

Abstract
Engineering stabilized proteins is a fundamental challenge in the development of industrial and pharmaceutical biotechnologies. We present Stability Oracle: a structure-based graph-transformer framework that achieves SOTA performance on accurately identifying thermodynamically stabilizing mutations. Our framework introduces several innovations to overcome well-known challenges in data scarcity and bias, generalization, and computation time, such as: Thermodynamic Permutations for data augmentation, structural amino acid embeddings to model a mutation with a single structure, a protein structure-specific attention-bias mechanism that makes transformers a viable alternative to graph neural networks. We provide training/test splits that mitigate data leakage and ensure proper model evaluation. Furthermore, to examine our data engineering contributions, we fine-tune ESM2 representations (Prostata-IFML) and achieve SOTA for sequence-based models. Notably, Stability Oracle outperforms Prostata-IFML even though it was pretrained on 2000X less proteins and has 548X less parameters. Our framework establishes a path for fine-tuning structure-based transformers to virtually any phenotype, a necessary task for accelerating the development of protein-based biotechnologies.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Introduction
The ability to predict and understand the change in protein thermodynamic stability (ΔΔG) for an amino acid substitution is a core task for the development of protein-based biotechnology, such as industrial biocatalysts1,2,3 and pharmaceutical biologics4,5,6,7. Proteins with enhanced thermodynamic stability are less prone to unfolding and aggregation and are more engineerable8,9; stabilizing the scaffold of a protein enables downstream exploration of potentially destabilizing mutations that may improve a target function8. Thermodynamic stability is measured by the change in Gibbs free energy (ΔG) between the native and unfolded state and reflects the underlying integrity of the global structure. Engineering proteins is a very laborious process and bottlenecks the development of protein-based biotechnologies10,11, making the development of computational methods that can accurately predict ΔΔG of a point mutation, and in turn identify stabilizing mutations, a highly active research area12,13,14,15,16.
Deep learning is currently revolutionizing many physical and biological disciplines17,18,19,20,21, with AlphaFoldV2 leading the way as the scientific breakthrough of the year in 202122,23 and sparking an entire wave of deep learning structure prediction tools24,25. Although a variety of sequence-based26,27,28,29 and structure-based30,31,32 deep learning frameworks for stability prediction have been reported, the lack of data and machine learning engineering issues have prevented deep learning algorithms from having a similarly revolutionary impact on protein stability prediction (see Supplementary Section A for details on these current deep learning frameworks).
Systematic analyses13,15,16,33 of the state-of-the-art (SOTA) computational stability predictors published over the last 15 years highlight data scarcity, variation, bias, leakage, and poor metrics for evaluating model performance as the critical issues hindering meaningful progress (see Supplementary Section B for a summary on the primary data issues). As a result, the community still primarily relies on physics-based methods, such as Rosetta34 and FoldX35, or shallow machine learning approaches36,37,38,39,40,41,42,43,44. While current SOTA computational tools often report 75-80% accuracies upon publication, these accuracies primarily reflect their performance on destabilizing mutations, which make up a majority of the test set and their identification is key for identifying pathogenic variants13,14,33. However, when evaluated for predicting stabilizing mutations on the datasets by third parties, about 20% of predictions are actually stabilizing13,14,33. Although stabilizing mutations are naturally less common45, poor generalization to stabilizing mutations has been demonstrated to be a result of pervasive data leakage between train-test splits and severe class imbalance (destabilizing mutations make up >70%) in the current training sets used by the computational stability prediction community and needs to be addressed for machine learning-guided protein engineering13,14,16,33. Finally, current computational tools are evaluated with Pearson correlation and RMSE as the primary metrics. Due to the significant imbalance between stabilizing (<30%) and destabilizing mutations in training and test sets and the innate variations associated with measuring ΔΔG15, improvements in these metrics do not necessarily translate into improvements for identifying stabilizing mutations13,33. Thus, metrics such as precision, recall, area under receiver operating characteristic (AUROC), and Matthew’s correlation coefficient (MCC) should also be considered when developing a model for protein engineering applications13,14,33 (See Supplementary Section B for a summary of metric issues).
In this work, we address these key issues and develop a robust stability prediction framework for engineering proteins. We present Stability Oracle: a structure-based deep learning framework that makes use of several innovations in data and machine learning engineering specific to stability prediction. Stability Oracle uses a graph-transformer architecture that treats atoms as tokens and utilizes their pairwise distances to inject a structural inductive bias into the attention mechanism. The input to Stability Oracle consists of the local chemistry surrounding a residue with the residue deleted (the masked microenvironment) and two amino acid embeddings to represent a specific point mutation. This design decision enables Stability Oracle to generate all 380 possible point mutations from a single microenvironment, circumventing the need for computationally generated mutant structures and making deep mutational scanning inference computationally inexpensive. To improve the generalization capability of Stability Oracle, we introduce a data augmentation technique—thermodynamic permutations (TP). Thermodynamic permutations, similar to thermodynamic reversibility (TR)46, are based on the state-function property of Gibbs free energy47. For a specific position in a protein, TP expands n empirical ΔΔG measurements into n(n − 1) thermodynamically valid measurements, thus increasing the dataset by up to an order of magnitude based on the number of residues that have multiple amino acids experimentally characterized (See Supplementary Fig. 2). Compared to TR, TP enlarges the mutation types sampled across microenvironments in a training or test set, attenuating mutation type bias produced by alanine scanning experiments. Furthermore, unlike TR, it generates a balanced set of stabilizing and destabilizing mutations to non-wild-type amino acids. This allows us to better assess generalization for protein engineering, as the goal is to mutate away from the wild type. We curate three datasets (C2878, cDNA117K, T2837) to address data leakage issues. MMseqs248 was used to generate these three datasets and ensure the maximum overlap between proteins in the training and test set is below 30% sequence similarity, a threshold within the “twilight zone” where 95% of protein pairs will have different structural folds49. Concat 2878 (C2878) and Test 2837 (T2837) datasets are new training and test splits from previously established training and test sets, respectively. The third (cDNA117K) is a curated subset of the natural domains from cDNA display proteolysis dataset #150, a dataset of ~850 K thermodynamic folding stability measurements across 354 natural and 188 de novo mini-protein domains (40-72 amino acids). This latter dataset is especially interesting because of its size and because it relies upon proteolytic stability as a surrogate for thermodynamic stability. Finally, we generate a sequence-based counterpart for Stability Oracle by fine-tuning ESM225 on our curated training and test sets using the Prostata framework26 and train Prostata-IFML. With Prostata-IFML, we conduct head-to-head comparisons to demonstrate the advantage of structure over sequence-based methods. Overall we show that Stability Oracle and Prostata-IFML are state-of-the-art structure and sequence-based frameworks for computational stability prediction, respectively.
Results
Designing a graph-transformer framework for structure-based protein engineering
In prior work, we have experimentally shown that representations learned via self-supervised deep learning models on masked microenvironments (MutCompute51,52 and MutComputeX53) can identify amino acid residues incongruent with their surrounding chemistry. These models can be used to “zero-shot" predict gain-of-function point mutations51,52,53,54,55, including in protein active sites of computational structures53. Self-supervised models, however, generate mutational designs that are not biased towards a particular phenotype and do not update predictions based on experimental data19.
The MutCompute framework uses a voxelized molecular representation51,53. For protein structures, voxelization is a suboptimal molecular representation, as most voxels consist of empty space and rotational invariance is not encoded. Furthermore, the MutCompute frameworks use convolution-based architectures, which lag behind modern attention-based architectures in terms of representation learning and predictive power.
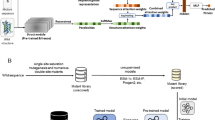
To develop a more powerful and generalizable framework for downstream tasks, we first built MutComputeXGT, a graph-transformer version of MutComputeX (Fig. 1a). Each atom is represented as a node with atomic elements, partial charges56, and SASA57 values as features with pairwise atomic distances labeling edges. Our graph-transformer architecture converts the pairwise distances into continuous and categorical attention biases to provide a structure-based inductive bias for the attention mechanism. To generate likelihoods for the masked amino acid, we average the final-layer hidden representations of all atomic tokens within 8Å of the masked Cα. The design decision to narrow the pooling to atoms in the first contact shell of the masked amino acid is based on insights from systematic variation of the microenvironment volume when training self-supervised 3DCNNs58. With a similar number of parameters and the same train-test splits, MutComputeXGT demonstrates superior representation learning capacity than MutComputeX, achieving wild-type accuracy of 92.98% ±0.26% compared to ~85%53.

a Self-supervised pre-training graph-transformer architecture (MutComputeXGT). b Fine-tuning of the pre-trained graph-transformer backbone for stability regression (Stability Oracle). In the regression head, we represent a mutation with “FromAA" and “ToAA" CLS tokens, which are the structural amino acid embeddings for the corresponding amino acids. c, d demonstrates how Stability Oracle combines structural amino acid embeddings and one masked microenvironment to generate thermodynamic permutations (TP) augmentation mutation inputs. Here, ΔΔG measurements at PDB:5UCE W43 (yellow transparent spheres) for mutations to both LEU and ARG enable the generation of the TP mutations c from LEU to ARG and d from ARG to LEU by simply swapping the order of the structural amino acid embeddings provided to the regression head. A diagram further describing TP is provided in Supplementary Fig. 2.
The Stability Oracle architecture makes use of both the feature-extractor and the classification head of MutComputeXGT for supervised fine-tuning (Fig. 1b). Previous structure-based stability predictors30,31,32,34,59,60 require two structures—either experimental or computational—to explicitly model the wild type and mutant amino acids. This second (mutant) structure is typically obtained using computational techniques such as AlphaFold or Rosetta. The drawbacks of this approach are (1) computational methods become expensive at inference time (as we describe below) and (2) it is difficult to evaluate the quality of computationally derived mutant structures. In contrast, Stability Oracle does not rely on a second structure. More specifically, structural features from the local chemistry surrounding a particular residue (the masked microenvironment) are extracted from a single initial structure, and a mutation is represented as a pair of “from” and “to” amino acid embedding vectors. To model the ΔΔG of a specific mutation, the microenvironment of the initial structure is used to contextualize the “from” and “to” amino acid embeddings in the regression head (as illustrated in Fig. 1b). This architectural design allows the framework to implicitly learn how “from” and “to” amino acids interact with the local chemistry, rather than relying on a computational structure prediction tool to provide chemical interactions. For a typical 300 amino acid protein, prior work would generate 5700 computational mutant structures (from Rosetta34 or AlphaFold22) in order to predict the ΔΔG of every possible single-point mutation during inference. Stability Oracle, on the other hand, requires only one structure to predict the ΔΔG for all 19 amino acid substitutions at every residue ( ~50 ms/residue). Runtime performance metrics on proteins of varying lengths are provided in Supplementary Table 1. The “from” and “to” amino acid embeddings are derived from the weights of the final layer of the MutComputeXGT classifier. This design decision is based on the insight that the weights of these 20 neurons represent the similarity of a microenvironment’s features to each of the 20 amino acids prior to being normalized into a likelihood distribution. Thus, they are structure-based contextualized embeddings of the 20 amino acids self-supervised pre-trained from a 50% sequence similarity representation of the Protein Data Bank (PDB)61, we will refer to them as Structural Amino Acid Embeddings.
We highlight several design decisions of the regression head used in Stability Oracle. Of note is the use of a Siamese62 attention architecture that treats the mutation embeddings as two classification (CLS) tokens (Fig. 1b). CLS tokens are commonly used in the natural language processing (NLP) community to capture the global context for downstream tasks63. Since atoms and amino acids are chemical entities at different scales, we designed the regression head so that a particular microenvironment contextualizes the “From” and “To” amino acid-level CLS tokens. Once contextualized for a given microenvironment, the two amino acid CLS tokens are then subtracted from each other to produce a mutation-hidden representation, which is then decoded into a ΔΔG prediction. This design enforces the state-function of the property of Gibbs free energy64, providing the proper inductive bias for Thermodynamic Reversibility and “self-mutations" (where ΔΔG = 0 kcal/mol).
Training Stability Oracle to generalize across protein chemistry
The Stability Oracle framework was designed to generalize across all 380 mutation types at all positions within a protein structure. The development of such a model has historically been limited by data scarcity, bias, and leakage. To address these issues, we curated training and test datasets and developed a data augmentation technique called thermodynamic permutations (TP).
It is well-known that a major issue with prior works is the inclusion of similar proteins between the training and test set (“data leakage”)42, resulting in poor evaluation of generalization13,14,33,42. It has been demonstrated that train-test splits at the mutation, residue, or protein level result in overfitting to the validation set, and strict sequence clustering is required to ensure proper evaluation of generalization13,14,33,42. Thus, we created new train-test splits based on a 30% sequence similarity threshold computed by MMSeqs248. First, we built the T2837 test set, which we then used to remove any homologous proteins from the remaining experimental data to produce the C2878 training set. The same procedure was used to construct the cDNA117K training set from the single mutant subset that had experimental structures available of the recently published cDNA-display proteolysis Dataset #150 (Fig. 2).

Even with the T2837 expanded test set, we are still unable to assess generalization performance on 14% of the 380 mutation types since they are not represented in T2837. Additionally, T2837 is heavily biased with mutations to alanine (Fig. 3a, bottom row), further hindering our ability to evaluate the generalization of our model. The community has traditionally relied on the data augmentation technique thermodynamic reversibility (TR)46 to generate datasets with expanded mutation type coverage (Supplementary Fig. 2). However, ~3% of mutation types in C2878 + TR and T2837 + TR still lack data (see Supplementary Fig. 3). More importantly, a major drawback of TR augmentation is that all stabilizing mutations it generates are to the wild-type amino acid, as shown in Fig. 3b. These mutations give no predictive power with respect to identifying non-wild-type stabilizing mutations, which is the main goal of thermodynamic stability prediction in the context of protein engineering. To improve the predictive power of deep learning frameworks for stabilizing proteins, additional data for stabilizing mutations not “to" the wild-type amino acid is required.

a Heatmap representation of the mutation type distribution present in C2878, cDNA117K, T2837. For C2878, the original, TP, and original + TP + TR dataset consist of 2878, 18,912, and 24,668 mutations that sample 86.8%, 100%, and 100% of the mutation types, respectively (first row). For cDNA117K, the original, TP, and original + TP + TR datasets consist of 116,641, 2,018,710, and 2,251,992 mutations, respectively, and each dataset samples 100% of the mutation types (second row). For T2837, the original, TP, and original + TP + TR datasets consist of 2837, 7720, and 13,394 mutations that sample 85.5%, 100%, and 100% of the mutation types, respectively (third row). b Comparing the ΔΔG distribution for the original training and test sets with their TP and TR augmentations. All three experimental datasets are biased towards destabilizing mutations (orig). TR augmentation provides additional data biased towards stabilizing mutations and TP augmentation provides additional data that is evenly distributed between stable and destabilizing mutations.
To address these issues and improve Stability Oracle’s ability to generalize, we introduce thermodynamic permutations (TP), a data augmentation technique. TP is based on the state-function property of Gibbs free energy, enabling the generation of thermodynamically valid point mutations at residues where multiple amino acids have been experimentally characterized. With TP, we can generate an additional 2.02M, 18.9K, and 7.7K point mutations that sample all 380 mutation types for cDNA117K, C2878, and T2837, respectively. Additionally, TP mitigates several sampling biases in all 3 datasets (Fig. 3a, middle column). First, it provides mutation data for the 13.2% and 14.5% mutation types absent in C2878 and T2837. TP generated data for C2878 and T2837 samples of the 380 mutation types, providing the first training and test sets with experimental ΔΔG measurements for all mutation types (the cDNA display proteolysis dataset does not directly measure ΔΔG but instead derives ΔΔG values from the next-generation sequencing data of multiplexed proteolytic experiments).
Figure 3a illustrates the improvement in sampling bias as a softening of red (oversampled) and blue (undersampled) toward white (balanced sampling). In the C2878 and T2837 datasets, this is most apparent for the “to” alanine bias. In cDNA117K, there is an oversampling bias of mutations “from" alanine, glutamate, leucine, and lysine and an undersampling bias for mutations “from" cysteine, histidine, methionine, and tryptophan. TP completely balances the cDNA117K mutation type distribution with each mutation type making up approximately 0.26% of the dataset (100%/380), depicted in Fig. 3a middle column, middle row as uniformly white. Thus, TP augmentation of cDNA117K provides the first large-scale ΔΔG dataset (>1M) that evenly samples all 380 mutation types across 100 protein domains. In contrast to TR, TP does not include stabilizing mutations to the wild-type amino acid and yields a balanced distribution (stabilizing vs. destabilizing) of ΔΔG measurements (Fig. 3b).
To develop Stability Oracle framework, we compared training on cDNA117K and/or C2878, with and without TP augmentation, using Structural Amino Acid Embeddings vs. one-hot encodings, and evaluated the performance on all test sets. We observed that Structural Amino Acid Embeddings significantly improve performance compared to the naïve one-hot encoding in Fig. 4a. UMAP visualization of the mutation-hidden representation for T2837 from the Structural Amino Acid Embeddings reveals that the “ToAA" CLS token drives the organization of the latent space and recover known biochemical relationships between the 20 amino acids as illustrated in Fig. 5a. We observe that 1) clustering of hydrophobes (LEU, VAL, ILE, MET), aromatics (PHE, TYR, TRP), and short polars (SER, THR and ASP, ASN) (right panel); 2) isolation of the unique amino acids (GLY, CYS, PRO) (right panel); 3) the unique situation of mutating away from GLY and adding a chiral side-chain (left panel). For a residue-specific case study of the 380 mutation types, see Supplementary Fig. 7. As for the training sets, training the self-supervised representations on cDNA117K + TP + TR provided the best performance overall on regression and classification metrics across the test sets (shown in Fig. 4b/c). While this might have been expected due to the sheer size and mutation-type balance compared to C2878 + TP + TR, it is interesting to note that proteolytic stability of single-domain natural proteins is in fact generally an excellent surrogate for thermodynamic stability (as was pointed out in the original publication50). From this data, the impact of TP on model generalization was unclear. To further examine how TP-augmented datasets impact generalization, we evaluated predictions at mutation types in T2837 + TP that were absent from C2878 + TR but in C2878 + TP + TR, namely the 12 mutation types with no experimental data (see Supplementary Fig. 3). For these mutation types, TP improves generalization: recall improves from 0.28 to 0.4 and precision improves from 0.47 to 0.67 (Fig. 6). We artificially expanded the mutation types that were missing data to 54 and observed similar, but attenuated improvements to both precision and recall (Fig. 6).

a Trained on cDNA117K, we demonstrate the performance with “Structural AA Embedding” and “One-Hot Encoding''. b Comparison of Stability Oracle’s performance when trained on the C2878 and cDNA117K training sets. c Comparison of Stability Oracle’s performance when trained on C2878 with and without a pre-trained backbone and Structural Amino Acid Embeddings and with and without TP augmentation. d Trained on T2837 + TP, Stability Oracle’s performance when tested on experimental or AlphaFold structures of T2837. We get p-value < 0.001 for all our reported correlation coefficients. Source data are provided as a Source Data file.

a UMAP visualization of the 128-dim mutation-hidden representation for T2837. Left and right panels are colored by the “FromAA” and “ToAA” in a mutation, respectively. b The experimental distribution of Stability Oracle’s stabilizing predictions (ΔΔG < −0.5 kcal/mol) on T2837 + TP test set for different “from" and “to" amino acid types. Here, stabilizing, neutral, and destabilizing mutations are defined by ΔΔG < −0.5 kcal/mol, ∣ΔΔG∣ ≤ 0.5 kcal/mol, and ΔΔG > 0.5 kcal/mol, respectively. Source data are provided as a Source Data file.

We report the accuracy, recall, and precision results (with 0 kcal/mol being the threshold) on two subsets of T2837 + TP, to demonstrate the effectiveness of permutation. On the left, we test the model on the 12 mutation types lacking experimental data in C2878 + TR (missing mutation types can be found in Supplementary Fig. 3). However, this analysis consists of only 54 mutations within T2837 + TP. On the right, we examine the impact of TP by artificially expanding the missing mutation types from 12 to 68 by removing mutation types from C2878 that had fewer than 8 training instances available. This filtered version of C2878 allowed us to evaluate the performance of TP on 663 mutations within T2837 + TP. Source data are provided as a Source Data file.
To prevent inflation of Stability Oracle’s classification performance, we focus our evaluations on T2837 + TP (10,557 mutations) and exclude all TR mutations, since these mutations are heavily biased with stabilizing mutations to the wild-type amino acid (see Supplementary Fig. 4). Here, Stability Oracle demonstrated a recall of 0.69, a precision of 0.70, and an AUROC of 0.83 (Fig. 4b). Surprisingly, further fine-tuning with C2878 + TP did not improve performance on T2837 or T2837 + TP. Our analysis reveals all proteins in C2878 are homologous (>30% sequence similarity) to at least one protein in cDNA117K and therefore C2878 does not expand the protein space available for training. This observation provides a rationale for the lack of performance improvement observed upon further fine-tuning on C2878. However, C2878 fine-tuning improves performance on the interface subset of T2837: the Pearson correlation improves from 0.30 to 0.35, and the Spearman correlation improves from 0.29 to 0.35 for the interface microenvironments. This improvement is expected since the cDNA dataset consists of monomeric single-domain structures, lacking interfaces with other proteins, ligands, or nucleotides. However, meaningful improvements are limited due to the scarce amount of protein-protein (127 mutations), protein-ligand (94 mutations), and protein-nucleotide (9 mutations) data in C2878.
Since experimental structures are often unavailable, we examined Stability Oracle’s ability to generalize to structures generated by Alphafold222 with the WT, “From”, and “To” amino acid present. We used ColabFold to generate template-free predicted structures for each protein in T283765. ColabFold failed to fold one protein and two structures were removed due to TM-align66 having US-scores < 0.566,67. This resulted in the removal of 50 mutations from T2837. When evaluating T2837 and T2837 + TP using the AlphaFold WT structure we observed no changes in classification metrics and slightly lower performance on regression metrics on T2837 + TP (Fig. 4d). Next, we evaluated the impact on the “From” and “To” AlphaFold structures on the T2837 TP-only dataset (7720 mutations, 100% mutation type coverage) and observed a 2–4% drop in classification and regression metrics (Supplementary Table 4). Overall, these results demonstrate the ability of Stability Oracle to generalize to AlphaFold scaffolds when an experimental structure is unavailable.
We conducted several comparisons against the literature. First, we report Pearson correlation coefficients (PCC) (both forward and reverse) on T2837 and all the common test sets. For the common test sets, we compare against several community predictors in Fig. 7 and provide all classification and regression metrics on literature test sets in Supplementary Table 5a. It is worth noting that Stability Oracle outperforms other predictors in the literature even with their documented data leakage issues14. To date, the most accurate and exhaustive thermodynamic stability dataset in the literature is the Gβ1 dataset45. We evaluate Stability Oracle’s performance on Gβ145 and, to the best of our knowledge, achieve SOTA on all 935 mutations (Pearson = 0.75, AUROC = 0.84) and the 835 mutation quantitative subset (Pearson = 0.67, AUROC = 0.81) (full results are provided Supplementary Table 6). Finally, we evaluated Stability Oracle’s structural sensitivity with a case study on p53, an issue previously documented for structure-based stability predictors68. We evaluated three p53 structures (PDB: 2OCJ, 3Q05, 2AC0) that differed in their protein length (94-312, 94-326, 94-293), resolution (2.05, 2.40, 1.80 Å), and biological assembly (homodimer with no DNA, homotetramer complexed with a DNA helix, homotetramer complexed with two DNA helices), respectively, visualized in Supplementary Fig. 1a. This case study demonstrates that Stability Oracle generalizes amid significant structural variations of p53, achieving a Pearson = 0.75 ± 0.02, Spearman = 0.76 ± 0.05, precision = 0.55 ± 0.07, and AUROC = 0.83 ± 0.02 (full results are provided in Supplementary Fig. 1b).

Evaluating Stability Oracle’s ability to identify stabilizing mutations
For computational stability predictors to accelerate protein engineering, it is critical that their predictions correctly identify stabilizing mutations. However, it is well documented that SOTA stability predictors can correctly predict stabilizing mutations with ~20% success rate and most stabilizing predictions are actually experimentally neutral or destabilizing13,33. While molecular dynamic-based methods, such as free energy perturbation (FEP), have demonstrated a 50% success rate at identifying stabilizing mutations, their computational demand prevents them from scaling to entire protein applications like computational deep mutational scans (DMS)33,69. Thus, there is a strong need for a method that can match the performance of FEP while being computationally inexpensive.
To evaluate Stability Oracle’s ability to identify stabilizing mutations, we filtered its predictions on T2837 and T2837 + TP at different ΔΔG thresholds and assessed the distribution of experimental stabilizing (ΔΔG < −0.5 kcal/mol), neutral (∣ΔΔG∣ ≤ 0.5 kcal/mol), and destabilizing (ΔΔG > 0.5 kcal/mol) mutations. The 0.5 kcal/mol cutoff was chosen based on the average experimental error70. With the ΔΔG < −0.5 kcal/mol prediction threshold, 1770 mutations were filtered with an experimental distribution of 74.0% stabilizing, 17.8% neutral, and 8.2% destabilizing and 48.1% of all stabilizing mutations were correctly identified. A systematic analysis of prediction thresholds is provided in Fig. 8 and Supplementary Table 8a. The success rate of predicting stabilizing mutations (74%) appears to surpass what is typically observed with FEP methods (~50%)33,69 with orders of magnitude less computational cost (Supplementary Table 1). We further examine Stability Oracle’s ability to identify stabilizing mutations by amino acid (Fig. 5b). Here, we observe that Stability Oracle is able to correctly predict stabilizing mutations across most amino acids, whether mutating “from” or “to”. However, several amino acids lack sufficient “from” or “to” stabilizing predictions to draw meaningful conclusions. This data scarcity is even more apparent when looking at the 380 “from”-"to” pairs (see Supplementary Fig. 5), highlighting how data scarcity still hinders proper model evaluation.

In the first column, we compare T2837 and observe that Stability Oracle had the highest stabilization and lowest destabilization fraction with similar recall. In the second column, we compare T2837 + P and observe that Stability Oracle still has the highest stabilization and lowest destabilization fraction but Prostata-IFML has a better recall. Source data are provided as a Source Data file.
It has been pointed out by the community that experimentally characterized surface stabilizing mutations are biased towards hydrophobic amino acids33. An analysis by Broom et al. of the ProTherm database71 indicates that surface stabilizing mutations typically increase side-chain hydrophobicity (ΔΔGsolvation) with a median change of 0.8 kcal/mol. This hydrophobicity bias is equivalent to an alanine-to-valine mutation on the protein surface33. We examined if this underlying hydrophobic bias persisted within our training pipeline by computing the precision and recall of polar and hydrophobic mutations as a function of relative solvent accessibility (RSA) of the wild-type residue. Our precision and recall results across different RSA bins of T2837 and T2837+ TP indicate that the cDNA117K + TP training set does not produce models biased towards predicting hydrophobic amino acids on protein surfaces (Fig. 9).

The results demonstrate that there are no biases between polar and hydrophobic residues throughout a protein structure on both T2837 and T2837 + TP: we do a test of significance for the absolute value between polar and hydrophobic data examples on the whole dataset (not per RSA bin due to lack of data) and it is insignificant. Here, polar amino acids consist of S, T, N, Q, D, E, R, K, H, and Y and hydrophobic amino acids consist of L, M, I, V, F, W, A. Source data are provided as a Source Data file.
Comparing sequence and structure fine-tuned stability predictors
Over the last three years, self-supervised protein large language models (pLLMs or “sequence models”) have had a tremendous impact on the protein community25,72,73,74,75,76,77,78,79.
Understanding sequence vs. structure-based prediction models continues to be an active area of research in protein engineering and design19,80. We evaluated Stability Oracle against two computational stability deep learning frameworks: Prostata and RaSP. Prostata26 is a sequence-based framework that fine-tunes ESM2 embeddings ensembling five distinct regression heads (a SOTA protein language model)25 on common training and test sets. However, the Prostata was trained with homologous proteins (a sequence similarity cutoff of 75%) with respect to SSym and S669, resulting in inflated performance on T2837 and its subset test sets (breakdown of the performance and data leakage are provided in Supplementary Table 7). In order to address this data leakage and conduct a fair comparison, we fine-tuned ESM2’s representation using the same training and test sets as Stability Oracle and only the outer-product regression head architecture. We call our version of ESM2’s representations fine-tuned on thermodynamic stability Prostata-IFML. We also compare against the RaSP framework81: a structure-based 3DCNN model that follows a similar training pipeline. Briefly, RaSP is pre-trained with self-supervision on 18Å masked microenvironments sampled from 2315 structures clustered at a 30% sequence similarity and then fine-tuned on 35 DMS datasets computationally generated by the Rosetta Cartesian-ΔΔG program81. In our analysis, we modified the RaSP Colab notebook to generate DMS predictions on every protein in T2837 and every mutation in T2837 + TP.
Stability Oracle outperforms or matches Prostata-IFML on every metric (Fig. 10), even though Stability Oracle has 548 times fewer parameters ( ~658M vs. ~ 1.2M) and was pre-trained with 2000 times fewer proteins ( ~ 46M vs. ~ 23K) at the same sequence similarity (50%). As for RaSP, Stability Oracle significantly outperforms it on nearly every classification and regression metric on both T2837 and T2837 + TP. The performance is only comparable for Pearson on T2837 and precision on T2837 + TP.

We refer the readers to Supplementary Section A for detailed results. Source data are provided as a Source Data file.
In terms of identifying stabilizing mutations, Stability Oracle also achieves the best performance (Fig. 8). At each prediction threshold, Stability Oracle had the highest proportion of correctly identified stabilizing mutations and the lowest proportion of destabilizing mutations (we exclude −1.5 kcal/mol threshold for T2837 due to data scarcity). It is typical for precision to be inversely proportional to recall, and we observe this tradeoff with regard to Stability Oracle and Prostata-IFML on T2837 + TP, where Prostata-IFML has better recall. We suspect this difference is due to their loss: Stability Oracle uses a huber loss and Prostata uses mean square error. Nonetheless, both Stability Oracle and Prostata-IFML have superior performance on both correctly identifying stabilizing mutations (precision) and identifying more stabilizing mutations (recall) compared to RaSP. A detailed comparison is provided in Supplementary Table 8. In parallel to this work, ThermoMPNN–a deep learning framework that fine-tunes the ProteinMPNN82 representations also on the megascale cDNA proteolysis dataset–was developed. Using the publicly available checkpoint, we found that Stability Oracle outperforms ThermoMPNN on SSym, S669, myoglobin, and P53 across multiple regression and classification metrics (Supplementary Table 10).
Finally, we compare all three framework’s ability to predict self-mutations: the “from" and “to" amino acids are the same and the ΔΔG is 0 kcal/mol. Similar to the forward vs. reverse experiments, which assess the thermodynamic robustness of predictors, self-mutations evaluate generalization to trivial examples that were not present in the training set but are inherent in thermodynamics. For wild-type self-mutations on T2837 Stability Oracle, Prostata-IFML, and RaSP achieve RMSE of 0.0033, 0.0018, and 0.8370 kcal/mol, respectively. This demonstrates that Stability Oracle and Prostata-IFML implicitly learn to capture self-mutations. RaSP, however, is unable to generalize to self-mutations and this drop in performance is also observed for TR augmentation of T2837 (Supplementary Table 9c).
Discussion
Reliable prediction of stabilizing mutations is critical for the acceleration of protein-based biotechnologies. However, as of March 2023, all computational stability predictors prioritize improvements in the Pearson and RMSE metrics. Neither of these metrics, however, is appropriate for evaluating improvements in identifying stabilizing mutations; several studies explain this in great details13,14,33. We report these regression metrics but use classification metrics, such as AUROC, MCC, recall, and precision, to guide our model development. By using a 30% sequence similarity threshold for train-test splits, we expect the performance of Stability Oracle and Prostata-IFML to have superior generalization compared to models trained using traditional train-test splits, which suffer from data leakage. Stability Oracle and Prostata-IFML seem to correctly identify stabilizing mutations that potentially outperform FEP-based methods while being several orders of magnitude faster. However, systematic experimentation that directly compares FEP-based methods is needed for confirmation.
The literature has recently adopted thermodynamic reversibility (TR) to address the imbalance between stabilizing and destabilizing mutations in training and test sets. However, TR biases the training towards stabilizing mutations of wild-type amino acids. For computational models that leverage evolutionary features and protein structures as input, mutations to wild-type amino acids leak information and are of limited use in a protein engineering context where mutating away from the wild type is the primary goal. Our data augmentation technique, thermodynamic permutations (TP), generates mutations with a balanced ΔΔG distribution and does not generate mutations to wild-type amino acids (Supplementary Fig. 3b). This mitigates the above imbalance and expands the number of stabilizing mutations to non-wild-type amino acids in both the training and test sets. In addition, TP reduces bias to wild-type inherent in the self-supervised pre-training step. We speculate that during end-to-end fine-tuning, TP forces the feature-extractor to find additional chemical signatures in the microenvironment, not just those most relevant for wild-type identification. Further, TP also generates ΔΔG measurements for mutation types in microenvironments that would rarely be experimentally characterized. This expands the microenvironment/mutation type combinations available for training and testing and is highly likely to improve generalization across structural motifs found within a protein scaffold. However, we lack the necessary test data needed to examine this hypothesis. We expect TP to be of great use for the development of frameworks for higher-order mutations, where data scarcity is an even bigger issue.
Of note is the performance of Stability Oracle’s much smaller model (in terms of parameters) relative to Prostata-IFML. Stability Oracle’s ability to outperform or match Prostata-IFML is evidence that protein structures contain information beyond the amino acid sequence itself. Self-supervised deep learning models, such as MutComputeX, typically struggle with predicting mutations in the core, with a tendency to re-predict the wild-type residue in the closely packed environment. Stability Oracle is explicitly trained to learn stabilizing substitutions from the microenvironment, providing a structure-based method for predicting core mutations.
In terms of training set size, note that the ~ 25,000 ΔΔG measurements (C2878 + TP + TR) led to performance comparable to models trained on ~ 2.2 million proteolytic stability measurements (cDNA117K + TP + TR). While T2837 is still a limited evaluation set, this indicates that both the quality and quantity of data are of great importance for training. Additional experimental techniques directed towards producing larger datasets with detailed thermodynamic information, especially for residues at functional interfaces, may further improve generalization capabilities.
Accurate identification of stabilizing mutations will continue to impact a wide variety of areas, from predicting protein therapeutics and vaccines with greater stabilities and shelf lives to predicting enzymes that can work at higher temperatures for biomanufacturing and environmental bioremediation. While previous algorithms, such as MutCompute, have proven adept at improving the stabilities of proteins51,53,54, Stability Oracle will likely further improve the hit rate of functional, stable substitutions due to its improved accuracy across a variety of metrics. More importantly, Stability Oracle is attuned to thermodynamic effects, rather than just sterics. This allows for predictions across a wider class of substitutions, including those at protein: protein interfaces, such as antibody: antigen interactions, as well as those at protein: ligand and protein: nucleotide interfaces.
Methods
Model architecture
We build a transformer-based neural network for both mask amino acid self-supervised learning and ΔΔG supervised learning. We describe the model architectures for the self-supervised backbone model and for the ΔΔG regression head.
MutComputeXGT: self-supervised graph transformer for amino acid likelihoods
We first introduce our graph-transformer model for self-supervised tasks, outlining its key components and specifications. We describe the general pictures of our model, and then we list the model inputs and outputs. Finally, we elaborate on some key inductive bias designs in our model. Generally speaking, in our tasks, the input for the model is a local environment for the target amino acid. Given one amino acid, we set the Carbon-α as the center and grab all the atoms within radius n. In the context of self-supervised training, we apply masking to each atom within the target amino acid and predict the corresponding amino acid type. This approach leverages graph-based representations and self-supervised learning to capture important structural features and enhance our understanding of protein sequences. Denote N as the number of tokens, the coordinates \(\in {{\mathbb{R}}}^{N\times 3}\), atom types \(\in {{\mathbb{R}}}^{N\times 1}\) and physical properties \(p\in {{\mathbb{R}}}^{N\times P}\) are given to the neural network, we apply an embedding layer to convert categorical atom types into continuous representations \(e\in {{\mathbb{R}}}^{N\times E}\) where E = 20 represents the number of amino acid types and concatenate embeddings together with physical properties h = Concat(e, p). The coordinates \(\in {{\mathbb{R}}}^{N\times 3}\) are used to calculate atom-wise Euclidean distance \(D\in {{\mathbb{R}}}^{N\times N}\), which serves as the attention bias in the attention layers. The concatenated features h then pass through several attention blocks. In each attention block, we have two attention layers and one MLP layer. The MLP layers are the same as the standard attention block, while in each attention layer, after calculating the KQT, we add additional attention mask and attention-bias terms. The attention bias is calculated based on the distance matrix \(D\in {{\mathbb{R}}}^{N\times N}\). Given D, we create multiple feature vectors and output the attention bias for each head in one attention layer. Apply K = 16 radial basis functions (RBF) with different bandwidths, we get \(k\in {{\mathbb{R}}}^{N\times N\times K}\). Categorizing the distance into C = 4 class, we get \(c\in {{\mathbb{R}}}^{N\times N\times C}\). Put a linear transformation after concatenating c and k, we get \({{{{{{\rm{bias}}}}}}}\in {{\mathbb{R}}}^{N\times N\times {{{{{{\rm{Head}}}}}}}}\) where Head denotes the number of heads in one attention layer. Finally, we collect the hidden representations h for all the atoms and select those whose corresponding atom is close to the masked amino acid. Specifically, we set the carbon-α for the masked amino acid as the center and select all the atoms in an 8 Å radius. The features of these atom tokens are average pooled, and passed to the classifier, before being normalized into the 20 amino acid likelihoods. The classifier consists of a Linear-ReLU-Linear block, with 128 and 20 neurons at these linear layers, respectively.
Stability Oracle: graph-transformer fine-tuned for ΔΔG prediction
The Stability Oracle framework contains two parts, a backbone that encodes the masked local environment and a regression head. For the purpose of this discussion, we primarily focus on the architecture of the regression head, as the pre-trained weights from the self-supervised learning task are loaded into the backbone. We get one masked local environment together with two amino acid types (one is the wild-type and the other is the mutation), and target at predicting the ΔG changes. We first extract useful atomic features from the backbone model. Given a masked local environment, we pass it through the backbone and get the output \(h\in {{\mathbb{R}}}^{N\times H}\) where H denotes the hidden dimension. Given the wild-type and mutation amino acid type, we extract the corresponding amino acid embedding \({e}_{wt}\in {{\mathbb{R}}}^{1\times H}\) and \({e}_{mut}\in {{\mathbb{R}}}^{1\times H}\) in the final linear layer of the backbone. We then apply Concat(h, ewt) and Concat(h, emut) to get two hidden representations for wild-type local environments and mutated local environment, respectively.
Pass Concat(h, ewt) and Concat(h, emut) through the attention blocks, we extract the ewt and emut in the final layer, subtract them, apply a linear layer, and output the predicted ΔΔG.
Training configuration
During training, we shuffle the training data and use Huber loss with δ = 1 as the training loss. When training on large-scale datasets, e.g., cDNA117K, we end-to-end tune all the parameters, with a learning rate of 5 × 10−5 for regression head parameters and a learning rate of 2 × 10−5 for backbone parameters. We apply optimizer AdamW with batch size 960 (batch size 240 with accumulation step 4), weight decay 0.1, optimizer EMA with η = 0.99, and number of iterations 750. To fine-tune the CDNA-trained model on the small datasets (e.g., C2878), we freeze the backbone parameters and use AdamW with a learning rate 5 × 10−7, batch size 1, 024, weight decay 0.1, and number of iterations 500. Train from scratch on the small datasets (e.g., C2878), we freeze the backbone parameters and set optimizer AdamW with 5 × 10−5, batch size 1, 024, weight decay 0.1, and number of iterations 500.
Pre-training dataset
We pre-train our graph-transformer backbone using the same procedure as MutComputeX53. Briefly, this dataset consists of a 90:10 split of 2,569,256 microenvironments sampled from 22,759 protein sequences clustered at 50% sequence similarity and having a structure resolution of at least 3 Å from the RCSB (November 2021).
cDNA117K training set generation
To curate the cDNA117K dataset50, we downloaded the file K50_dG_Dataset1_Dataset2.csv from the version 1 deposit at the Zenodo repository https://zenodo.org/record/7401275#.Y6st59JBxD_. First, we removed all de novo domains and then filtered all single-point mutations on protein scaffolds that have a wild-type structure pdb id provided in the csv. Finally, using structure sequences, the remaining mini-proteins were filtered for 30% sequence overlap with T2837 via MMSeqs2 easy-search command with flags -c 0.3 -s 7.5 -seq-id-mode 1. The closest sequence similarity between cDNA117K and T2837 is 25.4%.
C2878 training set generation
To curate C2878 dataset50, we first concatenated Q174430, O256716, S264840, and FireProtDB83. We address duplicates between the datasets by taking the ΔΔG measurement with the highest absolute value. This was intended to address the neutral bias and move the ΔΔG predictions from a model away from 0. With duplicates removed, we generate C5266, which is then filtered for 30% sequence overlap with T2837. The structure sequences were used for the sequence similarity filtering. The same MMSeqs2 command was used as cDNA117K.
T2837 test set generation
To curate T2837 dataset50, we first concatenated P5330, Myoglobin30, SSym30, and S66912. We address duplicates between the datasets by taking the ΔΔG measurement with the highest absolute value. This was intended to address the neutral bias and move the ΔΔG predictions from a model away from 0. With duplicates removed, we generate T1187. The 30% sequence overlap with C5266 was removed to produce T2226. T2226 was then merged and deduplicated in the same manner to produce T2837. The structure sequences were used for the sequence similarity filtering.
Generation of T2837 AlphaFold structure
We ran ColabFold 2.3 on the full UniProt sequence associated with each PDB ID. For the TP-only “From” and “To” datasets, We modified the wild-type UniProt sequence to reflect each mutation. We used ColabFold’s default parameters and chose the folded PDB structure with the highest pLDDT score for each sequence.
Test set
To fully test different models, we collect and clean related datasets in the literature and make our test set. To get more comparisons, we report the numbers on all the literature datasets here. In summary, we test our model on S-sym, P53, Myoglobin and S669. S-sym, P53, and Myoglobin datasets are proposed in ThermoNet30 and are widely used by the follow-up works as the testing benchmarks. S669 includes curated data dissimilar at 25% sequence identity to S2648 training data. Additionally, we create several more domains from the original test data. ① Swap the mutation and wild-type amino acids, and this test domain is marked as TR. ② Once we have multiple single-point mutations in the same position, we randomly pick two amino acids as the wild-type and the mutation. We mark this test domain as TP.
Evaluation metrics
We evaluate regression and classification metrics. First, for a fair comparison, we report Pearson correlation and RMSE for our regressor. Furthermore, in practice, instead of RMSE, we have more interest in the performance of stabilizing mutations. Therefore, we report the precision and recall for the stabilizing mutations and the AUROC values for the binary classification problems.
Prostata-IFML details
We construct a strong sequence-based baseline from Prostata26. This baseline model takes the wild-type and mutant sequences as input. Both sequences are first tokenized and independently passed through the same pre-trained ESM2 backbone. The features at the mutation position are selected from both the wild-type and mutant sequences. An outer product enables interactions between the wild-type and mutant embeddings. The result is flattened and a small decoder predicts a change in thermodynamic stability. This is referred to as the outer-product variant in PROSTATA. For training, we employ the identical cDNA dataset that was employed to train Stability Oracle. Given the relatively shorter length of cDNA protein sequences, we were able to increase the batch size significantly to 64 mutations, as opposed to the original batch size of 1. We trained our model using the Adam optimizer for 3 epochs. During the training, we followed a one-cycle learning rate schedule. In the first epoch, the learning rate was linearly increased from 0 to 6.4 × 10−4. The learning rate was gradually annealed to 0 in the subsequent epochs. We fine-tune the same training sets and evaluate our models on the same test sets as above.
ThermoMPNN details
We use the official codebase on GitHub at https://github.com/Kuhlman-Lab/ThermoMPNN with the official checkpoint at https://github.com/Kuhlman-Lab/ThermoMPNN/blob/main/models/thermoMPNN_default.pt to generate predictions for model comparisons.
RaSP details
We use the codebase on GitHub at https://github.com/KULL-Centre/_2022_ML-ddG-Blaabjerg to generate predictions for model comparisons.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All relevant data supporting the key findings of this study are available within the article and its Supplementary Information files. Datasets are available on the Zenodo repository84 at the GitHub repository: https://github.com/danny305/StabilityOracle. Source data are provided in this paper.
Code availability
Code used to generate figures and the Stability Oracle model weights are available on a Zenodo repository84 and at the GitHub repository: https://github.com/danny305/StabilityOracle.
References
Wu, S., Snajdrova, R., Moore, J. C., Baldenius, K. & Bornscheuer, U. T. Biocatalysis: enzymatic synthesis for industrial applications. Angew. Chem. Int. Ed. 60, 88–119 (2021).
Bell, E. L. et al. Biocatalysis. Nat. Rev. Methods Prim. 1, 1–21 (2021).
Adams, J. P., Brown, M. J., Diaz-Rodriguez, A., Lloyd, R. C. & Roiban, G. D. Biocatalysis: a pharma perspective. Adv. Synth. Catal. 361, 2421–2432 (2019).
Jay, S. M. & Lee, R. T. Protein engineering for cardiovascular therapeutics: Untapped potential for cardiac repair. Circ. Res. 113, 933–943 (2013).
Gebauer, M. & Skerra, A. Engineered protein scaffolds as next-generation therapeutics. Annu. Rev. Pharmacol. Toxicol. 60, 391–415 (2020).
Meghwanshi, G. K. et al. Enzymes for pharmaceutical and therapeutic applications. Biotechnol. Appl. Biochem. 67, 586–601 (2020).
Banta, S., Dooley, K. & Shur, O. Replacing antibodies: engineering new binding proteins. Annu. Rev. Biomed. Eng. 15, 93–113 (2013).
Chen, K. & Arnold, F. H. Tuning the activity of an enzyme for unusual environments: sequential random mutagenesis of subtilisin e for catalysis in dimethylformamide. Proc. Natl Acad. Sci. USA 90, 5618–5622 (1993).
Ebo, J. S., Guthertz, N., Radford, S. E. & Brockwell, D. J. Using protein engineering to understand and modulate aggregation. Curr. Opin. Struct. Biol. 60, 157–166 (2020).
Arnold, F. H. Design by directed evolution. Acc. Chem. Res. 31, 125–131 (1998).
Giver, L., Gershenson, A., Freskgard, P.-O. & Arnold, F. H. Directed evolution of a thermostable esterase. Proc. Natl Acad. Sci. USA 95, 12809–12813 (1998).
Pancotti, C. et al. Predicting protein stability changes upon single-point mutation: a thorough comparison of the available tools on a new dataset. Brief. Bioinforma. 23, bbab555 (2022).
Benevenuta, S., Birolo, G., Sanavia, T., Capriotti, E. & Fariselli, P. Challenges in predicting stabilizing variations: an exploration. Front. Mol. Biosci. 9, 1–10 (2023).
Pucci, F., Schwersensky, M. & Rooman, M. Artificial intelligence challenges for predicting the impact of mutations on protein stability. Curr. Opin. Struct. Biol. 72, 161–168 (2022).
Sanavia, T. et al. Limitations and challenges in protein stability prediction upon genome variations: towards future applications in precision medicine. Comput. Struct. Biotechnol. J. 18, 1968–1979 (2020).
Caldararu, O., Mehra, R., Blundell, T. L. & Kepp, K. P. Systematic investigation of the data set dependency of protein stability predictors. J. Chem. Inf. Model. 60, 4772–4784 (2020).
Fedik, N. et al. Extending machine learning beyond interatomic potentials for predicting molecular properties. Nat. Rev. Chem. 6, 653–672 (2022).
Bender, A. et al. Evaluation guidelines for machine learning tools in the chemical sciences. Nat. Rev. Chem. 6, 428–442 (2022).
Diaz, D. J., Kulikova, A. V., Ellington, A. D. & Wilke, C. O. Using machine learning to predict the effects and consequences of mutations in proteins. Curr. Opin. Struct. Biol. 78, 102518 (2023).
Xu, Y. et al. Deep dive into machine learning models for protein engineering. J. Chem. Inf. Model. 60, 2773–2790 (2020).
Bordin, N. et al. Novel machine learning approaches revolutionize protein knowledge. Trends Biochem. Sci. 48, 345–359 (2023).
Jumper, J. et al. Highly accurate protein structure prediction with alphafold. Nature 596, 583–589 (2021).
Ahdritz, G. et al. Openfold: retraining alphafold2 yields new insights into its learning mechanisms and capacity for generalization. bioRxiv https://doi.org/10.1101/2022.11.20.517210 (2022).
Baek, M. et al. Accurate prediction of protein structures and interactions using a three-track neural network. Science 373, 871–876 (2021).
Lin, Z. et al. Evolutionary-scale prediction of atomic-level protein structure with a language model. Science 379, 1123–1130 (2023).
Umerenkov, D. et al. PROSTATA: a framework for protein stability assessment using transformers. Bioinformatics 39, btad671 (2023).
Benevenuta, S., Pancotti, C., Fariselli, P., Birolo, G. & Sanavia, T. An antisymmetric neural network to predict free energy changes in protein variants. J. Phys. D Appl. Phys. 54, 245403 (2021).
Chen, T. et al. Hotprotein: a novel framework for protein thermostability prediction and editing. In: The Eleventh International Conference on Learning Representations (OpenReview.net, 2023).
Hie, B. L. et al. Efficient evolution of human antibodies from general protein language models. Nat. Biotechnol. 42, 275–283 (2024).
Li, B., Yang, Y. T., Capra, J. A. & Gerstein, M. B. Predicting changes in protein thermodynamic stability upon point mutation with deep 3d convolutional neural networks. PLoS Comput. Biol. 16, e1008291 (2020).
Cao, H., Wang, J., He, L., Qi, Y. & Zhang, J. Z. Deepddg: predicting the stability change of protein point mutations using neural networks. J. Chem. Inf. Model. 59, 1508–1514 (2019).
Wang, S., Tang, H., Shan, P. & Zuo, L. Pros-GNN: predicting effects of mutations on protein stability using graph neural networks. Comput. Biol. Chem. 107, 107952 (2023).
Broom, A., Trainor, K., Jacobi, Z. & Meiering, E. M. Computational modeling of protein stability: quantitative analysis reveals solutions to pervasive problems. Structure 28, 717–726.e3 (2020).
Kellogg, E. H., Leaver-Fay, A. & Baker, D. Role of conformational sampling in computing mutation-induced changes in protein structure and stability. Proteins Struct. Funct. Bioinforma. 79, 830–838 (2011).
Schymkowitz, J. et al. The foldx web server: an online force field. Nucleic Acids Res. 33, W382–W388 (2005).
Capriotti, E., Fariselli, P. & Casadio, R. I-mutant2. 0: predicting stability changes upon mutation from the protein sequence or structure. Nucleic Acids Res. 33, W306–W310 (2005).
Pires, D. E., Ascher, D. B. & Blundell, T. L. mcsm: predicting the effects of mutations in proteins using graph-based signatures. Bioinformatics 30, 335–342 (2014).
Pires, D. E., Ascher, D. B. & Blundell, T. L. Duet: a server for predicting effects of mutations on protein stability using an integrated computational approach. Nucleic Acids Res. 42, W314–W319 (2014).
Laimer, J., Hofer, H., Fritz, M., Wegenkittl, S. & Lackner, P. Maestro-multi agent stability prediction upon point mutations. BMC Bioinforma. 16, 1–13 (2015).
Dehouck, Y., Kwasigroch, J. M., Gilis, D. & Rooman, M. Popmusic 2.1: a web server for the estimation of protein stability changes upon mutation and sequence optimality. BMC Bioinforma. 12, 1–12 (2011).
Worth, C. L., Preissner, R. & Blundell, T. L. Sdm-a server for predicting effects of mutations on protein stability and malfunction. Nucleic Acids Res. 39, W215–W222 (2011).
Wainreb, G., Wolf, L., Ashkenazy, H., Dehouck, Y. & Ben-Tal, N. Protein stability: a single recorded mutation aids in predicting the effects of other mutations in the same amino acid site. Bioinformatics 27, 3286–3292 (2011).
Romero, P. A., Krause, A. & Arnold, F. H. Navigating the protein fitness landscape with Gaussian processes. Proc. Natl Acad. Sci. USA 110, E193–E201 (2013).
Cheng, J., Randall, A. & Baldi, P. Prediction of protein stability changes for single-site mutations using support vector machines. Proteins Struct. Funct. Bioinforma. 62, 1125–1132 (2006).
Nisthal, A., Wang, C. Y., Ary, M. L. & Mayo, S. L. Protein stability engineering insights revealed by domain-wide comprehensive mutagenesis. Proc. Natl Acad. Sci. USA 116, 16367–16377 (2019).
Thiltgen, G. & Goldstein, R. A. Assessing predictors of changes in protein stability upon mutation using self-consistency. PLoS One 7, e46084 (2012).
Atkins, P., Atkins, P. W. & de Paula, J. Atkins’ Physical Chemistry (Oxford University Press, 2014).
Steinegger, M. & Soding, J. Mmseqs2 enables sensitive protein sequence searching for the analysis of massive data sets. Nat. Biotechnol. 35, 1026–1028 (2017).
Rost, B. Twilight zone of protein sequence alignments. Protein Eng. 12, 85–94 (1999).
Tsuboyama, K. et al. Mega-scale experimental analysis of protein folding stability in biology and design. Nature 620, 434–444 (2023).
Shroff, R. et al. Discovery of novel gain-of-function mutations guided by structure-based deep learning. ACS Synth. Biol. 9, 2927–2935 (2020).
Lu, H. et al. Machine learning-aided engineering of hydrolases for pet depolymerization. Nature 604, 662–667 (2022).
d’Oelsnitz, S. et al. Biosensor and machine learning-aided engineering of an amaryllidaceae enzyme. Nat. Commun. 15, 2084 (2024).
Paik, I. et al. Improved bst DNA polymerase variants derived via a machine learning approach. Biochemistry 62, 410–418 (2021).
Liu, Y. et al. Asymmetric synthesis of α-chloroamides via photoenzymatic hydroalkylation of olefins. J. Am. Chem. Soc. 146, 7191–7197 (2024).
Raček, T. et al. Atomic charge calculator II: web-based tool for the calculation of partial atomic charges. Nucleic Acids Res. 48, W591–W596 (2020).
Mitternacht, S. Freesasa: an open source c library for solvent accessible surface area calculations. F1000Research 5, 1–10 (2016).
Kulikova, A. V. et al. Learning the local landscape of protein structures with convolutional neural networks. J. Biol. Phys. 47, 435–454 (2021).
Guerois, R., Nielsen, J. E. & Serrano, L. Predicting changes in the stability of proteins and protein complexes: a study of more than 1000 mutations. J. Mol. Biol. 320, 369–387 (2002).
Kortemme, T., Kim, D. E. & Baker, D. Computational alanine scanning of protein-protein interfaces. Sci. STKE 2004, pl2–pl2 (2004).
wwPDB consortium. Protein Data Bank: the single global archive for 3D macromolecular structure data. Nucleic Acids Res. 47, D520–D528 (2018).
Bertinetto, L., Valmadre, J., Henriques, J. F., Vedaldi, A. & Torr, P. H. Fully-convolutional siamese networks for object tracking. In: Computer Vision–ECCV 2016 Workshops: Amsterdam, The Netherlands, October 8–10 and 15–16, 2016, Proceedings, Part II 14, 850–865 (Springer, 2016).
Vaswani, A. et al. Attention is all you need. Adv. Neural. Inf. Process. Syst. 30, 5998–6008 (2017).
Pucci, F., Bernaerts, K. V., Kwasigroch, J. M. & Rooman, M. Quantification of biases in predictions of protein stability changes upon mutations. Bioinformatics 34, 3659–3665 (2018).
Mirdita, M. et al. Colabfold: making protein folding accessible to all. Nat. Methods 19, 679–682 (2022).
Zhang, C., Shine, M., Pyle, A. M. & Zhang, Y. Us-align: universal structure alignments of proteins, nucleic acids, and macromolecular complexes. Nat. Methods 19, 1109–1115 (2022).
Zhang, Y. & Skolnick, J. Scoring function for automated assessment of protein structure template quality. Proteins Struct. Funct. Bioinforma. 57, 702–710 (2004).
Caldararu, O., Blundell, T. L. & Kepp, K. P. A base measure of precision for protein stability predictors: structural sensitivity. BMC Bioinforma. 22, 1–14 (2021).
Scarabelli, G., Oloo, E. O., Maier, J. K. & Rodriguez-Granillo, A. Accurate prediction of protein thermodynamic stability changes upon residue mutation using free energy perturbation. J. Mol. Biol. 434, 167375 (2022).
Capriotti, E., Fariselli, P., Rossi, I. & Casadio, R. A three-state prediction of single point mutations on protein stability changes. BMC Bioinforma. 9, 1–9 (2008).
Bava, K. A., Gromiha, M. M., Uedaira, H., Kitajima, K. & Sarai, A. Protherm, version 4.0: thermodynamic database for proteins and mutants. Nucleic Acids Res. 32, D120–D121 (2004).
Rives, A. et al. Biological structure and function emerge from scaling unsupervised learning to 250 million protein sequences. Proc. Natl Acad. Sci. USA. https://doi.org/10.1101/622803 (2019).
Meier, J. et al. Language models enable zero-shot prediction of the effects of mutations on protein function. Adv. Neural Inf. Process. Syst. 34, 29287–29303 (2021).
Rao, R. M. et al. MSA transformer. In: International Conference on Machine Learning, 8844–8856 (PMLR, 2021).
Riesselman, A. J., Ingraham, J. B. & Marks, D. S. Deep generative models of genetic variation capture the effects of mutations. Nat. Methods 15, 816–822 (2018).
Frazer, J. et al. Disease variant prediction with deep generative models of evolutionary data. Nature 599, 91–95 (2021).
Notin, P. et al. Tranception: protein fitness prediction with autoregressive transformers and inference-time retrieval. In: International Conference on Machine Learning, 16990–17017 (PMLR, 2022).
Notin, P. M. et al. Trancepteve: Combining family-specific and family-agnostic models of protein sequences for improved fitness prediction. bioRxiv https://doi.org/10.1101/2022.12.07.519495 (2022).
Nijkamp, E. et al. Progen2: exploring the boundaries of protein language models. Cell Syst. 14, 968–978 (2023).
Kulikova, A. V. et al. Sequence and structure based deep learning models represent different aspects of protein biochemistry. bioRxiv. https://doi.org/10.1101/2023.03.20.533508 (2023).
Blaabjerg, L. M. et al. Rapid protein stability prediction using deep learning representations. eLife 12, e82593 (2023).
Dauparas, J. et al. Robust deep learning–based protein sequence design using proteinmpnn. Science 378, 49–56 (2022).
Stourac, J. et al. Fireprotdb: database of manually curated protein stability data. Nucleic Acids Res. 49, D319–D324 (2021).
Diaz, D. danny305/StabilityOracle: v0.0.1 https://doi.org/10.5281/zenodo.12636946 (2024).
Montanucci, L., Capriotti, E., Frank, Y., Ben-Tal, N. & Fariselli, P. Ddgun: an untrained method for the prediction of protein stability changes upon single and multiple point variations. BMC Bioinforma. 20, 1–10 (2019).
Rodrigues, C. H., Pires, D. E. & Ascher, D. B. Dynamut2: assessing changes in stability and flexibility upon single and multiple point missense mutations. Protein Sci. 30, 60–69 (2021).
Acknowledgements
This work was sponsored by grants from the Defense Threat Reduction Agency (HDTRA1201001), the Welch Foundation (F-1654) and the NSF AI Institute for Foundations of Machine Learning (IFML). We would like to thank AMD for the donation of critical hardware and support resources from its HPC Fund.
Author information
Authors and Affiliations
Contributions
D.J.D., A.K., and A.G.D. conceived the project. D.J.D. designed the data engineering pipeline for pre-training and fine-tuning with support from J.M.L. and J.W. D.J.D. developed the training and test set splits. D.J.D. and C.G. developed the graph-transformer architecture and trained Stability Oracle. J.O.Z. trained and evaluated Prostata-IFML. D.Y. generated the AlphaFold datasets. The manuscript was written primarily by D.J.D. and C.G. with support from J.O.Z., A.K., A.G.D., and A.D.E. D.J.D., C.G. and A.K. supervised all aspects of the study.
Corresponding author
Ethics declarations
Competing interests
D.J.D. and J.M.L have financial relationships with Intelligent Proteins LLC. C.G., J.Z., J.W., D.Y., A.D.E., A.D., and A.R.K. declare that they have no conflict of interest.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Diaz, D.J., Gong, C., Ouyang-Zhang, J. et al. Stability Oracle: a structure-based graph-transformer framework for identifying stabilizing mutations. Nat Commun 15, 6170 (2024). https://doi.org/10.1038/s41467-024-49780-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-49780-2
- Springer Nature Limited
This article is cited by
-
Zero-shot transfer of protein sequence likelihood models to thermostability prediction
Nature Machine Intelligence (2024)