

Abstract
Circumventing the conventional two-electron oxygen reduction pathway remains a great problem in enhancing the efficiency of H2O2 photosynthesis. A promising approach to achieve outstanding photocatalytic activity involves the utilization of redox intermediates. Here, we engineer a polyimide aerogel photocatalyst with photoreductive carbonyl groups for non-sacrificial H2O2 production. Under photoexcitation, carbonyl groups on the photocatalyst surface are reduced, forming an anion radical intermediate. The produced intermediate is oxidized by O2 to produce H2O2 and subsequently restores the carbonyl group. The high catalytic efficiency is ascribed to a photocatalytic redox cycle mediated by the radical anion, which not only promotes oxygen adsorption but also lowers the energy barrier of O2 reduction reaction for H2O2 generation. An apparent quantum yield of 14.28% at 420 ± 10 nm with a solar-to-chemical conversion efficiency of 0.92% is achieved. Moreover, we demonstrate that a mere 0.5 m2 self-supported polyimide aerogel exposed to natural sunlight for 6 h yields significant H2O2 production of 34.3 mmol m−2.
Similar content being viewed by others
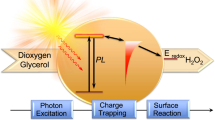


Introduction
Hydrogen peroxide (H2O2), which serves as an eco-friendly oxidant and a versatile energy carrier, has widespread applications in healthcare, disinfection, wastewater treatment, and chemical synthesis1. The global market demand for H2O2 is estimated to possibly increase at a compound annual growth rate of 4.6%, reaching 5.7 million tons by 20282. Currently, more than 95% of the H2O2 in the market is predominantly produced through the anthraquinone process, which relies on noble palladium-based catalysts and consumes substantial energy resources. Moreover, the conventional homogeneous anthraquinone process has been criticized for extracting produced H2O2 and generating toxic byproducts3. Consequently, exploration of environmentally friendly approaches for the synthesis of H2O2 is desirable.
Artificial photosynthesis of H2O2 using organic semiconductors represents an advanced approach toward a sustainable and environmentally friendly future2,4. To date, powder photocatalysts that can harness water, oxygen, and sunlight to generate H2O2 have been reported5. For example, g-C3N4 derivatives, resorcinol-formaldehyde resins, conjugated polymers, covalent triazine frameworks, and covalent organic frameworks have exhibited H2O2 production6. However, previously established photocatalytic systems have significant limitations due to the direct collision of photogenerated electrons with oxygen, which results in rapid charge carrier recombination and diverse photoreduction pathways7,8,9. Given the sluggish nature of the two-electron reduction of O2, photoinduced electrons are prone to recombine with holes before reacting with dissolved oxygen in water, significantly decreasing the quantum efficiency. In addition, the O2 reduction process involves two competing reactions, i.e., one-electron reduction and four-electron reduction of O2, severely limiting the selectivity for H2O2. These issues result in insufficient activity for photocatalytic H2O2, which is far from sufficient for large-scale production. Hence, the development of an innovative reaction pathway for the photosynthesis of H2O2 is imperative.
In green plants, photogenerated electrons are effectively channeled through redox species such as plastoquinone and NADP, which is advantageous for preventing electron-hole recombination and enhancing quantum efficiency10,11. The utilization of sufficiently reducing species as reaction intermediates to drive the reduction of O2 is an ideal approach for the photosynthesis of H2O2. Inspired by the high activity and selectivity advantages of the industrial anthraquinone process, polymer photocatalysts incorporating anthraquinone units as reductive sites or cocatalysts have been employed to promote two-electron oxygen reduction for H2O2 synthesis12,13,14. Nonetheless, in-depth insights into the anthraquinone photocatalytic mechanism for solar-driven H2O2 production are lacking. Polyimides with anthraquinone-like structures have been used as photoreceptors or electronic materials due to their combination of photostability, electronic structure tuning, and redox properties15,16. These qualities align well with the requirements of the photocatalytic system that we want to design. In particular, anion radicals of produced aromatic imides and diimides have been identified as potent electron reductants17,18,19,20. However, a conceptually related mechanism involving photoinduced anion radical intermediates has not yet been comprehensively revealed for H2O2 photosynthesis.
In this work, we developed a covalently crosslinked polyimide aerogel photocatalyst featuring a reductive C = O group, designated PI-BD-TPB. A notable H2O2 concentration of 2.85 mM h−1 coupled with an apparent quantum yield (AQY) of 14.28% at 420 ± 10 nm was attained. Impressively, even a mere 0.5 m2 self-supported polyimide aerogel exposed to natural sunlight displayed a remarkable H2O2 yield of 34.3 mmol m−2, rendering it conducive to separation and recycling in large-scale applications. More importantly, we revealed the pathway of an intermediate-induced photocatalytic redox cycle for H2O2 synthesis. Under photoexcitation, carbonyl groups on the polyimide aerogel surface were reduced to anion radicals, which were oxidized by O2 to generate H2O2, followed by reversion to carbonyl groups. Through comprehensive in situ experimental investigations along with theoretical calculations, we deeply demonstrated that the redox cycle mechanism enhanced O2 adsorption and lowered the energy barrier of the O2 reduction reaction, thereby significantly boosting the overall photosynthetic production of H2O2.
Results and discussion
Photocatalytic performance and scalable production of H2O2
The PI-BD-TPB aerogel photocatalyst for solar-driven H2O2 production was prepared via condensation of the triangular aromatic triamine 1,3,5-tris[4-amino(1,1-biphenyl-4-yl)]-benzene (TPB) as the donor unit and the linear 3,3′,4,4′-biphenyltetracarboxylic dianhydride (BD) as the acceptor unit by the sol-gel-thermal imidization route (Supplementary Figs. 3, 4). The photocatalytic performance of PI-BD-TPB for non-sacrificial H2O2 production was assessed under simulated sunlight illumination. The average H2O2 concentration for PI-BD-TPB in water under a saturated O2 atmosphere was 2833 μM h−1, which largely exceeded that of most reported polymeric photocatalysts (Fig. 1a). The H2O2 yield in O2-saturated pure water was 3.78 times greater than that in air-saturated pure water (748.47 μM h−1), strongly indicating the indispensable role of oxygen in the reaction system. Nevertheless, a remarkable accumulation amount was exhibited, up to 14.34 mM H2O2 after 12 h (Supplementary Figs. 8). In addition to exhibiting high activity, PI-BD-TPB displayed ultradurable performance for a continuous 144 h photocatalytic run, demonstrating satisfactory photostability (Fig. 1b). According to Fourier transform infrared (FTIR) spectroscopy, solid-state 13C NMR spectroscopy, X-ray diffraction (XRD), scanning electron microscopy (SEM), transmission electron microscopy (TEM) and UV-vis absorption results, the PI-BD-TPB aerogel exhibited no significant structural, morphological or absorption edge changes after long-term reaction (Supplementary Figs. 9–13).

a The stable H2O2 production with O2 or air atmosphere of the PI-BD-TPB aerogel under AM 1.5 illumination. Reaction conditions: 15 mg aerogel photocatalyst, 20 mL H2O, 298 K. b The stable cyclic photocatalytic H2O2 production of the PI-BD-TPB aerogel. c SCC efficiency comparison of the PI-BD-TPB aerogel with other reported photocatalysts, See Supplementary Table S1 for more detailed information including references. d The wavelength-dependent apparent quantum yield (AQY) of the PI-BD-TPB aerogel in the photocatalytic H2O2 production, 400 ± 10 nm,420 ± 10 nm, 435 ± 10 nm, 500 ± 10 nm, 530 ± 10 nm, 600 ± 10 nm, and 650 ± 10 nm, Error bars on mean values are standard deviations of three independent H2O2 production tests. e Practical performance test based on the PI-BD-TPB aerogel under irradiation intensity of 100 mW cm−2. f Digital image of the photocatalytic H2O2 production under natural sunlight on Jiangnan University campus using the PI-BD-TPB aerogel membrane from a side view with the 0.5 m2 panel reactor containing 10 L H2O.
The solar-to-chemical conversion efficiency (SCC) of PI-BD-TPB was measured to be 0.92%, surpassing the typical plant efficiency (0.10%) (Fig. 1c and Supplementary Fig. 14). As shown in Fig. 1d, the AQY of PI-BD-TPB exhibited a wavelength dependence, which was in accordance with its absorption spectrum. The maximum AQY of PI-BD-TPB at 420 ± 10 nm reached 14.28%. Moreover, PI-BD-TPB exhibited negligible activity for H2O2 decomposition under continuous irradiation for 24 h (Supplementary Fig. 15). The macroscopic polyimide aerogel photocatalyst not only has good photostability and photocatalytic reactivity, but also is convenient for separation and recyclability. Benefiting from the facile process, macroscopic PI-BD-TPB aerogel with membrane shapes was also prepared, confirming the feasibility of large-scale fabrication. We performed an activity test under 100 mW cm−2 irradiation for 8 h, and PI-BD-TPB exhibited a remarkable H2O2 yield of 88.6 mmol m2 (Fig. 1e). Motivated by the excellent photocatalytic performance of PI-BD-TPB, a scalable test for photocatalytic H2O2 production in an outdoor environment was conducted under natural sunlight as the energy source. A 0.5 m2 polyimide aerogel membrane photocatalyst effectively produced a H2O2 yield of 34.3 mmol m2, demonstrating its practical value for large-scale hydrogen peroxide production (Fig. 1f and Supplementary Figs. 16–18).
Carbonyl group photo-reduction to anion radical intermediate
To understand the excellent photocatalytic performance in H2O2 production, we investigated the structural characteristics of the PI-BD-TPB aerogel. We successfully fabricated covalently crosslinked polyimide aerogels with π-conjugated and π-stacked donor-acceptor structures. PI-BD-TPB, which had a low density (ca. 37.78 mg/cm3), was so light that it could rest on top of a dandelion (Fig. 2a). FTIR spectroscopy was employed to monitor the chemical structure (Fig. 2b). Bands were clearly observed in the PI-BD-TPB curve at 1777.5, 1708.9 and 738.6 cm−1, representing the asymmetric stretching, symmetric stretching and bending vibrations of carbonyl groups, respectively. The band at 1358.1 cm−1 was assigned to the stretching vibration of -C − N, indicating the complete condensation of BD and TPB monomers21. Solid-state 13C NMR spectroscopy (Fig. 2c) was used to further confirm the formation of the polyimide aerogel. The observed signal at 166.5 ppm indicated the presence of a carbonyl carbon atom in the imide ring. Additional chemical shifts in the range of 100–150 ppm were attributed to phenyl carbon atoms22. Moreover, PI-BD-TPB showed a new band located at 288.5 eV in the high-resolution C 1 s spectrum, corresponding to the chemical bond of -C = O (Supplementary Fig. 19)23. Collectively, the FTIR, solid-state 13C NMR and XPS spectra demonstrated that the prepared PI-BD-TPB aerogel photocatalyst is rich in carbonyl groups.

a Schematic diagram of synthesis of the PI-BD-TPB aerogel with π-conjugated and π-stacked D-A structure. b FTIR spectra of the monomer BD and TPB, with the PI-BD-TPB aerogel. c Solid-state 13C NMR spectra of the PI-BD-TPB aerogel. d Schematic diagram of anion radical intermediate formation via photogenerated electron transfer and reduction. e In situ X-ray photoelectron spectroscopy spectrum of O 1 s on the PI-BD-TPB aerogel. f Electron paramagnetic resonance spectra of the PI-BD-TPB before and after light irradiation (300 W Xe lamp). g Ultraviolet-visible absorption spectra of BD-TPB and BD-TPB* anion radical in DMF solution with Na2S2O4 as electron donor under Ar atmosphere.
Additional characterizations were carefully performed to understand the features of the PI-BD-TPB aerogel. PAA is the precursor of polyimide, whose molecular weight distribution determines the molecular weight of the final product. Due to the insolubility of PI-BD-TPB, we measured the molecular weight of the PAA gel powders via gel permeation chromatography in NMP. The average molecular weight (Mw) and corresponding polydispersity index (PDI, PDI = Mw/Mn, Mn = 3.4 kDa) were 8.0 kDa and 2.35 (Supplementary Fig. 20). SEM and TEM images of the PI-BD-TPB aerogel (Supplementary Figs. 21–23) revealed cross-linked spherical particles with an average diameter of ~ 1 μm. High-resolution transmission electron microscopy (HRTEM) images revealed that PI-BD-TPB was locally crystalline in nature. The interlayer distance was measured to be 0.43 nm (Supplementary Fig. 24). Furthermore, the XRD peak of PI-BD-TPB was located at 2θ ≈ 20.3°, representing the 0.43 nm interlamellar d-spacing of π–π stacking (Supplementary Fig. 25). The porosity of PI-BD-TPB was assessed using N2 sorption measurements at 77.3 K, and the Brunauer‒Emmett‒Teller surface area was calculated to be 372.8 m2 g−1. By employing a nonlocal density functional theory model, its pore size was determined to be ~ 1.5 nm (Supplementary Fig. 26). The macroscopic pore size distribution on the surface of the BD-TPB aerogel was analyzed by mercury intrusion porosimetry (Supplementary Fig. 27). The macroscopic pore size on the surface of BD-TPB was ~ 2 μm.
Thermogravimetric analysis revealed that the PI-BD-TPB aerogel had a high thermal stability up to 550 °C (Supplementary Fig. 28). The chemical stability of the PI-BD-TPB aerogel was investigated by immersing it in diverse solvents. Notably, the FTIR spectra of PI-BD-TPB after soaking in different solvents were almost unchanged, confirming its excellent chemical stability (Supplementary Fig. 29). The good chemical and thermal stability are ascribed to the strong imide linkage and highly conjugated structure. The average ζ potential of PI-BD-TPB was − 55.1 mV in H2O aqueous solution, indicating strong adsorption of H+ (Supplementary Fig. 30). PI-BD-TPB displayed a hydrophilic surface with a contact angle of 40.4° (Supplementary Fig. 31). The macroscopic polyimide aerogel was cylindrical, with a diameter of 3 cm and a height of 2 cm. Compared to powder with the same weight, it was characterized by a low density (ca. 37.78 mg/cm−3). (Supplementary Figs. 32–34).
Adsorption is a special feature of aerogel materials. The 0.53 g PI-BD-TPB aerogel rapidly absorbed 13.89 g of H2O after 5 min, which was approximately 25 times its weight, exhibiting an effective adsorption capacity (Supplementary Figs. 35, 36). In addition, the PI-BD-TPB aerogel with excellent mechanical behavior could hold 200 times its own weight (Supplementary Fig. 37). A compression–recovery test was performed to estimate the mechanical durability, and the PI-BD-TPB aerogel, as an elastic material, exhibited excellent reversible compressibility at strains of 10%, 20%, 30%, 40%, 50% and 60% (Supplementary Fig. 38). Benefiting from the facile synthetic route, a macroscopic polyimide membrane was also prepared, further exhibiting high operability (Supplementary Fig. 39). In summary, the covalently crosslinked polyimide aerogel has a low density, hydrophilicity, an effective absorption ability, high chemical and thermal stability, excellent resilience and good mechanical behavior.
Theoretical calculations showed that the highest occupied molecular orbital (HOMO) was mainly distributed on the TPB unit, while the lowest unoccupied molecular orbital (LUMO) was distributed on the BD unit, suggesting transfer of photogenerated electrons from the TPB unit to the BD unit (Supplementary Fig. 40). The -C = O groups on BD-TPB had electron affinity characteristics according to the electrostatic potential distribution (Supplementary Fig. 41). The electrostatic potential of PI-BD-TPB under photoexcitation indicated that the carbonyl group had a strong ability to extract electrons (Supplementary Fig. 42). Meanwhile, the related photophysical experiments containing electrochemical impedance spectroscopic spectra, photocurrent spectra, surface photovoltage spectra and transient fluorescence spectra were performed. We confirm that PI-BD-TPB aerogel with donor-acceptor structure has excellent charge separation efficiency and extended carrier lifetime. (Supplementary Fig. 46–49).
To investigate the reductive property of -C = O in PI-BD-TPB, we performed cyclic voltammetry measurements (Supplementary Fig. 43). Two pairs of peaks were observed at approximately 0.509 V / 1.674 V and 0.009 V / 0.905 V (vs. RHE), which could be assigned to enolization of carbonyl oxygen, indicating that the R-C = O in the imide ring stores electrons24,25. Mott–Schottky plots, X-ray photoelectron spectroscopy valence band spectra, and ultraviolet-visible absorption spectra were obtained to determine the band gap and band position of PI-BD-TPB (Supplementary Figs. 44, 45). The band gap of PI-BD-TPB was estimated to be 2.21 eV. The conduction band edge (ECB) of PI-BD-TPB was calculated to be − 0.19 V (vs. RHE), and the valence band edge (EVB) was 2.02 V (vs. RHE). These results confirmed that the PI-BD-TPB aerogel provided a thermodynamically favorable driving force for the reduction of R-C = O and two-electron oxidation of H2O. Note that the anion radical is highly likely to form on the surface of PI-BD-TPB during photoexcitation due to the hyper-conjugated structure (Fig. 2d).
The structural variation of the C = O group in PI-BD-TPB was monitored by in situ X-ray photoelectron spectroscopy (Fig. 2e). Initially, in the dark, the O 1 s XPS peak was resolved into one major component (532.1 eV) for the C = O group. Under light irradiation, a prominent new peak at 530.8 eV attributed to the C − O bond emerged26,27. This observation indicated that the C = O group in the imide ring underwent structural transformation, in which it received photogenerated electrons to form a C − O group. In contrast, the C = O signal decreased. After the light was turned off, the O 1 s peak reverted to the original state, proving the transformation of C = O bonds to C − O bonds. To gain further insight into the structural variation of the C = O group, electron paramagnetic resonance (EPR) spectroscopy was performed on PI-BD-TPB (Fig. 2f). There was no obvious signal at g = 2.003 in the dark. Upon light irradiation, the intensity of the signal at g = 2.003 significantly increased, indicating that photoinduced electrons were transferred to carbonyl groups, corresponding to imide radical formation28. These changes demonstrated that the reduction of R-C = O to the anion radical occurred on the PI-BD-TPB surface during the photocatalytic process. Moreover, the presence of superoxide radicals was excluded based on the EPR measurement results (Supplementary Fig. 50). The above results confirmed that the R-C = O groups in the photocatalyst structure were reduced by photogenerated electrons, forming anion radicals in our system.
UV‒vis spectroscopy and fluorescence tests were conducted to probe the formation of anion radicals. In the UV‒vis absorption spectrum (Fig. 2g), the 250–300 nm absorption peak was from the carbonyl group on the imide ring. The carbonyl group received electrons provided by Na2S2O4 as the electron donor upon light illumination29, and the structure changed; thus, the absorption appeared to be red-shifted. The new peak at 418 nm corresponded to the carbon‒oxygen anion radical formed by the carbonyl group that obtained electrons30,31. Meanwhile, we performed the fluorescence test of BD-TPB under dark and light conditions31,32. Under dark conditions, PI-BD-TPB exhibited an obvious signal at 460 nm. The signal intensity at 460 nm decreased after light irradiation, indicating that the carbonyl group received photogenerated electrons and formed the anion radical, leading to a decrease in the fluorescence signal (Supplementary Fig. 51). Thus, the reduction of the -C = O group in our system generates the anion radical under light irradiation, which is consistent with the experimental results. We also confirmed that the photogenerated holes in the PI-BD-TPB aerogel could be consumed via a direct two-electron water oxidation pathway for H2O2 generation on the C atom in the TPB donor unit (Supplementary Figs. 52–56). Therefore, the photogenerated electrons and holes in PI-BD-TPB were used for the reduction of the -C = O group and oxidation of H2O, respectively.
Enhanced oxygen adsorption by anion radical intermediate
To thoroughly investigate the role of the anion radical intermediate in the oxygen reduction reaction, we conducted in situ FTIR spectroscopy under operando conditions. The entire process was carried out in a 2-propanol solution with a saturated O2 atmosphere. As shown in Fig. 3a, b, no oxygen adsorption signal was observed under dark conditions, while the characteristic peak for the O − O stretching signal at 948.22 cm−1 and the O − O adsorption signal at 1023.7 − 1196.7 cm−1 were detected during the illumination process33,34. In particular, a weak O − O adsorption signal could still be detected 5 min after the light was turned off, indicating that the anion radical intermediate significantly enhanced oxygen adsorption. In situ Raman spectra were also obtained to provide direct evidence (the test conditions were the same as those in the in situ FTIR spectroscopy measurements). The 761.2 cm−1 band was assigned to the in-plane bending mode of the imide moiety35. No signal was detected in the dark, indicating that the O2 adsorption of the PI-BD-TPB aerogel was weak. However, a new broad band appeared at 942 cm−1 during the irradiation process, corresponding to the O − O stretching of the imide ring (Fig. 3c)36,37. These findings, which align with the results of in situ FTIR spectroscopy measurements, strongly support that the anion radical intermediate promotes O2 adsorption.

a Experimental in situ FTIR spectra and (b) The peak intensity of O2 adsorption recorded during photoreaction in a 2-propanol aqueous solution with saturated oxygen (2-propanol as electron donor, 10% v/v). c In situ Raman spectra of the PI-BD-TPB aerogel recorded during photoreaction in a 2-propanol aqueous solution with saturated oxygen, a 785 nm laser was taken as light source to shine directly on the photocatalyst surface, and a computer synchronously collected the Raman signals. d Different adsorption site a of O2 on -C = O state (BD-TPB) and radical anion state (BD-TPB*) respectively. e The adsorption energy of O2 on different sites of C = O state (BD-TPB) and radical anion state (BD-TPB*), respectively.
The adsorption of oxygen molecules on photocatalysts has been recognized as the most critical step of the O2 reduction process38. The O2 adsorption property of the anion radical intermediate was investigated by first-principles calculations to understand the correlation between the structure and performance. We calculated the adsorption of O2 in the -C = O state (BD-TPB) and radical anion state (BD-TPB*). Initially, the oxygen adsorption on different sites in the initial PI-BD-TPB state was weak. In contrast, the oxygen adsorption energy on each site was significantly decreased in the protonated anion intermediate state (Fig. 3d, e). In particular, the two symmetrical carbon atoms on the imide ring (site 2) were found to be particularly favorable for the Yeager-type (side-on) O2 adsorption, which was consistent with the results of the experimental in situ FTIR and Raman spectra. According to the literature, the Yeager-type (side-on) adsorption with enhanced adsorption affinity for O2 is extremely conducive to the oxygen reduction reaction for H2O2 synthesis34.
Redox cycle mechanism via anion radical intermediate
To elucidate the O2 reduction pathway for H2O2 production, we devised a continuous “Light-Ar/Dark-O2” experiment similar to plant photosynthesis (Fig. 4a)39. The system was purged with Ar gas to exhaust O2 gas in the pure water before turning on the light. Approximately 129 μM H2O2 was generated by PI-BD-TPB under the “Light-Ar” condition, showing that the photogenerated holes oxidized water to synthesize H2O2. Significantly, when the light was turned off and the system was subsequently purged with O2 gas, approximately 23 μM H2O2 continued to be detected, revealing that the formed intermediate could combine with O2 to produce H2O2 within a certain time. This result implied the importance of the radical intermediate in the dark-driven reaction for H2O2 production. Multiple experiments verified that the amount of H2O2 continuously increased within 10 min under the “Dark-O2” condition. As a result, we speculate that the carbonyl group on the catalyst surface stored photogenerated electrons during the photoreaction and formed the anion intermediate. In the dark, the anion intermediates released the stored electrons and directly reduced O2 to H2O2. The O2 reduction reaction pathways for H2O2 generation via the redox intermediate greatly improved the utilization rate of photogenerated electrons.

a H2O2 production on the PI-BD-TPB aerogel under different control conditions, Error bars on mean values are standard deviations of three independent tests, reaction conditions: catalyst (50 mg), water (25 mL), Ar atmosphere in the light or O2 atmosphere in the dark. b In situ FTIR spectrum of the PI-BD-TPB aerogel with Ar atmosphere under 300 W Xe lamp and O2 gas purging in the dark at 1200–2000 cm−1 and 2700–2900 cm−1, the peak intensity of H2O2 species on “Light-Ar /Dark-O2” condition. c Isotopic experiments with 18O2 or H218O for H2O2 production on the PI-BD-TPB photocatalyst, See Supplementary Table 2 for more detailed information. d 1H spectrum of H2O2 in D2O. e 1H spectrum of the photocatalytic D2O2 production of the BD-TPB in D2O. f Calculated free energy diagrams of H2O2 production from O2 reduction pathway by model systems of C = O state (BD-TPB) and radical anions state (BD-TPB*), respectively. g Key steps of H2O2 production from O2 reduction pathway on the PI-BD-TPB aerogel photocatalyst.
In situ FTIR spectra confirmed the above hypothesis. In situ FTIR spectra of PI-BD-TPB were continuously recorded during the light / dark reaction to verify the structural variation of the PI-BD-TPB photocatalyst. As shown in Fig. 4b, the signal intensity of the C = O groups at 1650 cm−1 decreased under the light-Ar condition and reverted to the original state under the dark-O2 condition, and the formation and disappearance of the peak at 1283 cm−1 was attributed to the C − O group40. The peak at ~ 1354 cm−1 corresponding to O − H bending displayed a similar trend to that of the C − O group41, indicating that the photogenerated electrons reduced the carbonyl group to the protonated anion intermediate. This observation aligns with the prior results of zeta potential experiments. The new infrared vibration signal at 2849 cm−1 can be attributed to the typical (ν2 + ν6) / 2ν6 − OH bending feature of H2O242, which gradually increased due to the rapid structural transformation. The O − O characteristic peak at 946 cm−1 was clearly detected in the dark reaction, indicating that the protonated anion intermediate promoted oxygen adsorption through the Yeager-type (side-on) absorption (Supplementary Fig. 57). These phenomena confirmed that the anion radical intermediate combined with O2 to produce H2O2 and then reverted to the carbonyl group, accomplishing a catalytic redox cycle. The entire process is similar to plant photosynthesis involving light and dark reactions.
Isotopic labeling experiments were conducted to investigate photocatalytic H2O2 production (Fig. 4c and Table S2). With the H216O and 18O2 system,18O signals were evidently observed in the produced H2O2, indicating the existence of the O2 reduction pathway for H2O2 synthesis in our catalytic system43,44. The amount of H218O2 generated via reduction of 18O2 and the total H2O2 production amount were 0.0652 mmol and 0.1269 mmol, respectively, indicating that half of the H2O2 was mainly produced by the O2 reduction pathway. Similarly, to detect the O in H2O2 produced from the H2O oxidation reaction, we used H218O and 16O2 as the reaction system. The isotopic results showed that 18O signals were also observed in the produced H2O2, confirming H2O2 production through H2O oxidation43,44. The amount of H218O2 generated via the oxidation of H218O and the total H2O2 production amount were 0.0512 mmol and 0.1040 mmol, respectively, demonstrating that half of the H2O2 was mainly produced by the H2O oxidation pathway. Therefore, the amounts of H2O2 produced by oxidation reaction and reduction reaction are relatively equivalent. Moreover, we also proved that the proton (H+) source for H2O2 generation was H2O (Fig. 4d, e). We measured the 1H spectrum of H2O2 with D2O as a solvent to qualitatively observe its chemical shift. In D2O, considering the concentration of the products, we prepared a 15 mM H2O2 solution with D2O as the solvent. Affected by the solvent, the chemical shift (1H) of H2O2 in D2O was 11.04 ppm. These results were consistent with the 1H spectrum of H2O2 in a previous publication45. Then, we carried out the photocatalytic H2O2 production experiment in D2O. After the reaction, the suspension was monitored with 1H nuclear magnetic resonance (NMR). No H2O2 signal was observed at 11.04 ppm in the sample, indicating that the proton source for H2O2 generation was H2O. Elemental analysis was conducted for the PI-BD-TPB photocatalyst before and after the photoreaction, and the H content was basically stable (Supplementary Fig. 58). Thus, photogenerated holes oxidize water to produce H2O2, and then, O2 combines with H+ induced by the holes and subsequently releases H2O2 in our system. Nine photocatalysts with carbonyl groups were prepared and evaluated to verify the anion radical intermediate-mediated H2O2 synthesis strategy (Supplementary Figs. 59–64).
The O2 reduction process over the PI-BD-TPB photocatalyst was simulated to understand the reaction pathways from a thermodynamic point of view. As depicted in Fig. 4f, we compared the Gibbs free energy diagrams of the conventional one-step 2e− pathway (Pathway I) and the pathway of the photocatalytic redox cycle via the redox intermediate (Pathway II) for H2O2 production. In Pathway II, the O2* species had a lower Gibbs free energy (ΔG = − 0.99 eV) at the two symmetric C atoms of the carbonyl group on the imide ring. The calculation results revealed that the anion radical intermediate-mediated photocatalytic redox cycle was beneficial for O2 adsorption, providing a powerful thermodynamic driving force. Moreover, Pathway II exhibited a lower ΔG for *HOOH formation (− 2.60 eV) than Pathway I (− 2.30 eV) and was more favorable for H2O2 desorption from the photocatalyst surface. A rotating disk electrode test was performed to determine the average electron transfer number involved in the O2 reduction reaction. The average electron transfer number was calculated to be approximately 1.976, indicating that the PI-BD-TPB aerogel had a high two-electron selectivity for the O2 reduction reaction (Supplementary Fig. 65). To investigate the H2O2 desorption capability of the PI-BD-TPB aerogel, electrochemical H2O2 reduction reaction measurements were conducted in an Ar-saturated 0.5 M H2SO4 electrolyte containing 15 mM H2O2. We demonstrated that BD-TPB under light irradiation had poorer H2O2 reduction reaction activity (Supplementary Fig. 66). We also tested the electrochemical performance for H2O2 generation on PI-BD-TPB photocatalyst via the constant potential method. These results showed that the PI-BD-TPB had excellent two-electron oxygen reduction for H2O2 electrosynthesis (Supplementary Fig. 67).
Through comprehensive in situ spectroscopic studies correlated with theoretical calculations, we elucidated the reaction pathways over the PI-BD-TPB photocatalyst (Fig. 4g). Under the excitation of simulated sunlight, photoelectrons and holes were effectively separated. Photogenerated holes oxidized water to produce H2O2 and proton source. Moreover, the C = O group in the imide ring stored photogenerated electrons was converted into the anion radical intermediate, and subsequently combined with H+ produced by holes, transforming into the protonated anion intermediate. The intermediate spontaneously adsorbed O2 to once again release H2O2 and reverted to the C = O group as in the original state, achieving a catalytic redox cycle.
Discussion
In summary, a crosslinked polyimide aerogel photocatalyst incorporating reductive C = O groups was engineered for the effective synthesis of H2O2 from H2O and O2. With an AQY of 14.28% at 420 ± 10 nm, the polyimide aerogel under 100 mW cm−2 irradiation for 8 h produced 88.6 mmol m−2 H2O2. More importantly, our research introduces the approach for H2O2 photosynthesis involving an aromatic anion radical intermediate that mediates the photocatalytic redox cycle with O2. This redox cycle pathway not only dramatically improves O2 adsorption but also thermodynamically favors the oxygen reduction reaction, consequently enhancing the efficiency of H2O2 photosynthesis.
Methods
Synthesis of polyimide BD-TPB aerogel
All chemicals used in the research were purchased from Tansoole Co, Ltd., without additional purification. The polyimide BD-TPB photocatalyst presented in this research were prepared by the imidization of aromatic triamines 1,3,5-tris[4-amino(1,1-biphenyl-4-yl)] benzene (TPB) with aromatic dianhydrides 3,3′,4,4′-biphenyltetracarboxylic dianhydride (BD). Typically, BD (26.47 mg, 0.90 mmol) dissolved in 1.0 mL of 1-methayl-2-pyrrolidinone (NMP) solution, then TPB (34.78 mg, 0.60 mmol) dispersed in 1.0 mL of Mesitylene solution, then Isoquinoline (0.1 mL) was added. After being sonication with a power of 100 W for 10 min, and luminous yellow solution was quickly obtained. Then poured into a mold and allowed to gel within 60 min in 0 °C. The gel was upon further solvothermal treatment at 180 °C for 48 h. The products were washed in a solution of 75% NMP in ethanol for 24 hours. Subsequently, the solvent was exchanged in 24 h intervals with 25% NMP in ethanol, and then 100% ethanol for three times. Finally, the macroscopic BD-TPB aerogel was rinsed with deionized water and was freeze-dried (yield: ~ 92%).
Synthesis of poly (amic acid) (PAA)
The precursor of polyimide was synthesized from the polycondensation reaction between the imidization of the dianhydrides BD and the triamines TPB. BD (26.47 mg, 0.90 mmol) was dissolved in 2.0 mL NMP solution, then TPB (34.78 mg, 0.60 mmol) was also added. The mixture solution was stirred at 0 °C for 24 h to obtain the yellow solution named PAA solution. Then PAA solution was precipitated with deionized water and the precipitate was washed several times with ethanol and dried under vacuum (yield: ~ 65%).
Scalable synthesis of polyimide BD-TPB aerogel membrane
Typically, BD (2.64 g) dissolved in 200 mL NMP solution, then TPB (3.47 g) dispersed in 200 mL Mesitylene solution, then Isoquinoline (1.0 mL) was added. After being sonication with a power of 100 W for 10 min, and luminous yellow solution was quickly obtained. Then poured into the mold and allowed to gel within 12 h in 0 °C. The gel was upon further solvothermal treatment at 180 °C for 48 h. The products were washed in a solution of 75% NMP in ethanol for 24 hours. Subsequently, the solvent was exchanged in 24 h intervals with 25% NMP in ethanol, and then 100% ethanol for three times. Finally, the macroscopic BD-TPB aerogel membrane was dried (yield: ~88%).
Photocatalytic H2O2 production
15 mg of PI-BD-TPB photocatalyst and 20 mL of deionized water were put in a quartz square bottle (100 mL). O2 was bubbled into the suspension for 30 min in the dark. During the reaction, O2 was kept bubbling to maintain the O2-rich environment. A 300 W Xe lamp was utilized as the light source, and all photocatalytic experiments were performed in the same experimental condition. The light average intensity is 325 mW·cm−2. The H2O2 concentration was determined by a potassium titanium oxalate method46.
Determination of AQY efficiency
The apparent quantum yield (AQY) of photocatalyst was measured under 300 W Xe lamp irradiation. The photocatalytic reaction was carried out with 40 mg photocatalyst powder in 25 mL water at 50 °C. The active area of the reactor was ~ 0.785 cm−2. The light intensity at 420 nm ± 10 nm was calculated to be 3.72 mW cm−2. Then, AQY was calculated by the following equation:
Measurement of SCC efficiency
The solar-to-chemical energy conversion (SCC) efficiency was determined by the photocatalytic experiment47. 80 mg photocatalyst powder and 60 mL water were added into a flask and bubbled with O2 for 30 min, the reaction was carried out at 50 °C in the water bath. The SCC efficiency was calculated via the following equation: where the free energy (ΔG) for H2O2 formation is 117 kJ mol−1, the irradiance of the spectrum is 1000 W m−2 and the irradiated area is 0.785 × 10−4 m2. The total input energy was therefore 0.0785 W. reaction time is 1200 s. The SCC was calculated as follows:
The stability test
The PI-BD-TPB photocatalyst and 20 mL of deionized water were used in a quartz square bottle. The suspension was well dispersed by ultrasonication for 15 min and O2 was bubbled into the suspension for 30 min in the dark. During the reaction, oxygen was kept bubbling to maintain the O2-rich environment. Other conditions were kept the same. Between each test, H2O2 and H2O were removed by evaporation. After that, the left macroscopic PI-BD-TPB photocatalyst was further dried in a vacuum dry box at 60 °C for 12 h to remove the possible residual of H2O2.
Photocurrents and photoelectrochemical measurements
The Mott–Schottky plots and electrochemical impedance of the catalysts were measured on an electrochemical workstation (CHI660E, CHI Instruments, Shanghai, China)48. A 300 W Xe lamp was utilized as the light source and Na2SO4 (0.5 M) aqueous solution was used as the supporting electrolyte49. A platinum wire and Ag/AgCl electrode were used as counter electrode and reference electrode. 50 μL of Nafion, dry ethanol (1 mL) and photocatalyst (5 mg) were sonicated for 30 min. Then 100 μL of the suspension was dripped onto an ITO glass substrate and dried. The application potential was converted to RHE potentials with respect to Ag/AgCl using the following equation:
Rotating disk electrode measurement
A glassy carbon rotating disk electrode was served as the substrate for the working electrode. The working electrode was prepared as follows: 20 mg of power photocatalysts was dispersed in 2 mL ethanol containing 20 μL Nafion. 20 μL of the above slurry was put onto the disk electrode and dried at room temperature. The linear sweep voltammogram curves were recorded in an O2-saturated 0.1 M phosphate buffer solution at room temperature and a scan rate of 10 mV s−1 with different rotation speeds.
Rotating ring-disk electrode measurement
A ring-disk electrode was served as the substrate for the working electrode. The voltammograms were obtained in a 0.1 M phosphate buffer solution under Ar atmosphere at a scan rate of 10 mV s−1 and a rotation rate of 1000 rpm. The potential of the ring electrode was set to − 0.23 V (vs. Ag/AgCl) to detect O2. The potential of the ring electrode was set to 0.6 V (vs. Ag/AgCl) to detect H2O2.
Electron paramagnetic resonance measurement
The EPR measurement was carried out to detect superoxide radical or hydroxide radical by adding 5,5-dimethyl-1-pyrroline N-oxide (DMPO) as a spin-trapping reagent. A 300 W Xe lamp was used as the light source. The measurements were conducted as follows: photocatalyst were dispersed in water or MeOH containing DMPO with a Pyrex glass tube which was sealed with a rubber septum cap.
Isotopic labeling experiment
18O in the produced H218O2 was determined by converting H218O2 to H218O46.18O in the converted H2O was analyzed by Liquid Water Isotope Analyzer (Los Gatos Research, USA). The conversion method was as follows: First, the collected potassium titanium oxalate-H2O2 complex was re-dissolved and dried; then, the potassium titanium oxalate-H2O2 complex was reduced by KI to convert the contained H2O2 species to H2O; finally, the converted H2O was collected by distillation and detected by Liquid Water Isotope Analyzer.
Data availability
The authors declare that all data supporting the findings of this study are available within the paper, Supplementary Information files and source data at the figshare link.
References
Sun, Y., Han, L. & Strasser, P. A comparative perspective of electrochemical and photochemical approaches for catalytic H2O2 production. Chem. Soc. Rev. 49, 6605–6631 (2020).
Freese, T., Meijer, J. T., Feringa, B. L. & Beil, S. B. An organic perspective on photocatalytic production of hydrogen peroxide. Nat. Catal. 6, 553–558 (2023).
Gopakumar, A. et al. Lignin-supported heterogeneous photocatalyst for the direct generation of H2O2 from seawater. J. Am. Chem. Soc. 144, 2603–2613 (2022).
Wu, Q. et al. A metal-free photocatalyst for highly efficient hydrogen peroxide photoproduction in real seawater. Nat. Commun. 12, 483 (2021).
Yong, Z. & Ma, T. Solar-to-H2O2 catalyzed by covalent organic frameworks. Angew. Chem. Int. Ed. 62, e202308980 (2023).
Cheng, H., Cheng, J., Wang, L. & Xu, H. Reaction pathways toward sustainable photosynthesis of hydrogen peroxide by polymer photocatalysts. Chem. Mater. 34, 4259–4273 (2022).
Yu, W. et al. Photocatalytic hydrogen peroxide evolution: what is the most effective strategy? Nano Energy 104, 107906 (2022).
Wu, S. & Quan, X. Design principles and strategies of photocatalytic H2O2 production from O2 reduction. ACS EST Engg. 2, 1068–1079 (2022).
Shiraishi, Y. et al. Resorcinol-formaldehyde resins as metal-free semiconductor photocatalysts for solar-to-hydrogen peroxide energy conversion. Nat. Mater. 18, 985–993 (2019).
Barber, J. Photosynthetic energy conversion: natural and artificial. Chem. Soc. Rev. 38, 185–196 (2009).
Zhang, L. & Wang, Y. Decoupled artificial photosynthesis. Angew. Chem. Int. Ed. 62, e202219076 (2023).
Xu, X. et al. Conjugated organic polymers with anthraquinone redox centers for efficient photocatalytic hydrogen peroxide production from water and oxygen under visible light irradiation without any additives. ACS Catal. 12, 12954–12963 (2022).
Zhao, C. et al. Molecular level modulation of anthraquinone-containing resorcinol-formaldehyde resin photocatalysts for H2O2 production with exceeding 1.2% efficiency. Angew. Chem. Int. Ed. 62, e202218318 (2023).
Zhang, X. et al. Keto-enamine-based covalent organic framework with controllable anthraquinone moieties for superior H2O2 photosynthesis from O2 and water. Chem. Eng. J. 466, 143085 (2023).
Rao, K. V., Haldar, R., Kulkarni, C., Maji, T. K. & George, S. J. Perylene based porous polyimides: tunable, high surface area with tetrahedral and pyramidal monomers. Chem. Mater. 24, 969–971 (2012).
Kobaisi, M. A., Bhosale, S. V., Latham, K., Raynor, A. M. & Bhosale, S. V. Functional naphthalene diimides: synthesis, properties, and applications. Chem. Rev. 116, 11685–11796 (2016).
Song, Z., Zhan, H. & Zhou, Y. Polyimides: promising energy-storage materials. Angew. Chem. Int. Ed. 49, 8444–8448 (2010).
Li, L. et al. Molecular engineering of aromatic imides for organic secondary batteries. Small 17, e2005752 (2021).
Greenfield, S. R., Svec, W. A., Gosztola, D. & Wasielewski, M. R. Multistep photochemical charge separation in rod-like molecules based on aromatic imides and diimides. J. Am. Chem. Soc. 118, 6767–6777 (1996).
Ghosh, I., Ghosh, T., Bardagi, J. I. & König, B. Reduction of aryl halides by consecutive visible light-induced electron transfer processes. Science 346, 725–728 (2014).
Maschita, J. et al. Ionothermal synthesis of imide-linked covalent organic frameworks. Angew. Chem. Int. Ed. 59, 15750–15758 (2020).
Fang, Q. et al. Designed synthesis of large-pore crystalline polyimide covalent organic frameworks. Nat. Commun. 5, 4503 (2014).
Kofuji, Y. et al. Graphitic carbon nitride doped with biphenyl diimide: efficient photocatalyst for hydrogen peroxide production from water and molecular oxygen by sunlight. ACS Catal. 6, 7021–7029 (2016).
Ma, Y., Guo, Z., Dong, X., Wang, Y. & Xia, Y. Organic proton-buffer electrode to separate hydrogen and oxygen evolution in acid water electrolysis. Angew. Chem. Int. Ed. 58, 4622–4626 (2019).
Yang, H., Liu, S., Cao, L., Jiang, S. & Hou, H. Superlithiation of non-conductive polyimide toward high-performance lithium-ion batteries. J. Mater. Chem. A 6, 21216–21224 (2018).
Luo, X. X. et al. Covalent organic framework with highly accessible carbonyls and pi-cation effect for advanced potassium-ion batteries. Angew. Chem. Int. Ed. 61, e202117661 (2022).
Yang, X. et al. Mesoporous polyimide-linked covalent organic framework with multiple redox-active sites for high-performance cathodic Li storage. Angew. Chem. Int. Ed. 61, e202207043 (2022).
Tu, F. et al. Photoinduced radical persistent luminescence in semialiphatic polyimide system with temperature and humidity resistance. Adv. Sci. 10, e2301017 (2023).
Hasegawa, M., Sensui, N., Shindo, Y. & Yokota, R. Structure and properties of novel asymmetric biphenyl type polyimides. homo- and copolymers and blends. Macromolecules 32, 387–396 (1999).
Che, Y. et al. Enhancing one-dimensional charge transport through intermolecular π-electron delocalization: conductivity improvement for organic nanobelts. J. Am. Chem. Soc. 129, 6354–6355 (2007).
Jiao, Y., Liu, K., Wang, G., Wang, Y. & Zhang, X. Supramolecular free radicals: near-infrared organic materials with enhanced photothermal conversion. Chem. Sci. 6, 3975–3980 (2015).
Hasegawa, M. et al. Photophysical processes in aromatic polyimides. studies with model compounds. J. Polym. Sci., Part B Polym. Phys. 31, 1617–1625 (1993).
Chang, J. N. et al. Oxidation-reduction molecular junction covalent organic frameworks for full reaction photosynthesis of H2O2. Angew. Chem. Int. Ed. 62, e202218868 (2023).
Luo, Y. et al. Sulfone-modified covalent organic frameworks enabling efficient photocatalytic hydrogen peroxide generation via one-step two-electron O2 reduction. Angew. Chem. Int. Ed. 62, e202305355 (2023).
Tsai, W. H., Boerio, F. J. & Jackson, K. M. Characterization of interphases between PMDA/4-BDAF polyimides and silver substrates using surface-enhanced Raman scattering and reflection-absorption infrared spectroscopy. Langmuir 8, 1443–1450 (2002).
Shiraishi, Y. et al. Sunlight-driven hydrogen peroxide production from water and molecular oxygen by metal-free photocatalysts. Angew. Chem. Int. Ed. 53, 13454–13459 (2014).
Teng, Z. et al. Atomically dispersed antimony on carbon nitride for the artificial photosynthesis of hydrogen peroxide. Nat. Catal. 4, 374–384 (2021).
Zhang, X. et al. Unraveling the dual defect sites in graphite carbon nitride for ultra-high photocatalytic H2O2 evolution. Energy Environ. Sci. 15, 830–842 (2022).
Yan, H. et al. Spontaneous exciton dissociation in organic photocatalyst under ambient conditions for highly efficient synthesis of hydrogen peroxide. Proc. Natl. Acad. Sci. USA 119, e2202913119 (2022).
Ye, Y. X. et al. A solar-to-chemical conversion efficiency up to 0.26% achieved in ambient conditions. Proc. Natl. Acad. Sci. USA 118, e2115666118 (2021).
Yue, J. Y. et al. Thiophene-containing covalent organic frameworks for overall photocatalytic H2O2 synthesis in water and seawater. Angew. Chem. Int. Ed. 62, e202309624 (2023).
Chen, L. et al. Acetylene and diacetylene functionalized covalent triazine frameworks as metal-free photocatalysts for hydrogen peroxide production: a new two-electron water oxidation pathway. Adv. Mater. 32, e1904433 (2020).
Pan, C. et al. Efficient and stable H2O2 production from H2O and O2 on BiPO4 photocatalyst. Appl. Catal. B Environ. 316, 121675 (2022).
Zhang, Y. et al. H2O2 generation from O2 and H2O on a near-infrared absorbing porphyrin supramolecular photocatalyst. Nat. Energy 8, 361–371 (2023).
Stephenson, N. A. & Bell, A. T. Quantitative analysis of hydrogen peroxide by 1H NMR spectroscopy. Anal. Bioanal. Chem. 381, 1289–1293 (2005).
Chi, W. et al. Boosting H2O2 photosynthesis by accumulating photo-electrons on carbonyl active site of polyimide covalent organic frameworks. Appl. Catal. B Environ. 355, 124077 (2024).
Liu, L. et al. Linear conjugated polymers for solar-driven hydrogen peroxide production: The importance of catalyst stability. J. Am. Chem. Soc. 143, 19287–19293 (2021).
Kou, M. et al. Photocatalytic CO2 conversion over single-atom MoN2 sites of covalent organic framework. Appl. Catal. B Environ. 291, 120146 (2021).
Wang, Y. et al. The construction of an alkynyl-containing porous polymer for enhanced photocatalytic H2O2 generation. New J. Chem. 48, 9043 (2024).
Acknowledgements
The work is supported by the National Natural Science Foundation of China (22136002, 22172064, 22376083), Special Fund Project of Jiangsu Province for Scientific and Technological Innovation in Carbon Peaking and Carbon Neutrality (BK20220023). The authors would also like to thank Yaning Zhang, Yong Liu, Yunfan Yang, and Yujie Ling from Jiangnan University for their help.
Author information
Authors and Affiliations
Contributions
W.C. and Y.D. contributed equally. Y.D. and Y.Z. conceived the idea. W.C. conducted performance evaluations, designed experiments, and fabrication experiments. B.L. and Z. L. conducted the theoretical calculation. C.P., J.Z., and H.Z. participated in the paper discussions. W.C., Y.D. and Y.Z. planned the research and wrote the paper. All the authors revised and approved the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Hangxun Xu, Yao Zheng, and the other, anonymous, reviewer for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Chi, W., Dong, Y., Liu, B. et al. A photocatalytic redox cycle over a polyimide catalyst drives efficient solar-to-H2O2 conversion. Nat Commun 15, 5316 (2024). https://doi.org/10.1038/s41467-024-49663-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-49663-6
- Springer Nature Limited