (2014).
Xu, Y. et al. Engineering of a UDP-glycosyltransferase for the efficient whole-cell biosynthesis of Siamenoside I in Escherichia coli. J. Agric. Food Chem. 70, 1601–1609 (2022).
Zhang, P., Zhang, Z., Zhang, L., Wang, J. & Wu, C. Glycosyltransferase GT1 family: phylogenetic distribution, substrates coverage, and representative structural features. Comput. Struct. Biotechnol. J. 18, 1383–1390 (2020).
Tiwari, P., Sangwan, R. S. & Sangwan, N. S. Plant secondary metabolism linked glycosyltransferases: an update on expanding knowledge and scopes. Biotechnol. Adv. 34, 714–739 (2016).
Aubuchon, L. N., Schmiederer, K. & Lee, S. G. Comparative structural analysis of plant uridine diphosphate (UDP)-dependent glycosyltransferases (UGTs) in plant specialized metabolism: structures of plant UGTs for biosynthesis of steviol glycosides. Phytochem. Rev. 22, 385–406 (2023).
Lairson, L. L., Henrissat, B., Davies, G. J. & Withers, S. G. Glycosyltransferases: structures, functions, and mechanisms. Annu. Rev. Biochem. 77, 521–559 (2008).
Wang, X. Structure, mechanism and engineering of plant natural product glycosyltransferases. FEBS Lett. 583, 3303–3309 (2009).
Hui, S. et al. Crystal structures of a multifunctional triterpene/flavonoid glycosyltransferase from Medicago truncatula. Plant Cell 17, 3141–3154 (2005).
Wendy, O. et al. Structure of a flavonoid glucosyltransferase reveals the basis for plant natural product modification. EMBO J. 25, 1396–1405 (2006).
Lee, S. G., Salomon, E., Yu, O. & Jez, J. M. Molecular basis for branched steviol glucoside biosynthesis. Proc. Natl Acad. Sci. 116, 13131–13136 (2019).
Brazierhicks, M. et al. Characterization and engineering of the bifunctional N- and O-glucosyltransferase involved in xenobiotic metabolism in plants. Proc. Natl Acad. Sci. 104, 20238–20243 (2007).
Huang, W., He, Y., Jiang, R., Deng, Z. & Long, F. Functional and structural dissection of a plant steroid 3-O-glycosyltransferase facilitated the engineering enhancement of sugar donor promiscuity. ACS Catal. 12, 2927–2937 (2022).
Lariviere, L. & Morera, S. A base-flipping mechanism for the T4 phage beta-glucosyltransferase and identification of a transition-state analog. J. Mol. Biol. 324, 483–490 (2002).
Wang, H. et al. Synthetic inositol phosphate analogs reveal that PPIP5K2 has a surface-mounted substrate capture site that is a target for drug discovery. Chem. Biol. 21, 689–699 (2014).
He, B. et al. Glycosyltransferases: mining, engineering and applications in biosynthesis of glycosylated plant natural products. Synth. Syst. Biotechnol. 7, 602–620 (2022).
Liu, Z., Li, J., Sun, Y., Zhang, P. & Wang, Y. Structural Insights into the catalytic mechanism of a plant diterpene glycosyltransferase SrUGT76G1. Plant Commun. 1, 100004–100015 (2020).
Rahimi, S. et al. Triterpenoid-biosynthetic UDP-glycosyltransferases from plants. Biotechnol. Adv. 37, 107394–107412 (2019).
Nazor, J., Liu, J. & Huisman, G. Enzyme evolution for industrial biocatalytic cascades. Curr. Opin. Biotechnol. 69, 182–190 (2021).
Wang, Q.-S. et al. Upgrade of macromolecular crystallography beamline BL17U1 at SSRF. Nucl. Sci. Tech. 29, 68 (2018).
Zhang, W.-Z. et al. The protein complex crystallography beamline (BL19U1) at the Shanghai Synchrotron Radiation Facility. Nucl. Sci. Tech. 30, 170 (2019).
Kabsch, W. Xds. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132 (2010).
Kabsch, W. Integration, scaling, space-group assignment and post-refinement. Acta Crystallogr. D Biol. Crystallogr. 66, 133–144 (2010).
Adams, P. D. et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 (2010).
He, J. B. et al. Molecular and structural characterization of a promiscuous C-glycosyltransferase from Trollius chinensis. Angew. Chem. Int. Ed. Engl. 58, 11513–11520 (2019).
Emsley, P. & Cowtan, K. Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 (2004).
Schrödinger, L. The PyMOL molecular graphics system, version 1.3r1. (2010).
Rohl, C. A., Strauss, C. E. M., Misura, K. M. S. & Baker, D. Protein structure prediction using Rosetta. Methods Enzymol. 383, 66 (2003).
Wang, Z. et al. Comprehensive evaluation of ten docking programs on a diverse set of protein–ligand complexes: the prediction accuracy of sampling power and scoring power. Phys. Chem. Chem. Phys. 18, 12964–12975 (2016).
O’Boyle, N. M. et al. Open Babel: an open chemical toolbox. J. Cheminformatics 3, 1–14 (2011).
Pronk, S. et al. GROMACS 4.5: a high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics 29, 845–854 (2013).
Van Der Spoel, D. et al. GROMACS: fast, flexible, and free. J. Comput. Chem. 26, 1701–1718 (2005).
Maier, J. A. et al. ff14SB: improving the accuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theory Comput. 11, 3696–3713 (2015).
Jakalian, A., Jack, D. B. & Bayly, C. I. Fast, efficient generation of high-quality atomic charges. AM1-BCC model: II. Parameterization and validation. J. Comput. Chem. 23, 1623–1641 (2002).
Li, J. et al. Efficient O-glycosylation of triterpenes enabled by protein engineering of plant glycosyltransferase UGT74AC1. ACS Catal. 10, 3629–3639 (2020).
Wang, J., Wolf, R. M., Caldwell, J. W., Kollman, P. A. & Case, D. A. Development and testing of a general amber force field. J. Comput. Chem. 25, 1157–1174 (2004).
Jorgensen, W. L., Chandrasekhar, J., Madura, J. D., Impey, R. W. & Klein, M. L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79, 926–935 (1983).
Bussi, G., Donadio, D. & Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 126, 014101 (2007).
Parrinello, M. & Rahman, A. Polymorphic transitions in single crystals: a new molecular dynamics method. J. Appl. Phys. 52, 7182–7190 (1981).
Darden, T., York, D. & Pedersen, L. Particle mesh Ewald: an N⋅ log (N) method for Ewald sums in large systems. J. Chem. Phys. 98, 10089–10092 (1993).
Essmann, U. et al. A smooth particle mesh Ewald method. J. Chem. Phys. 103, 8577–8593 (1995).
Hess, B., Bekker, H., Berendsen, H. J. & Fraaije, J. G. LINCS: a linear constraint solver for molecular simulations. J. Comput. Chem. 18, 1463–1472 (1997).
Acknowledgements
We thank the staffs at the Shanghai Synchrotron Radiation Facility (Shanghai, China) for technical support during diffraction data collection. The project was funded by the National Natural Science Foundation of China (U20A2004 to X.M.), the Strategic Priority Research Program of CAS (XDB37020101 to M.L.), the CAMS Innovation Fund for Medical Sciences (CIFMS) (2021-I2M-1-071 to X.M.), the National Laboratory of Biomacromolecules (2023kf08 to X.M. and M.L.), the CAS Project for Young Scientists in Basic Research (#YSBR-015 to X.S.) and the Youth Innovation Promotion Association at the Chinese Academy of Sciences (Y2022038 to X.S.).
Author information
Authors and Affiliations
Contributions
M.L., X.M. and Z.L. conceived the project; S.C., S.Z. and M.L. designed the research; S.C. performed the expression, purification, crystallization, and the activity assays; S.Z. performed data processing and structure determination; N.W. and S.C. performed MD simulation; X.S. and Z.L. helped in experiments and data analysis; M.L., S.C. and S.Z. analyzed the data and wrote the manuscript with input from all authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Ramon Hurtado-Guerrero and the other, anonymous, reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Cui, S., Zhang, S., Wang, N. et al. Structural insights into the catalytic selectivity of glycosyltransferase SgUGT94-289-3 towards mogrosides. Nat Commun 15, 6423 (2024). https://doi.org/10.1038/s41467-024-50662-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-50662-w
- Springer Nature Limited