

Abstract
Histone H2AX plays a key role in DNA damage signalling in the surrounding regions of DNA double-strand breaks (DSBs). In response to DNA damage, H2AX becomes phosphorylated on serine residue 139 (known as γH2AX), resulting in the recruitment of the DNA repair effectors 53BP1 and BRCA1. Here, by studying resistance to poly(ADP-ribose) polymerase (PARP) inhibitors in BRCA1/2-deficient mammary tumours, we identify a function for γH2AX in orchestrating drug-induced replication fork degradation. Mechanistically, γH2AX-driven replication fork degradation is elicited by suppressing CtIP-mediated fork protection. As a result, H2AX loss restores replication fork stability and increases chemoresistance in BRCA1/2-deficient tumour cells without restoring homology-directed DNA repair, as highlighted by the lack of DNA damage-induced RAD51 foci. Furthermore, in the attempt to discover acquired genetic vulnerabilities, we find that ATM but not ATR inhibition overcomes PARP inhibitor (PARPi) resistance in H2AX-deficient tumours by interfering with CtIP-mediated fork protection. In summary, our results demonstrate a role for H2AX in replication fork biology in BRCA-deficient tumours and establish a function of H2AX separable from its classical role in DNA damage signalling and DSB repair.
Similar content being viewed by others


Introduction
The advent of poly(ADP-ribose) polymerase inhibitors (PARPi) has revolutionized the treatment landscape for cancers harboring homologous recombination (HR) deficiencies, particularly those resulting from BRCA1 and BRCA2 mutations1. These mutations impair the cell’s ability to repair DNA double-strand breaks (DSBs) via HR2, thus compromising genomic stability. PARPi exploit this vulnerability by inducing synthetic lethality in BRCA-deficient cells3,4, which are unable to repair DNA damage effectively. Despite the clinical successes of PARPi, resistance invariably arises, posing significant challenges to durable therapeutic outcomes. The mechanisms underlying this resistance, which range from mutations restoring BRCA activity or HR to alterations in drug efflux mechanisms5, continue to be a focal point of research. Recent studies have challenged the prevailing model of PARPi action which posits that lethality arises primarily through “trapping” PARP enzymes onto DNA6,7,8. Indeed, work by Petropoulos et al. 9. suggests that the disruption of transcription-replication conflicts may play a pivotal role, indicating that our understanding of PARPi mechanisms is still evolving.
Functional CRISPR-Cas9 screens have emerged as powerful tools to identify mechanisms of drug resistance, including resistance to PARP inhibitors. By enabling the systematic knockout of genes across the genome in a high-throughput manner, these screens allow to uncover genes that, when lost, confer survival advantages to cancer cells under treatment pressure. This approach has proven instrumental in delineating the complex networks of genetic interactions and pathways that contribute to the emergence of resistance, providing valuable insights that can guide the development of next-generation therapies and combination treatments to circumvent resistance. The study of resistance mechanisms requires robust in vivo models that can recapitulate tumour dynamics and drug responses accurately. In this context, genetically engineered mouse models (GEMMs) of BRCA1- and BRCA2-associated breast cancer have proven invaluable advantages5,10,11. These models, including those developed with large intragenic deletions in Brca1 or Brca2 that preclude reactivation of protein function after secondary mutations, allow for the detailed investigation of PARPi resistance mechanisms that are BRCA-independent. For instance, these models have elucidated distinct patterns of resistance arising in BRCA1- versus BRCA2-deficient tumours12, such as the loss of 53BP1-RIF1-REV7-CST-SHLD complex driving HR restoration in BRCA1-deficient tumours13,14,15,16,17, and the loss of PARG leading to restoration of PARP signaling in BRCA2-deficient tumour18. Moreover, we and others have previously identified the restoration of replication fork protection as a key mechanism to protect genome stability and acquire chemotherapy resistance in BRCA-deficient tumour cells19,20. However, the precise genetic alterations in chemoresistant tumours underpinning restored fork protection have not been identified.
In this work, we carry out multiple functional CRISPR-Cas9 genetic screens and identify a critical role for the histone H2AX in mediating chemotherapy response. Importantly, we identify a role of H2AX in modulating replication fork stability which appears to contribute to the sensitivity of BRCA-deficient tumours to PARPi. By examining these dynamics, we find that γH2AX-dependent replication fork degradation is triggered by the inhibition of CtIP-mediated fork protection. Hence, we uncover additional molecular insights into both the mechanism of action of PARPi and the molecular basis of therapy resistance. This work promises to not only enhance our understanding of DNA damage response in cancer therapy but also to guide the development of more effective strategies to overcome resistance in BRCA-mutated cancers.
Results
Functional genetic screens and complementary in vivo analysis using BRCA1/2 models identify H2AX as a critical mediator of chemotherapy response
To identify new genes involved in the cellular response to PARPi in BRCA2;p53-deficient tumours, we performed a genome-wide CRISPR-Cas9 screen in our KB2P3.4 (K14cre;Trp53-/-;Brca2-/-) mouse mammary tumour cell line21. Cells were transduced with the mouse sgRNA GeCKO_V2 library targeting 20,628 genes22, and then treated with a nearly lethal dose of 200 nM of the AZD2461 PARPi for 3 weeks (Fig. 1a). At the end of the treatment, extracted genomic DNA from surviving cells was subjected to NGS and analyzed with the MAGeCK MLE algorithm23 (Fig. 1a). To increase the confidence of our screening data, we crossed the hits of this screen with the results obtained from four other genetic screens: two screens for PARPi resistance carried with a targeted DDR shRNA library in KB2P3.4 cells treated with AZD2461 or olaparib18; a genome-wide CRISPR-Cas9 screen performed in human RPE1-hTERT TP53-/-;BRCA1-/- cells selected with olaparib16; a genome-wide CRISPR-Cas9 screen performed in KB2P1.21 cells (K14cre;Trp53-/-;Brca2-/-) treated with a lethal dose of cisplatin (Fig. 1b). This data processing allowed us to identify general chemoresistance mechanisms independent of: (1) the specific PARPi, (2) BRCA1 or BRCA2 deficiency, (3) the type of screen (shRNA- or CRISPR-based), (4) the species (mouse or human), or (5) the anti-cancer agent used (PARPi or cisplatin). Our analysis revealed that sgRNA/shRNA against the histone H2afx/H2AFX were greatly enriched in all the analyzed screens and scored among the top identified hits (Fig. 1b, c). Conversely, sgRNA against 53BP1, a known key modulator of chemoresistance in BRCA1-deficient tumours, were enriched specifically in the RPE1-hTERT TP53-/-;BRCA1-/- screen but not in the screens performed in BRCA2-deficient cells (Fig. 1c). These results suggest that H2AX loss is associated with PARPi resistance through a mechanism different than 53BP1 inactivation which only occurs in BRCA1- but not in BRCA2-deficient cells24.

a Design of the genome-wide CRISPR-Cas9 genetic screen carried out in KB2P3.4 cells treated with the poly(ADP-ribose) polymerase (PARP) inhibitor AZD2461. b Venn diagram showing the overlap of potential gene candidates identified in each individual screen. c Violin plots showing the Z-Normalized Beta-Enrichment Score from 5 different genetic screens for chemoresistance carried out in different BRCA1/2-deficient cell lines. Data were all analyzed using the MAGeCK MLE algorithm to allow for cross comparison. d Design of an in vivo pipeline to query for genetic alterations and Homologous Recombination (HR) restoration in PARPi-resistant mammary tumours from KB1/2P mice. e Pie charts showing 45 and 34 PARPi-resistant mammary tumours from KB1P and KB2P mice, respectively. Tumours are grouped based on RAD51 IRIF+, Parg mutational status and H2afx gene expression (see Methods text).
Considering the key role for γH2AX in DNA damage signalling25,26, it was surprising that H2afx loss would promote PARPi resistance. For this reason, we tested whether reduced H2afx gene expression would also occur in two cohorts of PARPi-resistant mammary tumours from KB1P and KB2P mice (K14cre;Trp53F/F;Brca1F/F and K14cre;Trp53F/F;Brca2F/F, respectively), that acquired resistance in vivo following repeated PARPi cycles12,18 (Fig. 1d). In these tumours, we also determined RAD51 ionizing radiation-induced foci (IRIF) to distinguish Homologous Recombination (HR)-dependent from -independent mechanisms of PARPi resistance. We previously reported that none of the PARPi-resistant BRCA2-deficient tumours restored RAD51 IRIF12, whereas 64% (29/45) of the BRCA1-deficient tumours became RAD51 IRIF+ (Fig. 1e). Conversely, in the KB2P cohort, a large fraction of the tumours analyzed presented a major genomic structural change in the Parg locus, leading to the loss of Parg gene product (14/34) (Fig. 1e). Strikingly, we noticed that in both BRCA1/2-deficient tumour cohorts, H2afx gene expression was significantly reduced in a large fraction of the tumours (11/34 in the KB2P tumours and 12/45 in the KB1P tumours) with only a small overlap with Parg loss or HR restoration (Fig. 1e), which can be explained by the intra-tumoural heterogeneity of the resistance mechanisms11,27. Therefore, we concluded that H2afx downregulation frequently occurs in PARPi-resistant mammary tumours and that the underlying mechanism is likely to be different from previously reported mechanisms of resistance.
To validate our finding, we then depleted H2afx by CRISPR-Cas9 in KB2P3.4 (Trp53-/-;Brca2-/-) and KB1P-G3 (Trp53-/-;Brca1-/-) cells and studied PARPi response in vitro (Supplementary Fig. 1a-d). Consistent with our previous genetic analysis, H2afx depletion rescued the cellular sensitivity to the PARP inhibitors olaparib, AZD2461, and talazoparib in both KB2P3.4 and KB1P-G3 cells (Fig. 2a; Supplementary Fig. 1e, f). We also examined the impact of H2afx status on tumour growth in vivo27 (Fig. 2b). For this purpose, we transduced our BRCA2-deficient organoid line ORG-KB2P26N.1 with a non-targeting (NT) or a H2afx gRNA and verified the frameshift mutation rate by TIDE analysis (Fig. 2c). Next, we injected the modified organoids orthotopically into the inguinal mammary fat pad of female nude mice and waited until the tumour reached a palpable size (50–100 mm3). Mice were then randomized and 100 mg olaparib per kg or vehicle was administered daily by intraperitoneal injection. Strikingly, mice with H2afx-depleted tumours responded worse to the olaparib treatment than the H2afx wildtype counterparts and had to be sacrificed earlier (Fig. 2d; Supplementary Fig. 2e). We also generated stable H2AFX-/- clones in human RPE1-hTERT TP53-/-;BRCA1-/- cells and all showed full resistance to olaparib (Supplementary Fig. 2a). Moreover, shRNA-mediated depletion of H2AX caused olaparib resistance in the triple-negative breast cancer cell line MDA-MB-436, that carries the BRCA1 c.5277+1 G > A mutation and shows complete loss of BRCA1 protein product28 (Supplementary Fig. 2b, c). The H2AX-dependent PARPi sensitivity fully depended on Serine 139 phosphorylation, as we could fully complement PARPi response in KB1P-G3 cells with a wild-type H2AX but not with the H2AXS139A mutant (Fig. 2e; Supplementary Fig. 2d). Moreover, we also noticed that H2AX-depleted cells had reduced cellular sensitivity to the crosslinking agent cisplatin but not to ionizing radiation (IR) (Supplementary Fig. 1g, h), indicating that the mechanism of resistance is likely not associated with general DNA damage signalling.

a Clonogenic survival assay of KB2P3.4-derived cells treated, or mock treated, with the indicated concentrations of the PARPi olaparib and AZD2461 for 12 days. Plotted values express the mean ± SD of clonogenic survival (n = 3 independent experiments). P-values were calculated with two-way Anova test and adjusted for multiple comparisons. Source data are provided as a Source Data file. b Schematic design of the in vivo experiment. c Allelic modification rate of H2AX-deficient ORG-KB2P26N.1 organoids evaluated by TIDE analysis prior to transplantation. d Kaplan-Meier curves showing the survival of vehicle- or olaparib-treated mice bearing H2AX-proficient or deficient KB2P tumours. Each group contained 5 animals. P-value was calculated with the Mantel-Cox test. Source data are provided as a Source Data file. e Clonogenic survival assay of KB1P-G3-derived cells expressing the indicated H2AX variants and treated as in (a). Plotted values express the mean ± SD of clonogenic survival (n = 3 independent experiments). P-values were calculated with two-way Anova test and adjusted for multiple comparisons. Source data are provided as a Source Data file. f Therapy history of the CHIOVAR59 patient. On the right, H2AX IHC was performed on two tissue biopsies, primary tumour at diagnosis and colon metastasis after therapy resistance. Two areas of the slide are shown for each biopsy. Scale bar is 50 µm.
Based on these results we tested H2AX expression by immunohistochemistry using paired biopsies that we obtained from a high-grade serous ovarian carcinoma patient carrying a pathogenic somatic mutation in BRCA2 (c.3376 G > T/ p.Glu1126Ter) (Fig. 2f). The CHIOVAR59 patient received neo- and adjuvant platinum-based therapy. At first relapse, she was treated with olaparib combined with anti-VEGF and anti-PD-L1 (Fig. 2f), resulting in complete response. Fifteen months later, the patient had a second relapse with a metastasized peritoneal tumour resistant to all subsequent therapies. Compared to the sample taken before chemotherapy, we found a clear H2AX reduction in tumour cells of the drug-resistant colon metastasis (Fig. 2f). Although anecdotal, this case indicates that H2AX loss is a clinically relevant mechanism associated with chemotherapy resistance in BRCA-deficient tumours.
H2AX loss restores replication fork protection in BRCA-deficient tumour cells
We then dissected the molecular mechanism how H2AX loss promotes chemotherapy resistance. H2afx-depleted cells had reduced levels of micronuclei after olaparib treatment (Supplementary Fig. 3a), indicating reduced levels of genomic instability. Reduction in micronuclei formation is usually associated with the restoration of conservative HR repair, particularly in BRCA1-deficient cells29. Given the role of γH2AX in stabilizing 53BP1 near DNA lesions30,31, we tested whether H2AX loss increases PARPi resistance by preventing 53BP1 complex formation and restoring competent HR. Consistent with the function of γH2AX in recruiting 53BP1 near DNA damage sites, we observed that 53BP1 IRIF were significantly reduced in KB1P-G3 H2afx-depleted cells (Fig. 3a). Nevertheless, in contrast with 53BP1-deficient cells, H2afx depletion failed to restore RAD51 IRIF (Fig. 3b). This stark difference in RAD51 IRIF formation also reflected a different cellular response to olaparib, which was stronger in 53BP1-depleted cells as shown by their increased cell survival and the increased IC50 values after olaparib treatment (Supplementary Fig. 3b, c). However, to our surprise, we noticed that H2AX-deficient cells had increase cellular resistance to the replication fork stalling agent hydroxyurea (HU), which instead was not observed in 53BP1-depleted cells (Supplementary Fig. 3d). These results confirm that H2AX and 53BP1 control drug resistance via two genetically separable mechanisms (Supplementary Fig. 3e). We therefore inferred that H2AX loss may increase chemotherapy resistance via another proposed mechanism, that is the restoration of replication fork protection20. Consistent with this hypothesis, single molecule analysis of replication tracts showed that, in marked contrast with BRCA1/2-deficient cells where stalled forks undergo extensive nucleolytic degradation32,33,34,35,36,37, H2AX loss restored fork integrity in both KB1P-G3 and KB2P3.4 cells in response to hydroxyurea and camptothecin (Fig. 3c, d; Supplementary Fig. 4a). Importantly, a similar phenotype was not observed in 53BP1-deficient cells, where stalled forks still undergo extensive nucleolytic degradation (Supplementary Fig. 4b). Importantly, H2AX loss did not alter normal fork elongation rates (Supplementary Fig. 3f) which we recently observed in Mdc1-deleted cells38. H2AX loss also restored replication fork stability in the BRCA1-deficient MDA-MB-436 cell line (Supplementary Fig. 4c, d). Consistent with the critical role of H2AX Serine 139 phosphorylation for PARPi resistance (Fig. 2c), fork degradation was entirely abolished in KB1P-G3 H2AX-deficient cells complemented with the H2axS139A variant (Fig. 3d), indicating that H2AX phosphorylation on Serine 139 plays a critical role at stressed replication forks in BRCA-deficient cells.

a 53BP1 Ionizing Radiation-Induced Foci (IRIF) analysis in KB1P-G3 cells 4 h after 10 Gy exposure. Plotted values show the median of 53BP1 IRIF/cell from at least 600 cells (n = 2 independent experiments). P-values were calculated with one-way Anova test. Source data are provided as a Source Data file. b RAD51 IRIF analysis in KB1P-G3 cells treated as in (a). The presented data are the mean ± SD (n = 2 and n = 3 independent experiments for 53bp1-depleted and the other cell lines, respectively). Source data are provided as a Source Data file. c DNA fiber analysis in KB2P3.4 cells treated according to the depicted scheme. Hydroxyurea (HU) was used at 8 mM for 6 h. Plotted values show the median of individual IdU/CldU ratios from at least 200 fibers (n = 3 independent experiments). P-values were calculated with one-way Anova test and adjusted for multiple comparisons. Source data are provided as a Source Data file. d DNA fiber analysis in the indicated KB1P-G3 cells treated as in (c). Plotted values show the median of individual IdU/CldU ratios from at least 300 fibers (n = 3 independent experiments). P-values were calculated with two-way Anova test and adjusted for multiple comparisons. Source data are provided as a Source Data file. e Electron Microscopy (EM) analysis of reversed fork intermediates following HU treatment as in (c). The electron micrograph represents a reversed replication fork. P parental strand, D daughter strand, R regressed arm. The presented data are the mean ± SD (n = 3 independent experiments). P-values were calculated with the unpaired two-tailed Student’s t-test. Source data are provided as a Source Data file. f EM analysis showing the median number of forks with detectable ssDNA at the junction in cells treated as in (e). P-value was calculated with the unpaired two-tailed Student’s t-test. Source data are provided as a Source Data file. g CtIP SIRF in KB2P3.4-derived cells treated as in (c). Plotted values show the median of CtIP SIRF foci/cell from at least 140 cells (n = 3 independent experiments). P-values were calculated with one-way Anova test. Source data are provided as a Source Data file. h KB2P3.4 and KB1P-G3-derived cells were pretreated for 24 h with the indicated CtIP peptides prior to analog in vivo labeling according to the depicted scheme. HU was used at 8 mM and the CtIP peptides were used at 10 µM. Plotted values show the median of individual IdU/CldU ratios from at least 250 fibers (n = 2 independent experiments). P-values were calculated with one-way Anova test and adjusted for multiple comparisons. Source data are provided as a Source Data file.
The reduced fork resection observed in H2AX-deficient cells may be explained by a defect in fork reversal34,35,36,37,38, or by an increased protection of the regressed arms19,20. To understand which of the fork transaction steps is controlled by H2AX, we directly looked at fork architecture by electron microscopy (EM) analysis39. Notably, we reproduced the lack of reversed fork intermediates in BRCA2-deficient cells treated with HU (Fig. 3e), which has been previously attributed to the unrestrained nuclease activity at stalled forks34,35,36,37. In contrast, KB2P3.4 H2afx-depleted cells showed a significantly higher number of reversed fork intermediates after HU treatment (Fig. 3e), indicating that H2AX is not involved in the initial reversal step but rather in the protection of the newly formed regressed intermediates. Our EM analysis also revealed fewer replication intermediates with detectable single stranded DNA (ssDNA) at the fork junction in KB2P3.4 H2afx-deleted cells (Fig. 3f), likely indicating a less pronounced degradation from the reversed fork end. Suppression of ssDNA gaps, another recently proposed mechanism of PARPi tolerance40,41,42, was also assessed using the DNA fiber assay followed by in vitro S1 nuclease digestion43. This approach confirmed that the IdU/CldU ratio in H2AX-deficient cells is not altered (Supplementary Fig. 4e), corroborating our EM findings. Notably, these short ssDNA patches observed at the fork junction differ from the recently described post replicative gaps, as these intermediates were not detected far away from the fork, which would be indicative of aberrant Okazaki fragment processing in H2AX-deficient cells.
MRE11 has been proposed to be the main nuclease active at stalled forks, particularly in BRCA2-deficient cells32,33,34,35,36,37,44. However, we did not detect any major reduction in the MRE11 recruitment on nascent DNA after replication stress in KB2P3.4 H2afx-deleted cells by in situ analysis of protein interactions at DNA replication forks (SIRF)45 (Supplementary Fig. 4g). Given these results, we tested whether H2AX loss could increase replication fork protection without affecting nuclease dynamics. During the G0/G1 phase of the cell cycle, H2AX has been shown to inhibit the activity of CtIP46, a protein with crucial roles in DNA end resection but also with a key role in replication fork protection47,48. Therefore, we examined whether H2AX may similarly counteract CtIP-mediated fork protection. In agreement with previous findings46, we observed a stronger association of CtIP with stalled forks in KB2P3.4 and MDA-MB-436 H2AX-depleted cells (Fig. 3g; Supplementary Fig. 4f). To verify whether the increased association of CtIP at stalled forks coincides with increased fork protection, we monitored fork stability in BRCA-deficient H2afx-deleted cells depleted for CtIP. Since stable CtIP depletion significantly affects the proliferation of BRCA1/2-deficient cells48, we transiently treated KB2P3.4 and KB1P-G3 H2AFX-depleted cells with a hydrocarbon-stapled peptide (SP) that specifically interferes with CtIP functions by targeting its tetramerization49. Strikingly, pretreatment with the CtIP-SP, but not with a linear peptide (LP), restored fork degradation in both KB2P3.4 and KB1P-G3 H2AX-depleted cells (Fig. 3h). We infer from these data that the effect of H2AX loss in PARPi resistance in BRCA-deficient cells is mediated by the increased association of CtIP with stalled forks. Consistently, treatment with a partial inhibitory concentration of the CtIP-SP of 5 µM resensitised KB1P-G3 H2AX-depleted cells to PARPi (Supplementary Fig. 4h).
Sensitivity to ATM inhibitors is an acquired vulnerability of H2AX-deficient tumours
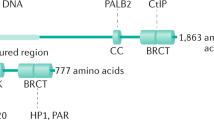
The DNA Damage Response (DDR) kinases ATM and ATR are known to phosphorylate CtIP at several conserved S/T-Q residues (Fig. 4a)50,51,52. ATM-, but not ATR-, mediated phosphorylation stimulates CtIP endonuclease activity in vitro51, a function which has latter been shown to dampen nucleolytic degradation of stalled forks48. We asked whether CtIP phosphorylation by DDR kinases is required for replication fork protection. To test this, we generated doxycycline-inducible U-2OS lines that express a siRNA-resistant CtIPWT, a CtIP8A variant where all its S/T-Q sites were mutated to alanine, a CtIPT859A variant with a point mutation only in the ATR phosphorylation site, and a CtIPL27E variant entirely lacking its tetramerization capacity51 (Supplementary Fig. 5a). Strikingly, CtIPWT expression complemented the lack of fork protection elicited by endogenous CtIP depletion, while the CtIP8A variant failed to complement the loss of endogenous CtIP in a similar fashion to the catalytically inactive CtIPL27E mutant (Fig. 4b). Interestingly, the CtIPT859A mutant is proficient in fork protection (Fig. 4b), consistent with previous results showing intact nuclease activity of CtIP-T859A mutant. Next, we asked whether these results could be recapitulated by using the clinically relevant ATMi AZD0156 and the ATRi AZD6738. Remarkably, the ATMi AZD0156, but not the ATRi AZD6738, robustly restored fork degradation in BRCA- H2AX-deficient cells (Fig. 4c), indicating that the ATR signalling branch is dispensable for fork protection in these tumours. We also observed loss of CtIP localization at stalled forks by SIRF upon AZD0156 treatment (Supplementary Fig. 5b), suggesting that ATM controls CtIP function at stalled forks by promoting its catalytic activity and its proper localization. Next, we investigated the effects of the same DDRi on PARPi resistance. Consistent with the above results, and in agreement with the idea that fork stabilization is the underlying mechanism of resistance in BRCA- H2AX-deficient tumours, partially inhibitory ATMi concentrations (AZD0156, 5-20 nM) entirely rewired PARPi response in BRCA- H2AX-deficient cells (Fig. 4d; Supplementary Fig. 5c). In parallel, we also tested the same partially inhibitory concentrations of the ATRi AZD6738, which failed to restore PARPi sensitivity in these cells (Fig. 4d). Interestingly, we have found that H2AFX scored as one of the strongest hits in loss-of-function CRISPR-Cas9 screens performed in distinct human cancer cell lines treated with the ATMi AZD0156 (Supplementary Fig. 5d), as also previously found. Based on these results, we selected the hypothesis that the chemotherapy resensitization by the ATMi could be particularly important in tumours which have lost H2AX. In line with this idea, we found that AZD0156 failed to restore PARPi sensitivity in the chemoresistant KB2P1.21 Mdc1-/- cells (Supplementary Fig. 5e), where PARPi resistance does not arise from restored replication fork protection (Supplementary Fig. 5f). These results strongly indicate that ATMi-induced resensitisation to chemotherapy is not due to the general inhibition of DNA repair transactions or due to the increase of replication stress levels, but rather on the specific deregulation of CtIP function at stressed forks. Finally, we explored whether ATMi restores PARPi sensitivity also in H2AX-deficient tumours in vivo (Fig. 4e). To address this question, we performed a similar experiment as shown in Fig. 2e and treated mice with the brain-penetrant ATMi AZD139053 (5 mg/kg, every 2nd day) for the whole duration of the olaparib treatment (Fig. 4e). Again, we observed that in the animals with the H2afx-depleted organoids, tumours did not respond to the olaparib treatment and grew over the therapy course compared to the tumours in the control group (Fig. 4f). However, consistent with our in vitro data, the AZD1390 combination restored PARPi response and impaired tumour growth throughout the duration of the experiment (Fig. 4f). Together, these data indicate that ATM inhibitors in combination with PARPi can be effectively used to overcome drug resistance in H2AX-deficient tumours and control tumour growth.

a Human CtIP protein map with highlighted the 8 ATM and ATR phosphorylation sites. b DNA fiber analysis in U-2OS-derived cells transfected with CtIP siRNA and treated according to the depicted scheme. 24 h before the experiment, CtIPWT, CtIPL27E, CtIP8A or CtIPT859A expression was induced by doxycycline treatment. Plotted values show the median of individual IdU/CldU ratios from at least 300 fibers (n = 2 independent experiments). P-values were calculated with one-way Anova test and adjusted for multiple comparisons. Source data are provided as a Source Data file. c DNA fiber analysis in KB1P-G3 and KB2P3.4-derived cells treated according to the depicted scheme. HU was used at 8 mM, AZD0156 and AZD6738 were used at 10 µM. Plotted values show the median of individual IdU/CldU ratios from at least 170 fibers (n = 3 independent experiments). P-values were calculated with one-way Anova test and adjusted for multiple comparisons. Source data are provided as a Source Data file. d Clonogenic survival assay of KB1P-G3-derived cells treated, or mock treated, with the indicated concentrations of olaparib, AZD0156, and AZD6738 for 12 days. Plotted values express the mean ± SD of clonogenic survival (n = 4 independent experiments). P-values were calculated with two-way Anova test and adjusted for multiple comparisons. Source data are provided as a Source Data file. e Schematic design of the in vivo experiment with the ATMi AZD1390. f Plotted values express the median size (mm3) of individual tumours from five animals transplanted with the indicated organoid lines and subjected to the indicated treatment for 28 consecutive days after tumour formation. P-values were calculated with two-way Anova test corrected with the Bartlett’s test. Source data are provided as a Source Data file.
Discussion
In this manuscript, we have shown a role for the histone H2AX in replication fork biology in BRCA1/2-deficient tumours. Previous work in BRCA-proficient cells reported a function for the histone H2AX in maintaining the stability of reversed replication forks54. However, we would like to emphasize that Schmid et al. 54. have analyzed the contribution of the H2AX-ATM checkpoint axis on fork slowing/reversal during unstressed replication, while we have focused on the role of H2AX-ATM in response to replication stress. This distinction is critical, as it aligns with emerging evidence suggesting that fork transactions may exhibit distinct genetic dependencies during unstressed replication compared to when exposed to genotoxic agents38,55. Thanks to the molecular characterization of the mechanisms behind the stabilization of stressed forks, we also identified an acquired genetic vulnerability of H2AX-deficient tumours, which is the inhibition of the ATM kinase. This discovery is of great interest and may have direct implications for the treatment of BRCA-mutated cancers at the metastatic stage, such as for the CHIOVAR59 case reported here. Future work on a larger scale will establish the clinical relevance of H2AX status for chemotherapy resistance and will help redirecting current therapeutic regimens towards more rationalized combination treatments.
Methods
Our research complies with all the relevant ethical regulations defined by the Swiss Federal Office of Public Health, the Ethics Committee of the Canton of Geneva (CCER, Switzerland), the Animal Ethics Committees of The Netherlands Cancer Institute (Amsterdam, The Netherlands), and the Canton of Bern (Switzerland). All the experiments were performed in full compliance with national laws.
Cell culture conditions
KB2P3.4 (Trp53-/-;Brca2-/-), KB2P1.21 (Trp53-/-;Brca2-/-), and KB1P-G3 (Trp53-/-;Brca1-/-) cells and all their derivative cell lines were grown at 37 °C in 3% O2 and cultured in Dulbecco’s modified Eagle medium Nutrient mixture F-12 (DMEM/F12; Gibco) supplemented with 10% Fetal Bovine Serum (FBS), pen/strep solution (50 U/ml), 5 ng/ml cholera toxin (Sigma Aldrich), insulin (5 µg/ml, Sigma Aldrich) and 5 ng/ml murine Epidermal Growth Factor (mEGF, Sigma Aldrich). ORG-KB2P17S.1 organoids were grown at 37 °C in normal oxygen embedded in Cultrex Reduced Growth Factor Basement Membrane Extract Type 2 (BME, Trevigen), seeded on 24-well suspension plates (Greiner Bio-One) and cultured in complete mouse mammary gland organoid medium: AsDMEM/F12 supplemented with 1 M HEPES (Sigma), GlutaMAX (Invitrogen), pen/strep (Gibco), B27 (Gibco), 125µM N-acetyl-L-cysteine (Sigma), 50 ng/ml murine epidermal growth factor (mEGF, Invitrogen). Human RPE1-hTERT TP53-/-, RPE1-hTERT TP53-/-;BRCA1-/-, and the RPE1-hTERT TP53-/-;BRCA1-/-;H2AFX-/- clones were grown at 3% O2 in Dulbecco’s modified Eagle medium Nutrient mixture F-12 (DMEM/F12; Gibco) and penicillin/streptomycin solution (100 U/ml). MDA-MB-436 cells were grown in RPMI supplemented with 10% FBS, 1% non-essential aminoacids. SW62O cells were grown in L-15 medium (Leibovitz), 10% FBS, 1X glutamax. HT29 cells were grown in DMEM, 10% FBS, 1% non-essential aminoacids, 1% L-glutamine. BT549 cells were grown in RPMI-1640, 10% FBS, 0.023 U/ml insulin. U-2OS-TeTON-eGFP-CtIPWT, U-2OS-TeTON-eGFP-CtIPL27E, U-2OS-TeTON-eGFP-CtIP8A, U-2OS-TeTON-eGFP-CtIPT859A were grown at 37 °C in DMEM supplemented with 10% FCS, 1% pen/strep, 5 µg/ml Blasticidin, 200 µg/ml Zeocin. CtIP expression was induced 24 h before the experiment with 1 µg/ml of doxycycline. Mission shRNA H2AX #1 (NM_002105), and H2AX #2 (NM_005657) were purchased from Sigma. All cell lines were authenticated by Brca1/2-specific PCR-based genotyping (mouse)13,21 and they were regularly tested for mycoplasma contamination (Mycoalert, Lonza).
Drugs and reagents
The following chemical reagents were used throughout the study: AZD2461 (kindly provided by AstraZeneca), olaparib (kindly provided by AstraZeneca and Syncom (Groningen, The Netherlands)), talazoparib (Selleckchem; #S7048), cisplatin (Teva; #7680479980428), hydroxyurea (Sigma; #H8627), AZD0156 (kindly provided by AstraZeneca), AZD6738 (Selleckchem; #S7693), AZD1390 (kindly provided by AstraZeneca), Mirin (Sigma; #M9948), camptothecin (Selleckchem; #S1288), 5-Iodo-2’-deoxyuridine (IdU) (Sigma; #I7125), 5-Chloro-2’-deoxyuridine (CldU) (Sigma; #C6891), Ethynyl-2’-deoxyuridine (EdU) (Sigma; #A10044). LP and SP (Bachem; #4143690 and #4111111, respectively).
Antibodies
The following primary antibodies were used: mouse anti-HA (clone 16B12, 1:1,000, #901533, Biolegend), mouse anti-γ-Tubulin (clone 6H3.1, 1:1,000, #5886, Cell Signalling), rabbit anti-GFP (1:1,000, #2555, Cell Signalling), rabbit anti-53BP1 (1:800, #A300-272A, Bethyl laboratories), rabbit anti-RAD51 (1:1,000, #70-012, Bioacademia), rat anti-BrdU (CldU) (clone [BU1/75 (ICR1)], 1:250, #ab6326, Abcam), mouse anti-BrdU (IdU) (1:40, #347580, BD Biosciences), rabbit anti-CtIP (1:500, #A300-488A, Bethyl Laboratories), mouse anti-Biotin (1:200, #200-002-211, Jackson Immuno Research), rabbit anti-MRE11 (1:500, a kind gift from Arnab Ray Chaudhuri19).
Genome-wide CRISPR/Cas9 screens
The PARPi resistance screens were performed in the KB2P3.4 tumour cell line, which was previously established from a KB2P tumour21. Mouse GeCKO_V2 library, pool B (62,804 gRNAs targeting 20,628 genes (3 gRNAs/gene) including 1,000 non-targeting gRNAs), was stably introduced into the cells by lentiviral transduction at multiplicity of infection (MOI) of 1.5. Mouse GeCKO_V2 CRISPR knockout pooled library was a kind gift from Feng Zhang22. 6 independent transductions were carried out to obtain mutagenized cells for biological replicates of the PARPi resistance screen. To perform the genetic screen with a 100 x library coverage, 6 × 106 mutagenized KB2P3.4 cells in each replicate were plated in 10 cm flasks, at low density (30,000 cells per flask) and grown in medium containing 200 nM AZD2461 for 3 weeks. The medium with the PARPi was refreshed twice a week. Cells were harvested before and after PARPi treatment for genomic DNA isolation. Subsequently, gRNA sequences were amplified from genomic DNA by two rounds of PCR amplification. Resulting PCR products were purified using MinElute PCR Purification Kit (Qiagen) and submitted for Illumina sequencing. Quality control was performed using R software (R Core Team, 2022) and package edgeR. Sequence alignment and enrichment analysis (day 0 vs PARPi-treated population) was carried out using the R package MAGeCKFlute56. Dataset of MAGeCK MLE analysis results of the CRISPR/Cas9 screen on RPE1-hTERT cells was extracted from the Supplementary Table 1 of Nordermeer et al. 16. For the CRISPR-Cas9 screens with the ATMi AZD0156, SW620, HT29, and BT549 cells were infected with lentiviral particles containing the whole-genome sgRNA library (Horizon Discovery), subjected to puromycin selection, and passaged to ensure loss of affected protein products. Puromycin-resistant cells were exposed to 10 nM ATMi (AZD0156) for 21 days (SW620, HT29) or 35 days (BT549), and remaining cell pools were isolated. Genomic DNA was extracted from these (QIAamp DNA Blood Maxi kit (Qiagen #51194)) and from parallel cell cultures treated in the absence of AZD0156, and DNA libraries were prepared and sequenced using an Illumina NextSeq next generation sequencing platform. Analysis of NGS data sets e.g. sgRNA abundance was achieved using Horizon Discovery’s data processing scripts, based on published analysis tool MAGeCK.
DDR shRNA-based genetic screens
PARPi resistance shRNA screens were previously described18. Briefly, a shRNA library targeting a DNA Damage Response (DDR) gene set was built based on a gene list described before and the NCBI search (terms: “DNA repair”, “DNA damage response”, “DNA replication”, “telomere-associated genes”)14. The shRNA library was stably introduced into the tumour cell line KB2P3.4, which was subsequently selected with the PARPi AZD2461 or olaparib for 3 weeks. Genomic DNA was purified before and after treatment, amplified and sequenced as described above. Sequence alignment and analysis were performed using the MAGeCK software, MAGeCK-VISPR Maximum Likelihood Estimation (MLE) module, and the R package MAGeCKFlute56.
Lentiviral transductions
Lentiviral stocks were generated by transient transfection of HEK293FT cells. On day 0, 6 × 106 HEK293FT cells were seeded in 150 cm cell culture dishes and on the next day transiently transfected with lentiviral packaging plasmids and the pLentiCRISPRv2 vector containing the respective H2afx-targeting gRNA or a non-targeting gRNA using 2 x HBS (280 nM NaCl, 100 mM HEPES, 1.5 mM Na2HPO4, pH 7.22), 2.5 M CaCl2 and 0.1 x TE buffer (10 mM Tris pH 8.0, 1 mM EDTA pH 8.0, diluted 1:10 with dH2O). After 30 h, virus-containing supernatant was concentrated by ultracentrifugation at 20,000 rcf for 2 h in a SW40 rotor and the virus pellet was finally resuspended in 100 μl PBS. The virus titer was determined using a qPCR Lentivirus Titration Kit (#LV900, Applied Biological Materials). For lentiviral transduction, 150,000 target cells were seeded in 6-well plates. 24 h later, virus at the MOI of 50 was applied with 8 μg/ml Polybrene (Merck Millipore). Virus-containing medium was replaced with medium containing puromycin (3.5 μg/ml, Gibco) 24 h later. Puromycin selection was performed for 3 days; subsequently cells were expanded and frozen down at early passage. Tumour-derived organoids were transduced according to a previously established protocol57. The target sites modifications of the polyclonal cell pools were analyzed by TIDE analysis.
Gene editing
For CRISPR/Cas9-mediated genome editing, KB2P3.4 and KB2P1.21 cells or KB2P17S.1 tumour-derived organoids were transduced with the pLentiCRISPRv2 vector encoding non-targeting gRNA, H2afx-targeting gRNA1 or H2afx-targeting gRNA2. The cells were then grown under puromycin (3 μg/ml) selection for 5 days. All constructs were verified with Sanger sequencing. For CRISPR/Cas9-mediated targeting of H2afx and Mdc1 genes in KB1P-G3, KB2P3.4, and KB2P1.21 cells, non-targeting gRNA, H2afx, and Mdc1-targeting gRNAs were cloned into the pX330 vector (Addgene; #42230). Sanger sequencing-verified pX330 plasmids containing the correct sequences of gRNAs were transfected in cells using the TransIT-LT1 transfection reagent (#MIR 6604, Mirus) according to the manufacturer’s protocol. The cells were then grown under Puromycin (3 μg/ml) selection for 5 days. CRISPR gRNA sequences were chosen from the GeCKO_V2 library. The gRNA sequences were as follows: mH2afx gRNA1: 5’-TCGTACACTATGTCCGGACG-3’; mH2afx gRNA2: 5’-GGCGCCGGCGGTCGGCAAGA-3’; Non-Targeting (NT) gRNA: 5’-TGATTGGGGGTCGTTCGCCA-3’; hH2AFX gRNA1: 5’-GACAACAAGAAGACGCGAATC-3’; mMdc1 gRNA1: 5’-GGTGTGTGGCGAATGGACAA-3’. H2ax reconstitution was performed using the pOZ-N-FH (a kind gift from Dipanjan Chowdury). The H2afx coding sequence from Mus musculus was ordered from Eurofins and cloned into the pOZ-N-FH backbone adding the 1 x HA tag at the N terminus using the in-fusion HD cloning kit (#12141, Takara). Full length wild type H2afx coding sequence was then mutagenized to obtain the desired S139A point mutation.
gDNA isolation, amplification, and TIDE analysis
To assess the modification rate at the gRNA-targeted region of H2afx, cells were pelleted, and genomic DNA was extracted using the QIAmp DNA mini kit (Qiagen) according to manufacturer’s protocol. Target loci were amplified using Phusion High Fidelity Polymerase (ThermoFisher Scientific) using a 3-step protocol: 98 °C for 30”, 35 cycles at 95 °C for 15”, 55 °C for 15” and 72 °C for 30”, 72 °C for 7’. Reaction mix consisted of 10 µl of 2 x Phusion Mastermix (ThermoFisher Scientific), 1 µl of 10 µM forward (5’-CAATCACTGGGCGCGTTC-3’) and reverse (5’-TGGCTCAGCTCTTTCTGTGAG-3’) primers and 100 ng of DNA in 20 µl total volume. PCR products were purified using the QIAquick PCR purification kit (Qiagen) according to manufacturer’s protocol and submitted with corresponding sequencing primers for Sanger sequencing to confirm target modifications using the TIDE algorithm. The sequencing primers used for analysis of the modification rate in mouse cells are the following: mgRNA1 seq. primer: 5’-CAATCACTGGGCGCGTTC-3’; gRNA2 seq. primer: 5’-GAGTACCTCACTGCCGAG-3’; hgRNA1 seq. primer: 5’-GACAACAAGAAGACGCGAATC-3’.
H2afx gene expression analysis in PARPi resistant mammary tumours
Differential H2afx gene expression analysis from normalized gene expression counts was evaluated using DIDS (version 0.10.1)58, selecting a threshold of P < 0.05 for statistical significance12,18,58.
Clonogenic assays
To assess the growth and survival upon treatment with PARPi, Cisplatin, Hydroxyurea, or irradiation, KB2P3.4 and KB1P-G3 cells were seeded in 6-well plates in the following densities: 3000 cells/well (KB2P3.4) and 4,000 cells/well (KB1P-G3). The treatment of cells with DMSO or indicated concentrations of PARPi olaparib or AZD2461 started at the day of plating the cells and lasted for the whole duration of the experiment. The medium with DMSO or PARPi was refreshed twice a week. The control, DMSO-treated plates were fixed 7 days after seeding, the PARPi-treated plates were fixed after 12 days. For the cisplatin treatment, cells were plated 24 h prior addition of cisplatin-containing media. After 24 h, medium was refreshed, and all the plates were fixed after 7 days. Irradiation was carried out in a fractionated manner using the indicated irradiation doses 24, 48 and 72 h following plating of the cells. Plates with non-irradiated cells were fixed 7 days post plating, the irradiated cells were fixed after 10 days. The fixation was done with 4% formalin and the surviving colonies stained with 0.1% crystal violet. The cell survival and growth were analyzed in an automated manner using the ImageJ ColonyArea plugin. For the competition assays, cells were collected before and after the experiment for gDNA isolation and TIDE analysis as described above. For clonogenic assays with the DDRi, the same experimental setup as in the clonogenic assays with PARPi was used. DDRi were added to the medium on the day of plating and the medium containing the drug was refreshed twice/week until the end of the experiment.
In vivo studies
All animal experiments were approved by the Animal Ethics Committee of the Netherlands Cancer Institute (Amsterdam, The Netherlands, license AVD30100202011584) and the Canton of Bern (Switzerland, license BE69/2021) and were performed in full compliance with national laws, which enforce Dir. 2010/63/EU (Directive 2010/63/EU of the European Parliament and of the Council of 22 September 2010 on the protection of animals used for scientific purposes). For tumour organoid transplantation, ORG-KB2P26N.1 organoids were collected, incubated with TripLE at 37 °C for 5’, dissociated into single cells, washed in PBS, resuspended in tumour organoid medium, and mixed in a 1:1 ratio of tumour organoid suspension and BME. Organoid suspension containing a total of 105 cells were injected in the fourth right mammary fat pad of 6–9 week-old female NMRI nude mice. Tumour size was measured by caliper and tumour volume was calculated ((length x width2)/2). Treatment of tumour bearing mice was initiated when tumours reached a detectable size of at least ~ 25–50 mm3, at which point mice were separated into a vehicle-treated group (NT gRNA n = 5, H2afx-targeting gRNA1 n = 5) and an olaparib-treated group (NT gRNA n = 5, H2afx-targeting gRNA n = 5). Olaparib (100 mg/kg) was administered orally for 28 consecutive days. The control tumour-bearing mice were dosed with vehicle following the same the schedule as the PARPi group. Animals were anesthetized with isoflurane, sacrificed with CO2 followed by cervical dislocation when tumours reached a volume of ~ 1000 mm3. Tumour sampling included cryopreserved tumour pieces, fresh frozen tissue, and formalin-fixed material (4% (v/v) formaldehyde in PBS). The maximum body weight loss permitted is 10% of the total animal weight.
H2AX immunohistochemistry on primary tumour samples
Patient CHIOVAR59 was enrolled in the biobank of ovarian cancer samples collected prospectively from ovarian cancer patients diagnosed at Hôpitaux Universitaires de Genève (HUG), Switzerland. CHIOVAR study was approved by the local ethics committee (CCER 2018-00407). The patient signed informed consent. Archival formalin-fixed paraffin embedded (FFPE) blocks were retrieved from the division of clinical pathology at HUG. Tumour and germline DNA was extracted and subjected to next-generation sequencing of a panel of 400 genes (NGS400v2 Agilent SureSelect XT HS) that revealed somatic mutation of BRCA2 p.Glu1126Ter and TP53 p.Ser166TyrfsTer4. Haematoxylin & Eosin sections from available FFPE blocks were reviewed by a board-certified pathologist (JCT). FFPE tissue sections were cut at 4 μm, dried overnight, dewaxed by immersion in xylene, rehydrated in ethanol of decreasing concentrations, subjected to heat-mediated antigen retrieval in pH6 citrate buffer for 10 min and incubated with a polyclonal anti-rabbit H2AX antibody (Novus Biologicals #NB100-383; 1:1000). H2AX immunohistochemistry was performed at the Translational Research Unit (University of Bern) with a BOND Research RX from Leica Biosystems as staining platform.
Immunofluorescence
Cells were seeded on coverslips in 24-well plates 3 days prior the experiment. To analyze 53BP1 and RAD51 foci formation in H2AX-deficient KB1P-G3 cells, DSB were induced by γ-irradiation (10 Gy) 4 h prior to fixation. Subsequently, cells were washed in PBS and fixed with 4% (v/v) PFA/PBS for 20’ at RT. Fixed cells were washed with PBS and permeabilized for 20’ in 0.2% (v/v) Triton X-100/PBS. Next, slides were washed three times with 0.2% Tween-20/PBS and blocked with staining buffer (PBS, BSA (2% w/v), glycine (0.15% w/v), Triton X-100 (0.1% v/v)) for 1 h at RT. Incubation with the primary rabbit polyclonal anti-53BP1 (#A300-272A, Bethyl Laboratories) and anti-RAD51 antibody (#70-012, Bioacademia) diluted 1:1000 in staining buffer was carried out for 2 h at RT. Slides were then washed four times for 5’ with 0.2% (v/v) PBS-Tween-20 and then incubated with Goat anti-rabbit IgG (H + L) Cross-Absorbed Secondary Antibody, Texas Red-X (# T-6391, ThermoFisher Scientific) diluted 1:2000 in staining buffer for 1 h at RT. Slides were washed three times for 5’ with 0.2% PBS-Tween-20, once with PBS and then mounted with Duolink In Situ mounting medium with DAPI (#DUO82040, Sigma Aldrich). Z-stack fluorescent images were acquired using the DeltaVision Elite widefield microscope (GE Healthcare Life Sciences). Multiple fields of view were imaged per sample with Olympus 100X/1.40, UPLS Apo, UIS2, 1-U2B836 objective and sCMOS camera at the resolution 2048 × 2048 pixels. Deconvolution of the acquired images was performed by the softWoRx DeltaVision software. Image analysis was performed using Fiji image processing package of ImageJ. Briefly, all nuclei were detected by the “analyze particles” command and all the foci per nucleus were counted with the “find maxima” command. Data were plotted with Prism software.
Analysis of micronuclei formation
KB2P3.4 cells were seeded on coverslips in 24-well plates and treated with DMSO or indicated concentrations of olaparib 24 h later. After 48 h of treatment, cells were washed with PBS and fixed with 4% (v/v) PFA/PBS for 20’ in RT. Cells were then washed 3 times in 0.2% (v/v) PBS-Tween-20 and permeabilized for 20’ in 0.2% (v/v) Triton X-100/PBS. Subsequently, slides were washed 3 times with PBS, counterstained with DAPI (1:50,000 dilution, #D1306, Life Technologies) and washed 5 more times with PBS before mounting in Fluorescence mounting medium (#S3023, Dako). Z-stack images were acquired using the DeltaVision Elite widefield microscope (GE Healthcare Life Sciences). Multiple fields of view were imaged per sample with Olympus 100X/1.40, UPLS Apo, UIS2, 1-U2B836 objective and sCMOS camera. The frequency of micronuclei positive cells was analyzed manually in Fiji.
Replication fork progression by DNA fiber analysis
Fork progression was measured as described previously43. Briefly, asynchronously growing subconfluent KB2P1.21 or KB2P3.4 cells were labeled with 25 μM thymidine analogue 5-chloro-2’-deoxyuridine (CIdU) (#C6891, Sigma-Aldrich) for 20’, washed three times with warm PBS and exposed to 250 μM of 5-iodo-2′-deoxyuridine (IdU) for 20’. All cells were collected by trypsinization and 2 μl of this cell suspension was then mixed with 8 μL of lysis buffer (200 mM Tris-HCl, pH 7.4, 50 mM EDTA, and 0.5% (v/v) SDS) on a positively charged microscope slide. After 9’ of incubation at RT, the slides were tilted at an ~ 30–45° angle to stretch the DNA fibers onto the slide. The resulting DNA spreads were air-dried, fixed in 3:1 methanol/acetic acid, and stored at 4 °C overnight. Next day, the DNA fibers were denatured by incubation in 2.5 M HCl for 1 h at RT, washed five times with PBS and blocked with 2% (w/v) BSA in 0.1% (v/v) PBST (PBS and Tween 20) for 40’ at RT while gently shaking. The newly replicated CldU and IdU tracks were stained for 3 h at RT using two different anti-BrdU antibodies recognizing CldU (#ab6326, Abcam) and IdU (#347580, BD Biosciences), respectively. After washing five times with PBS-T the slides were stained with goat the anti-mouse IgG (H + L) Cross-Adsorbed Secondary Antibody, Alexa Fluor 488 (#A-11029, ThermoFisher Scientific) diluted 1:600 in blocking buffer and with the Cy3 AffiniPure F(ab’)2 Fragment Donkey Anti-Rat IgG (H + L) antibody (#712-165-513, Jackson Immuno Research) diluted 1:150 in blocking buffer. Incubation with secondary antibodies was carried out for 1 h at RT in the dark. The slides were washed five times for 3’ in PBS-T, air-dried and mounted in Fluorescence mounting medium (#S3023, Dako). Fluorescent images were acquired using the DeltaVision Elite widefield microscope (GE Healthcare Life Sciences). Multiple fields of view from at least two slides (technical replicates) of each sample were imaged using the Olympus 60X/1.42, Plan Apo N, UIS2, 1-U2B933 objective and sCMOS camera at the resolution 2048 × 2048 pixels. To assess fork progression, the sum of individual CldU and IdU track lengths was measured using the segmented line tool in ImageJ software. Statistical analysis was carried out using Prism.
Replication fork stability by DNA fiber analysis
CldU and IdU pulse-labeling of asynchronously growing KB2P3.4 and KB1P-G3 cells expressing NT or H2afx-targeting gRNA was performed as described above. After pulse-labeling with IdU and three washes with PBS, medium containing 8 mM hydroxyurea (HU) was added for 6 h. Cells were then washed and harvested by trypsinization, and then processed as described above. Replication fork stability was analyzed by measuring the ratio between CldU and IdU tracks in ImageJ.
Detection of post-replicative ssDNA gaps by DNA fiber analysis
Detection of post-replicative ssDNA gaps was carried out as previously described43. In brief KB1P-G3 cells, were pulse labelled with 25 μM CldU (15’) followed by 250 μM IdU (45’) with or without 50 nM CPT. Cells were then harvested by trypsinization, washed with PBS and pre-extracted with CSK buffer (100 mM NaCl, 10 mM MOPS pH 7, 3 mM MgCl2, 300 mM sucrose and 0.5% Triton X-100 in water) for 10’ at RT. Next, isolated nuclei were harvested by centrifugation and incubated for 30’at 37 °C with S1 nuclease buffer containing or not 20 U/ml of S1 nuclease (#EN0321, Thermo Fisher Scientific). Nuclei were then pelleted, resuspended in PBS and spread onto microscope slides as previously described.
Immunoblotting
Cells were lysed for 40’ in RIPA buffer supplemented with halt protease and phosphatase inhibitor cocktail (100x) (#78420, Thermo Fisher Scientific) while briefly vortexed every 10’. Lysates were then centrifuged at 12,000 rcf for 10’ at 4 °C and the supernatant was collected to determine protein concentration using Pierce BCA Protein Assay Kit (#23225, Thermo Fisher Scientific). Before loading, protein lysates were denatured at 95 °C for 5’ in 6x SDS sample buffer. Proteins were separated by SDS/PAGE in 10% gel before wet transfer to 0.45μm nitrocellulose membranes (GE Healthcare) and blocked in 5% dry milk powder in TBS-T (100 mM Tris, pH 7.5, 0.9% NaCl, 0.05% Tween-20). Membranes were incubated with the mouse monoclonal anti-HA (1:1,000, #901533, Biolegend) and anti-γ-Tubulin (1:1,000, #5886, Cell Signalling), anti-GFP (1:1,000, #2555, Cell Signalling) primary antibodies for 2 h at RT. After three 5’ washes in TBS-T, anti-rabbit or anti-mouse horseradish peroxidase (HRP)-linked secondary antibodies (1:5,000, Cell Signalling) were applied for 1 h at RT. Images were acquired using Vilber FUSION FX chemiluminescent imager.
SIRF (in Situ analysis of protein Interactions at DNA Replication Forks)
SIRF assay was performed as previously reported45. Cells were seeded on coverslips and the following day they were pulsed with 25 μM EdU for 10’. After the EdU pulse, cells were initially pre-extracted with CSK buffer on ice for 5’ and then fixed with 3.7% Paraformaldehyde at RT for 10’. Coverslips were then washed with PBS and stored overnight at 4 °C. The following day cells were permeabilized in 0.2% Triton X-100 in PBS for 5’ and then the click reaction (100 mM Tris pH 8, 100 mM CuSO4, 2 mg/ml sodium-L-ascorbate, 10 mM biotin-azide) was performed for 90’ at 37 °C. Slides were then blocked for 1 h at 37 °C with blocking solution (PBS, BSA 2%, glycine 0.15%, Triton X-100 0.1%), followed by incubation with primary antibodies for 1 h at 37 °C (rabbit anti-CtIP 1:500, Bethyl Laboratories #A300-488A; mouse anti-Biotin 1:200, #200-002-211, Jackson Immuno Research; rabbit anti-MRE11 1:500, a kind gift from Arnab Ray Chaudhuri). After antibody incubation, coverslips were washed 2X with Buffer A for 5’ at RT (Duolink kit). Each coverslip was then incubated for 1 h at 37 °C with Duolink PLA probes (Thermo Fisher Scientific) diluted in blocking solution. After 2 washes with Buffer A for 5’ at RT, probes were ligated for 30’ at 37 °C and amplified by polymerase reaction for 100’ at 37 °C. Coverslips were then washed 2X with Buffer B for 5’ at RT (Duolink kit) and then mounted with DAPI on microscope slides. Images were acquired on multiple stacks using the DeltaVision Elite widefield microscope with a 60X objective. Deconvolution of the images was done using the softWoRx DeltaVision software. The number of foci in each cell was scored with ImageJ and the statistical analysis was performed using Prism.
Transmission electron microscopy of replication intermediates
The procedure was performed as described previously with minor modifications39. A total of 2.5–5.0 × 106 asynchronously growing KB2P3.4 cells expressing either NT or H2afx-targeting gRNA were treated with 8 mM hydroxyurea for 5 h, washed with PBS and then harvested by trypsinization and resuspended in 10 mL of cold PBS. DNA was cross-linked by exposing the living cells twice to 4,5′,8-trimethylpsoralen at a final concentration of 10 μg/mL followed by 3’ irradiation pulses with UV 365 nm monochromatic light (UV Stratalinker 1800, Agilent Technologies). The cells were then washed repeatedly with cold PBS and lysed with a cell lysis buffer (1.28 M sucrose, 40 mM Tris-Cl, pH 7.5, 20 mM MgCl2, and 4% (v/v) Triton X-100). The nuclei were then digested in a digestion buffer (800 mM guanidine-HCl, 30 mM Tris-HCl, pH 8.0, 30 mM EDTA, pH 8.0, 5% (v/v) Tween 20, and 0.5% (v/v) Triton X-100) supplemented with 1 mg/mL proteinase K at 50 °C for 2 h. Genomic DNA was extracted with a 24:1 Chloroform:Isoamyl alcohol mixture by phase separation (centrifugation at 8,000 rcf for 20’ at 4 °C) and precipitated by addition of equal amount of isopropanol to the aqueous phase, followed by another centrifugation step (8,000 rcf for 10’ at 4 °C). The obtained DNA pellet was washed once with 1 mL of 70% ethanol, air-dried at RT, and resuspended by overnight incubation in 200 μL TE (Tris-EDTA) buffer at RT. 12 μg of the extracted genomic DNA was digested for 5 h at 37 °C with 100U restriction enzyme PvuII-HF (#R3151S, New England Biolabs). The digest was cleaned up using a silica bead DNA gel extraction kit (#K0513, Thermo Fisher Scientific). The benzyldimethylalkylammonium chloride (BAC) method was used for native spreading of the DNA on a water surface and then loading it on carbon-coated 400-mesh magnetic nickel grids. After the spreading procedure, the electron density of the DNA was increased by platinum coating with the platinum-carbon rotary shadowing technique using the MED 020 High Vacuum Evaporator (Bal-Tec). The grids were then scanned in a semi-automated fashion using a transmission electron microscope (FEI Thalos 120, LaB6 filament) at high tension ≤ 120 kV and pictures were acquired with a bottom mounted CMOS camera BM-Ceta (4000 ×4000 pixels). The images were processed with MAPS Version 3.14 (Thermo Fisher Scientific) and analyzed using MAPS Offline Viewer Version 3.14.11 (Thermo Fisher Scientific). The percentage of RFs containing ssDNA stretches was evaluated by manual scoring. Samples from mock-treated and HU-treated cells were pooled together for the analysis of ssDNA-containing RFs.
Statistics & reproducibility
Statistical parameters including sample size, number of biological replicates, applied statistical tests and statistical significance are reported in the corresponding figure legends or materials and methods section. Power analysis (using a power of 0.8) was used to predetermine sample size for the mouse experiments shown in Figs. 2d and 4f. No data were excluded from the analyses. Except Figs. 1d, 1e, 2d, 4f, experiments were not randomized, and the investigators were not blinded to allocation during experiments and outcome assessment.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All data will be shared upon request by the lead contact with no restrictions. Source data are provided with this paper. Raw sequencing data of the genetic screens are available in European Nucleotide Archive (ENA) under accession number PRJEB75036. and PRJEB74933. Raw sequencing data of WE-seq and RNA-seq reported in this paper are available in European Nucleotide Archive (ENA) under accession number PRJEB61242. for KB1P(M) tumours and PRJEB61243. for KB2P tumours. The remaining data are available within the Article, Supplementary Information, or Source Data file. Source data are provided with this paper.
References
Lord, C. J. & Ashworth, A. PARP inhibitors: synthetic lethality in the clinic. Science 1158, 1152–1158 (2017).
Prakash, R., Zhang, Y., Feng, W. & Jasin, M. Homologous recombination and human health: the roles of BRCA1, BRCA2, and associated proteins. Cold Spring Harb. Perspect. Biol. 7, 1–27 (2015).
Farmer, H. et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 434, 917–921 (2005).
Bryant, H. E. et al. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 434, 913–917 (2005).
Gogola, E., Rottenberg, S. & Jonkers, J. Resistance to PARP inhibitors: lessons from preclinical models of BRCA-associated cancer. Annu Rev. Cancer Biol. 3, 235–254 (2019).
Murai, J. et al. Differential trapping of PARP1 and PARP2 by clinical Parp inhibitors. Cancer Res. 72, 5588–5599 (2013).
Murai, J. et al. Rationale for poly(ADP-ribose) polymerase (PARP) inhibitors in combination therapy with camptothecins or temozolomide based on PARP trapping versus catalytic inhibition. J. Pharm. Exp. Ther. 349, 408–416 (2014).
Pettitt, S. J. et al. Genome-wide and high-density CRISPR-Cas9 screens identify point mutations in PARP1 causing PARP inhibitor resistance. Nat. Commun. 9, 1849 (2018).
Petropoulos, M. et al. Transcription–replication conflicts underlie sensitivity to PARP inhibitors. Nature 628, 433–441 (2024).
Jonkers, J. et al. Synergistic tumor suppressor activity of BRCA2 and p53 in a conditional mouse model for breast cancer. Nat. Genet 29, 418–425 (2001).
Liu, X. et al. Somatic loss of BRCA1 and p53 in mice induces mammary tumors with features of human BRCA1-mutated basal-like breast cancer. Proc. Natl Acad. Sci. USA 104, 12111–12116 (2007).
Bhin, J. et al. Multi-omics analysis reveals distinct non-reversion mechanisms of PARPi resistance in BRCA1- versus BRCA2-deficient mammary tumors. Cell Rep. 42, 112538 (2023).
Jaspers, J. E. et al. Loss of 53BP1 causes PARP inhibitor resistance in BRCA1-mutated mouse mammary tumors. Cancer Discov. 3, 68–81 (2013).
Xu, G. et al. REV7 counteracts DNA double-strand break resection and affects PARP inhibition. Nature 521, 541–544 (2015).
Ghezraoui, H. et al. 53BP1 cooperation with the REV7–shieldin complex underpins DNA structure-specific NHEJ. Nature 560, 122–127 (2018).
Noordermeer, S. M. et al. The shieldin complex mediates 53BP1-dependent DNA repair. Nature 560, 117–121 (2018).
Barazas, M. et al. The CST complex mediates end protection at double-strand breaks and promotes PARP inhibitor sensitivity in BRCA1-deficient cells. Cell Rep. 23, 2107–2118 (2018).
Gogola, E. et al. Selective loss of PARG restores PARylation and counteracts PARP inhibitorlmediated synthetic lethality. Cancer Cell 33, 1078–1093 (2018).
Rondinelli, B. et al. EZH2 promotes degradation of stalled replication forks by recruiting MUS81 through histone H3 trimethylation. Nat. Cell Biol. 19, 1371–1378 (2017).
Chaudhuri, A. R. et al. Replication fork stability confers chemoresistance in BRCA-deficient cells. Nature 535, 382–387 (2016).
Evers, B. et al. Preclinical selective inhibition of BRCA2-deficient mammary tumor Cell growth by AZD2281 and Cisplatin. Clin. Cancer Res. 1, 3916–3925 (2008).
Sanjana, N. E., Shalem, O. & Zhang, F. Improved vectors and genome-wide libraries for CRISPR screening iPipet: sample handling using a tablet. Nat. Methods 11, 783–784 (2014).
Li, W. et al. Quality control, modeling, and visualization of CRISPR screens with MAGeCK-VISPR. Genome Biol. 16, 281 (2015).
Bouwman, P. et al. 53BP1 loss rescues BRCA1 deficiency and is associated with triple-negative and BRCA-mutated breast cancers. Nat. Struct. Mol. Biol. 17, 688–695 (2010).
Rogakou, E. P., Pilch, D. R., Orr, A. H., Ivanova, V. S. & Bonner, W. M. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J. Biol. Chem. 273, 5858–5868 (1998).
Bonner, W. M. et al. γH2AX and cancer. Nat. Rev. Cancer 8, 957–967 (2008).
Harvey-Jones, E. et al. Longitudinal profiling identifies co-occurring BRCA1/2 reversions, TP53BP1, RIF1 and PAXIP1 mutations in PARP inhibitor-resistant advanced breast cancer. Ann. Oncol. 35, 364–380 (2024).
Nacson, J. et al. BRCA1 mutation-specific responses to 53BP1 loss-induced homologous recombination and PARP inhibitor resistance. Cell Rep. 24, 3513–3527 (2018).
Bunting, S. F. et al. 53BP1 inhibits homologous recombination in brca1-deficient cells by blocking resection of DNA breaks. Cell 141, 243–254 (2010).
Paull, T. T. et al. A critical role for histone H2AX in recruitment of repair factors to nuclear foci after DNA damage. Curr. Biol. 10, 886–895 (2000).
Kleiner, R. E., Verma, P., Molloy, K. R., Chait, B. T. & Kapoor, T. M. Chemical proteomics reveals a g H2AX-53BP1 interaction in the DNA damage response. Nat. Chem. Biol. 11, 807–814 (2015).
Schlacher, K., Wu, H. & Jasin, M. A distinct replication fork protection pathway connects fanconi anemia tumor suppressors to RAD51-BRCA1/2. Cancer Cell 22, 106–116 (2012).
Schlacher, K. et al. Double-strand break repair-independent role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell 145, 529–542 (2011).
Mijic, S. et al. Replication fork reversal triggers fork degradation in BRCA2-defective cells. Nat. Commun. 8, 1–11 (2017).
Lemaçon, D. et al. MRE11 and EXO1 nucleases degrade reversed forks and elicit MUS81-dependent fork rescue in BRCA2-deficient cells. Nat. Commun. 8, 860 (2017).
Kolinjivadi, A. M. et al. Smarcal1-mediated fork reversal triggers mre11-dependent degradation of nascent DNA in the absence of Brca2 and stable Rad51 nucleofilaments. Mol. Cell 67, 867–881 (2017).
Taglialatela, A. et al. Restoration of replication fork stability in BRCA1- and BRCA2-deficient cells by inactivation of SNF2-family fork remodelers. Mol. Cell 68, 414–430 (2017).
Dibitetto, D. et al. DNA-PKcs promotes fork reversal and chemoresistance. Mol. Cell 82, 3932–3942 (2022).
Zellweger, R. & Lopes, M. Dynamic architecture of eukaryotic DNA replication forks in vivo, visualized by electron microscopy. Methods Mol. Bio. 1672, 261–294 (2017).
Panzarino, N. J. et al. Replication gaps underlie BRCA-deficiency and therapy response. Cancer Res. 81, 1388–1397 (2021).
Cong, K. et al. Replication gaps are a key determinant of PARP inhibitor synthetic lethality with BRCA deficiency. Mol. Cell 81, 3128–3144 (2021).
Dias, M. P. et al. Loss of nuclear DNA ligase III reverts PARP inhibitor resistance in BRCA1 / 53BP1 double-deficient cells by exposing ssDNA gaps. Mol. Cell 81, 4692–4708 (2021).
Quinet, A., Carvajal-Maldonado, D., Lemaçon, D. & Vindigni, A. DNA fiber analysis: mind the gap! Methods Enzym. 591, 55–82 (2017).
Mann, A. et al. POLQ prevents MRE11-NBS1-CtIP-dependent fork breakage in the absence of BRCA2 / RAD51 by filling lagging-strand gaps. Mol. Cell 82, 4218–4231 (2022).
Roy, S., Luzwick, J. W. & Schlacher, K. SIRF: Quantitative in situ analysis of protein interactions at DNA replication forks. J. Cell Biol. 217, 1553 (2018).
Helmink, B. A. et al. H2AX prevents CtIP-mediated DNA end resection and aberrant repair in G1-phase lymphocytes. Nature 469, 245–249 (2011).
Sartori, A. A. et al. Human CtIP promotes DNA end resection. Nature 450, 509–514 (2007).
Przetocka, S. et al. CtIP-mediated fork protection synergizes with BRCA1 to suppress genomic instability upon DNA replication stress. Mol. Cell 72, 568–582 (2018).
Kuster, A. et al. A stapled peptide mimetic of the CtIP tetramerization motif interferes with double-strand break repair and replication fork protection. Sci. Adv. 7, eabc6381 (2021).
Peterson, S. E. et al. Activation of DSB processing requires phosphorylation of CtIP by ATR. Mol. Cell 49, 657–667 (2013).
Makharashvili, N. et al. Catalytic and noncatalytic roles of the CtIP endonuclease in double-strand break end resection. Mol. Cell 54, 1022–1033 (2014).
Lanz, M. C., Dibitetto, D. & Smolka, M. B. D. N. A. damage kinase signaling: checkpoint and repair at 30 years. EMBO J. 38, e101801 (2019).
Durant, T. S. et al. The brain-penetrant clinical ATM inhibitor AZD1390 radiosensitizes and improves survival of preclinical brain tumor models. Sci. Adv. 4, eaat1719 (2018).
Schmid, J. A. et al. Histone ubiquitination by the DNA damage response is required for efficient DNA replication in unperturbed S phase. Mol. Cell 71, 897–910 (2018).
Mutreja, K. et al. ATR-mediated global fork slowing and reversal assist fork traverse and prevent chromosomal breakage at DNA interstrand cross-links. Cell Rep. 24, 2629–2642 (2018).
Wang, B. et al. Integrative analysis of pooled CRISPR genetic screens using MAGeCKFlute. Nat. Protoc. 14, 756–780 (2019).
Duarte, A. A. et al. BRCA-deficient mouse mammary tumor organoids to study cancer-drug resistance. Nat. Methods 15, 134–140 (2018).
de Ronde, J. J. et al. Identifying subgroup markers in heterogeneous populations. Nucleic Acids Res. 41, e200 (2013).
Acknowledgements
We wish to thank Hana Hanzlikova and Lea Lingg for critical reading of the manuscript. We thank all the members of the Rottenberg lab for helpful discussion. We thank Horizon Discovery for performing and analysing the CRISPR-Cas9 screens in SW620, HT29 and BT549 cells. We thank Daniel Durocher for sharing RPE1-hTERT TP53-/- and RPE1- hTERT TP53-/-;BRCA1-/- cells. We are grateful to Georgina Hayoz and Fabiana Steck for their support at the Vetsuisse Faculty mouse facility. We thank Denise Howald, Therese Waldburger, Cristina Graham Martinez, Aliaksandra Kakoinchankava, Valentine Dubois for technical assistance. Financial support came from the Swiss National Science Foundation (320030M_219453 to S.R. and J.J., 31BL30_189698 to S.R., 310030_208143 to A.A.S.), the European Union (ERC-2019-AdG-883877 to S.R.), the Swiss Cancer League (KFS-5519-02-2022 to S.R.), the ISREC foundation, the Boehringer Ingelheim Fonds (PhD fellowship to M.L.), the UZH Candoc Grant (no. FK-23-050 to F.V.), the Wilhelm Sander Foundation (no. 2019.069.1 to S.R.), and the Office of the Assistant Secretary of Defense for Health Affairs through the Ovarian Cancer Research Program under Award No. (W81XWH-22-1-0557 and W81XWH-22-1-0558 to S.R. and I.L.-G.). Opinions, interpretations, conclusions, and recommendations are those of the author and are not necessarily endorsed by the Department of Defense. Figures 1a, 1d, 2b, and supplementary fig. 5d were created with Biorender.
Author information
Authors and Affiliations
Contributions
D.D., M.L. and S.R. conceived the project. D.D., M.L., F.V., E.G., H.D., M.G.F., A.D., J.A.S., M.F.D., P.F., S.P., I.K., M.S., R.B., and N.A.M. performed and analyzed experiments. S.T.D. and J.V.F. performed the CRISPR screens with the ATMi AZD0156. M.D. performed the cisplatin resistance genetic screen in Fig. 1c. A.N.K. generated the U-2OS-TetON CtIP cell lines. J.T, M.U., I.S.L. were involved in the collection and characterization of tumour samples from patient CHIOVAR59. F.V. and H.D. equally contributed to this work. D.D., C.S.S., M.L., A.A.S, J.J. and S.R. were involved in supervision and funding acquisition. D.D. and S.R. wrote the manuscript. All the authors revised the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
S.T.D. and J.V.F. are or were full time employees and stockholders of AstraZeneca at the time of this study. The remaining authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Dibitetto, D., Liptay, M., Vivalda, F. et al. H2AX promotes replication fork degradation and chemosensitivity in BRCA-deficient tumours. Nat Commun 15, 4430 (2024). https://doi.org/10.1038/s41467-024-48715-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-48715-1
- Springer Nature Limited