

Abstract
Bacteriophages are widely recognised as rapidly evolving biological entities. However, knowledge about ancient bacteriophages is limited. Here, we analyse DNA sequence datasets previously generated from ancient palaeofaeces and human gut-content samples, and identify an ancient phage genome nearly identical to present-day Mushuvirus mushu, a virus that infects gut commensal bacteria. The DNA damage patterns of the genome are consistent with its ancient origin and, despite 1300 years of evolution, the ancient Mushuvirus genome shares 97.7% nucleotide identity with its modern counterpart, indicating a long-term relationship between the prophage and its host. In addition, we reconstruct and authenticate 297 other phage genomes from the last 5300 years, including those belonging to unknown families. Our findings demonstrate the feasibility of reconstructing ancient phage genome sequences, thus expanding the known virosphere and offering insights into phage-bacteria interactions spanning several millennia.
Similar content being viewed by others


Introduction
Bacteriophages diversified over billions of years through a co-evolutionary arms race with their microbial hosts1. Due to recent advancements in metagenomic sequencing2 and computational analysis3, the vast genomic diversity of phages can now be explored, catalogued4, or even tracked through space5 and time6. However, most studies only sample present-day phages, thus lacking an evolutionary perspective. The longest study of substitutions and recombinations in phage genomes spans nearly three decades7, representing only a partial glimpse into the complex evolutionary history of bacteriophages. To fully understand the phylogeny of these viruses and their effect on microbial ecosystems, it is essential to go further back in time.
Previous research on ancient virology has mainly focused on the reference-based reconstruction of human pathogens8, retroviral elements incorporated in animal genomes9, or eukaryotic viruses that have remained dormant since prehistory10. However, knowledge about ancient bacteriophages is limited11. The first reports on ancient phages described viruses from 14th-century faecal material in Belgium12 and the gut contents of pre-Columbian Andean mummies13. However, these studies lacked authentication based on terminal deamination patterns observed in ancient DNA (aDNA)14, which is used to distinguish ancient sequences from modern contaminants15. Furthermore, the small amount of sequenced DNA in these investigations resulted in the recovery of only fragmented phage genomes. To our knowledge, only one complete oral phage genome has been published that meets the criteria of authenticated aDNA reconstruction16.
While the recovery of hundreds of high-quality ancient bacterial genomes is now possible17,18, their phage counterparts remain largely unexplored. However, recent developments in computational tools for viral metagenomics3,4, combined with the extraction of exceptionally well-preserved ancient DNA samples17,19,20,21,22,23 offer new opportunities to unlock the mystery of past phage genome diversity.
Here, we analyse draft and complete genomes assembled from 150–5300 years old palaeofaeces and human gut-content samples17,19,20,21,22,23. We use this collection to (i) identify 298 ancient phage genomes, (ii) authenticate their ancient origin based on DNA damage patterns, (iii) determine their taxonomic assignments and relationships to modern viruses, (iv) predict hosts using state-of-the-art bioinformatic tools, and finally (v) characterise the particularly stable genome of one encountered virus, Mushuvirus mushu that is 97.7% identical to its present-day reference. Together, these results demonstrate that the large-scale recovery of virus genomes from ancient samples is possible and may provide unexpected insights into the evolutionary history of the human virome.
Results
Identification and validation of ancient phage genomes
We selected aDNA sequence datasets from 30 samples previously published in studies on the ancestral human gut microbiome17,19,20,21,22,23. These samples were derived from eight sites in Europe and North America, dating back between 150 and 5300 years ago, using the C14 method (Supplementary Data 1). The de novo assembly of selected libraries resulted in 72,693 high-quality contigs (mean length: 12,592 nucleotides ± 59.3 standard error; Fig. 1a). However, we deemed less than half of the assembled contigs as ancient based on deamination patterns observed at the 5′ ends of the sequencing reads (see Methods). A total of 2375 sequences were classified as viral by at least two methods (VIBRANT24, VirSorter225 and Jaeger; Supplementary Data 2) and were selected as bona fide virus contigs. Among these, 383 were at least 20 kb long or assessed by CheckV as either medium or better quality (>50% completeness; Fig. 1b and Supplementary Data 3) and selected for further analysis. We clustered the selected sequences into 298 species-level viral operational taxonomic units (hereafter referred to as aMGVs - ancient metagenomic gut viruses) based on 95% average nucleotide identity (ANI) with over 85% alignment fraction26. Despite fragmentation and degradation of the aDNA, our aMGV collection had size range from 5 kbp to 292 kbp, an estimated mean completeness of 50.3%, and comprised of 49 high-quality or complete genomes (Supplementary Data 3). Most of aMGVs (59%) are likely lytic, whereas the remaining 41% of viruses are potentially lysogenic (Supplementary Data 4). Although the mean damage at the first position of reads mapping to collected aMGVs was relatively low (0.025 frequency), it was 12 times higher than the control modern viral genomes from the human gut (0.002 frequency; Fig. 1c, d and Supplementary Data 3).

a Overview of workflow to identify ancient phage genomes. b Distribution of the genome sizes of 298 ancient metagenomic viruses (aMGVs) stratified by CheckV61 genome quality. c Distribution of C → T substitution frequencies at the first position of the 5’ end of sequencing reads from ancient (n = 298, red) and present-day (n = 943, blue) phage contigs. Box and whisker plots in c and d show median (centre line), upper and lower quartiles (represented by boxes), and highest (upper whisker) or lowest (lower whisker) value within a 1.5 inter-quartile range (IQR) while black dots indicate values outside of the IQR. d Comparison of damage patterns between selected modern viral genomes from MGV31 (blue) and aMGVs (red). The solid line shows the mean frequency of C → T substitutions and the shade indicates the standard deviation. Source data are provided as a Source Data file.
Human gut origin of ancient phage genomes
Deamination damage patterns is a well-established technique for ancient DNA validation, but the complexity of ancient metagenomic data can necessitate the implementation of additional authentication steps. In our case, we decided to rule out post-depositional contamination with DNA from the environment (e.g., cave sediments), which may also exhibit damage due to the same degradation processes as the aDNA of interest27. To estimate the risk of such contamination, we constructed a gene-sharing network using vContact228 to phylogenetically relate ancient and modern phages (Fig. 2a), including reference viruses from different environments in the IMG/VR database29 and prokaryotic viruses classified by the International Committee of Virus Taxonomy (ICTV)30. In the network (Supplementary Data 5), 151 ancient phages were distributed across 122 viral clusters (VCs). These VCs were dominated by mammalian-gut-associated viruses from IMG/VR (80%, see Fig. 2b), with a high representation (68%) of phages found in humans. The estimated contamination was relatively low; only 5% of aMGVs (n = 15) clustered with viruses from environmental or engineered ecosystems. We recognised that 49% of aMGVs (n = 147) did not cluster with any modern viruses. The mean ANI of all aMGVs to the closest IMG/VR and GenBank viral genome was 40% (± 24.7%, Supplementary Data 6 and 7). Only one outlier aMGV, Mushuvirus mushu, had more than 95% ANI, and it is described in further sections (Supplementary Fig. 1).

a vContact2 gene-sharing network28 of 151 ancient metagenomic gut virus (aMGV) genomes (red circles), 2198 selected close relatives from the IMG/VR database, and 3655 prokaryotic viruses classified by the International Committee of Virus Taxonomy. Distantly related aMGVs (n = 147) were outliers and not included in the gene-sharing network. b Waffle charts represent the proportion of contemporary viruses categorised by ecosystems in two data sets: clusters comprising aMGVs (left) and the entire IMG/VR database. Source data are provided as a Source Data file.
Further evidence supporting the ancient gut origin of the 298 analysed aMGVs came from the distribution of their predicted hosts (see Methods; Supplementary Fig. 2 and Supplementary Data 8). Over half of the ancient viruses were predicted to infect Clostridia and Bacteroidia hosts — two dominant classes in the gut microbiome31. The distribution of host bacteria assigned to ancient viruses was more similar to that of modern viruses from the digestive systems of different animals than viruses of other environments. For example, we observed a strong correlation (Pearson’s r = 0.95, P = 3.29 × 10−6) between the proportion of host classes of ancient phages and hosts of modern viruses in human large intestines (Supplementary Data 9). Hence, both the results of host prediction and the gene-sharing network analysis suggest that the ancient phage sequences primarily originated from the human gut rather than the surrounding environment.
Catalogue of the human gut virome expands into the past
We performed geNomad’s32 marker-based taxonomic assignment for all 298 ancient viral sequences, 293 of which (~98%) were assigned to the Caudoviricetes class, three to the Megaviricetes class (nucleo-cytoplasmic large DNA viruses - NCLDV), and two single-strand viruses to the Cressdnaviricota and Phixviricota phyla (Supplementary Data 10). Most aMGVs from the Caudoviricetes class were not classified at lower taxonomic ranks, except for nine that were assigned to the order Crassvirales and the families Autographiviridae, Straboviridae, Herelleviridae, and Rountreeviridae. To improve the taxonomic resolution, we selected 49 high-quality or complete ancient genomes and clustered them with RefSeq and IMG/VR virus genomes based on pairwise average amino acid identity (AAI) and gene sharing, followed by manual assignment to the ICTV taxonomy (see “Methods” section). Additionally, we constructed a proteomic tree to support the clustering results and illustrate the relationships between the modern and ancient viruses (Fig. 3). This analysis revealed that high-quality or complete aMGVs were distributed across 39 putative families and 46 putative genera (Supplementary Data 11).

A tree generated in ViPTreeGen70 encompassing 49 high-quality and complete ancient phages, their 265 closest relatives from IMG/VR29, and all 4703 prokaryote-infecting viruses from RefSeq76. The ancient phages are identified with red circles. Putative families were defined using a percentage of shared genes and amino acid identity between each pair of viruses. Clusters containing at least one ancient phage are highlighted in yellow and listed in Supplementary Data 11. The purple and blue highlighted clades represent putative families of Salmonella-infecting phages and T4 bacteriophage-related genomes, respectively. Outer rings provide additional information for each phage, such as its host assignment and genome size. Source data are provided as a Source Data file.
Most putative families (90%, 35 out of 39) and genera (72%, 33 out of 46) contained aMGVs and modern representatives from IMG/VR and/or RefSeq. Only a small proportion of those groups have been classified by ICTV, including six families (15%, Straboviridae, Peduoviridae, Casjensviridae, Microviridae, Chaseviridae, and Guelinviridae), and eight genera (17%, Tequatrovirus, Plaisancevirus, Pbunavirus, Astrithrvirus, Brucesealvirus, Loughboroughvirus, and Mushuvirus). Most of these putative taxa included a single ancient phage grouped with multiple contemporary viruses. An interesting example of such a grouping is a clade of Escherichia-infecting members of the Tequatrovirus genus (including bacteriophage T4, Supplementary Fig. 3a) with an ancient virus located on the ancestral branch. This ancient Tequatrovirus maintains genome organisation and conserved structural proteins, with ANI of 88.1% and AAI of 94.7% to its closest modern relative, Tequatrovirus cromcrrp10 (Supplementary Fig. 3b). Only a few putative taxa contained multiple ancient representatives. For example, two genus-like groups (“13” and “14” see Supplementary Data 11) formed a family-like unit that encompassed three ancient phages, 36 IMG/VR viruses and the Salmonella-infecting phage, Astrithrvirus astrithr.
In contrast to groups with multiple representatives, there were four putative families and 13 genera represented by only a single aMGV. While these phages could hypothetically represent extinct lineages, it is likely that modern representatives of those groups have yet to be discovered. Although it is beyond the scope of the current study, one way to address this question could be to identify conserved marker regions in the ancient genomes and attempt to amplify them in locations where many modern human gut phages come together, such as sewage systems5.
We also searched for ancient relatives of modern Crassvirales that are widespread in mammalian intestinal viromes. We found four such sequences. The two longest contigs are probably fragments of the same genome, similar to Bacteroides phage PhiCrAssBcn21 (Supplementary Fig. 4a). The remaining sequences are shorter (21 kb and 42 kb) and bear little resemblance to modern phages (less than 15% ANI; Supplementary Data S6 and Supplementary Fig. 4b).
Notably, our taxonomic analysis of high-quality ancient genomes was limited by the database-dependent tool CheckV, which has a bias for phages similar to known reference genomes. Consequently, the other 249 aMGVs could still include viral genomes unlike any sequence characterised to date. Despite this limitation, we have shown that our approach is promising for discovering previously unknown viruses and shedding light on dark matter lingering in ancient metagenomes.
Persistence of Mushuvirus mushu
Among the collection of ancient phages, we found the genome sequence of a bacteriophage from the Mushuvirus mushu species recently discovered in a prophage analysis of its host, Faecalibacterium prausnitzii33. In that study, the prophage could not be induced after DNA damage, but virions were observed in the gastrointestinal tract in a mouse model33. We believe that our aMGV represents an ancient sequence because the frequency of C → T substitutions at the first position of Mushuvirus mushu reads was high (0.042) compared to other sequences in our collection of ancient viruses (mean: 0.025; see Supplementary Fig. 5 and Supplementary Data 3). Moreover, the F. prausnitzii host of Mushuvirus mushu is a strict anaerobe that typically inhabits the mucosal surface in the gastrointestinal tract34, so it is unlikely it could have contaminated the original palaeofaeces sample21.
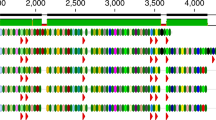
The ancient Mushuvirus mushu genome that was recovered from 1300-year-old faecal material from the Zape cave in Mexico was 97.7% identical to the modern reference genome FP_Mushu (RefSeq accession: NC_047913.1) that was extracted from wastewater in France33 (Fig. 4a, b). The mean sequence identity to ten IMG/VR genomes from the same species that were derived from the large intestine was 97.1% ± 0.6% (Fig. 4c and Supplementary Data 12). The ancient and modern genome sequences had similar lengths (36,623 bp and 36,636 bp, respectively) and perfect colinearity between 52 protein-coding genes (Fig. 4b). Phage mutation rates that are reported in the literature range from 1.976 × 10−4 to 4.690 × 10−3 nucleotide alterations per site per year due to substitutions and recombinations for virulent phages7,35, and 1.154 × 10−4 substitutions per site per year for temperate phages6. As shown in Fig. 4d, the probability of finding a 36 kbp long genome sequence with at least 97.7% identity approaches zero after a little over 200 years, even at the lowest mutation rates reported in the literature6,7,35. Notably, this calculation assumes direct ancestral relationship between both viruses, as encountering such similar genomes in two sister clades that accumulate mutations independently is even less likely. Surprised by such a low mutation rate, we estimated intra-population genetic diversity (microdiversity)36 of Mushuvirus mushu in modern Hadza hunter-gather’s gut37. However, we found that both nucleotide diversity and the number of divergent sites of Mushuvirus were higher than the median for other phage sequences (Fig. 4e and Supplementary Data 13). Thus, low mutation rates are unlikely to explain the observation of two nearly identical Mushuvirus mushu genomes in the span of 1300 years.

a Geographic and historical distances between the modern reference (France) and the ancient Mushuvirus mushu (from the Zape cave in Mexico) genomes. b Comparison of sequence conservation and gene organisation between the ancient and modern reference (NC_047913.1) genomes of Mushuvirus mushu. Black lines indicate alignments with small gaps. The height of the middle plot depends on the identity between genomes. The lowest similarities (grey colour) were concentrated in a ∼ 600-nucleotide variability region of the Hoc-like gene affected by the diversity-generating retroelement (DGR) mechanism. Two major structural differences in the ancient and modern genomes constituted an additional codon stop in a gene of unknown function (genome position: 9518) and 17 nucleotide insertion/deletions, which changed the open reading frame in the tail length tape measure protein. c Intergenomic similarity matrix calculated by VIRIDIC63 revealed high identity between ancient and modern representatives of the species Mushuvirus mushu deposited in RefSeq and IMG/VR v.4. d Probability of a 36,630-nucleotide phage genome having at least 97.7% unaltered nucleotides over a period of 1300 years. The probability was calculated using a binomial cumulative distribution function assuming different mutation rates reported in the literature6,7,35. The blue and green lines represent the mutation rates due to substitutions and recombinations in lytic phages, with rates of 4.690 × 10−3 and 1.976 × 10−4 altered nucleotides per site per year, respectively. The green line includes a rate of 1.154 × 10−4 altered nucleotides per site per year due to substitutions in temperate phage genomes. e Intra-population genetic diversity (microdiversity) of Mushuvirus mushu (red point) and other bacteriophages in modern Hadza hunter-gather’s gut37 based on calculation from InStrain36. f Table with Mushuvirus mushu hosts found using the BLASTn search against the Unified Human Gastrointestinal Genome collection45 and the Genome Taxonomy Database44. Source data are provided as a Source Data file.
A total of 22,059 sequencing reads were mapped to the ancient Mushuvirus mushu genome, resulting in 28x genome coverage. Our comparison of the ancient and modern genomes revealed 869 single nucleotide variants (SNVs). The distribution of these SNVs was concentrated in the specific section of the gene encoding the Hoc-like capsid decoration protein (Fig. 4b). This fragment is the target of a diversity-generating retroelement (DGR) that produces a large number of localised mutations through error-prone reverse transcription and is involved in host switching in some phages38,39,40. Indeed, the Hoc-like gene had a lower level of AAI to the reference FP_Mushu (91.8%) than other protein-coding genes (mean identity: 98.22%) and accounted for 16% of all SNVs in the genome. Notably, according to recently published RNA-seq data from cultured strains of F. prausnitzii, the Hoc-like gene is expressed at levels similar to a typical bacterial gene under most studied conditions41 (Supplementary Fig. 6). This finding suggests that the Hoc-like protein might play a role in lysogenized bacterial cells. Despite the potential significance of the Hoc-like gene in the evolution of Mushuvirus mushu, its function remains poorly understood with only homologues of the gene being characterised as enhancing binding to mucus on the mucosal surface of the intestine and potentially increasing encounters with mucosa-dwelling hosts42,43.
To analyse whether the ancient Mushuvirus mushu sequence represented an integrated prophage, we investigated the flanking regions of the contig (see “Methods” section). These regions differed between the ancient and modern viruses indicating a potential bacterial origin. The sequence similarity of the flanking regions of ancient Mushuvirus mushu points to Oscillospiraceae genus ER4, suggesting this may be a host for the ancient Mushuvirus mushu (Supplementary Data 13). Mushuvirus mushu appears to have a broad host range33; when comparing the ancient genome to bacterial sequences from the Genome Taxonomy Database (GTDB)44 and the Unified Human Gastrointestinal Genome (UHGG) collection45, we found prophage genomes with 97% sequence identity in seven different Oscillospiraceae genera, including Faecalibacterium (see Fig. 4f and Supplementary Data 15).
Discussion
In this study, we demonstrated the large-scale reconstruction of high-quality ancient phage genomes using state-of-the-art bioinformatic methods. To validate the authenticity of the reconstructed ancient genomes, we analysed their DNA damage patterns, relationships with contemporary viruses, and their host associations. There were no significant similarities to known viruses for approximately half of the reconstructed genomes. This indicates that our understanding of human gut virome history is limited. Nevertheless, advancements in viral metagenomics and access to well-preserved aDNA samples hold promise for expanding our understanding of virosphere evolution.
Bacteriophage genomes are often variable — shaped by rampant mutations, recombinations, and horizontal gene transfers1,7. Therefore, recent reports of near-identical phage genomes found over vast geographical distances46 and spanning several years in the same location47 were striking. Here, we reconstructed a high-quality ancient genome sequence of Mushuvirus mushu that was highly conserved despite at least 1300 years of evolution and a presence on different continents.
We propose three hypotheses pertaining to the unique preservation of the Mushuvirus mushu genome. First, the high conservation may arise from its replication strategy. The phage displays genomic characteristics similar to transposable “Mu-like” phages. Typically, these phages are associated with extensive rearrangement of the host’s genetic material and highly variable genome termini that result from prophage excision (the Mu abbreviation refers to mutator). However, transposable bacteriophages like Mushuvirus mushu lack their own replicase enzyme, relying instead on the host’s polymerase III for DNA replication48. This enzyme tends to have error rates orders of magnitude lower than these typical phage polymerases. Notably, this hypothesis seems to be at odds with our estimations of intra-population genetic diversity of phages from the gut of modern hunter-gatherers that indicated a relatively high microdiversity of Mushuvirus mushu.
The second hypothesis relates to the phage’s extensive host range, encompassing multiple Oscillospiraceae species. These species are prevalent in human populations ranging from hunter-gatherers to industrialised societies37. Thus, unrestricted genetic drift has the potential to disrupt specific adaptations for individual hosts, but this risk is counteracted by purifying selection. This mechanism may help to create the evolutionary stasis of viruses in long-term host relationships49 and results in obscuring molecular dating of viral lineages based solely on modern genomes50.
The third, perhaps most likely explanation for the conservation of Mushuvirus mushu is that it has existed mostly as an integrated prophage, and hence part of its evolution was similar to that of the host genome. Indeed, the identity between modern genomes of Faecalibacteria and their ancient counterparts found in human palaeofaeces (95%-97% ANI retained after 1000–2000 years)17 is comparable to this observed of modern and ancient strains of Mushuvirus. Some prophages of sporulating bacteria represent relics of a bygone era, that survived and spread by persisting within endospores and re-emerging in a relatively unchanged form51. However, none of the predicted hosts of Mushuvirus mushu have been reported to form endospores. As these hypotheses are not mutually exclusive, the observed conservation of the Mushuvirus mushu genome might result from different combinations of factors like replication strategy, genetic constraints imposed by the broad host range, and/or dormancy of the virus. Finally, there might be other reasons that we have not yet considered.
Regardless of the specific mechanism underlying the high conservation of Mushuvirus mushu genome, recent studies are increasingly reporting ancient phages that bear a remarkable resemblance to their modern counterparts. For instance, a recent preprint study on 2-million-year-old microbial and viral communities in North Greenland discovered three phage genomes that had high damage patterns and showed average nucleotide identity exceeding 96% when compared to contemporary phages52.
To sum up, our study highlights the utility of publicly accessible ancient metagenomes in investigating viruses associated with microorganisms. Our focus was on sequences from well-preserved gut and palaeofaeces samples. However, much of the currently available data comes from ancient teeth or dental plaque, and these samples await further exploration. In the future, we anticipate that similar studies of virome in ancient metagenomes will contribute to elucidating the complex history of viruses and their role in co-evolving with bacterial, animal and plant hosts.
Methods
Sample selection
Based on community-curated metadata from the AncientMetagenomeDir53, we selected 72 aDNA metagenomic libraries from 30 well-preserved human faeces or digestive gut contents. Data from palaeofaeces primarily originated from archaeological excavations in caves located in Boomerang Shelter, Arid West Cave or Zape in the USA and Mexico (n = 38) as well as the underground salt mines of the Hallstatt in Austria (n = 19). Moreover, we used the digestive contents (n = 15) from a biopsy of the Tyrolean Iceman mummy discovered more than 30 years ago in a melting glacier19. The samples represented material from 150 to 5300 years ago. Details about original publications, localisations (coordinates), and samples (sequencing depth and instruments) are described in the supplementary materials (Supplementary Data 1).
Sequence data preprocessing
Pair-ended Illumina reads were trimmed using Cutadapt v.4.154 with a quality cutoff = 25, minimum read-length = 30, and a minimum overlap with adapter sequence to reads = 1. A total of 2,628,045,312 clean reads from all 72 libraries were mapped to the Homo sapiens reference genome (hg37) using KneadData v.0.12.0 (with --bypass-trim option) to filter out human DNA (https://github.com/biobakery/kneaddata). The quality of the 2,352,455,887 remaining reads after preprocessing was controlled in Fastqc v.0.12.055.
De novo contig assembly
Filtered reads from each library were assembled into contigs using Metaspades v.3.15.556 with default settings. Only contigs longer than 4000 nucleotides with a minimum coverage of 20 were considered in the downstream analyses (Fig. 1).
aDNA authentication
After preprocessing, clean reads were mapped to the assembled contigs using Bowtie2 v.2.4.457 with default settings. It was observed that these default settings did not significantly affect the alignment compared to the --sensitive settings, likely due to the low aDNA damage (Supplementary Fig. 7). The resulting alignment was sorted and indexed with SAMtools v.1.1458. DNA damage patterns observed in reads were used to label corresponding contigs as ancient or modern. The analysis was run using PyDamage v.0.7015 - a programme that calculates the frequency of C to T transitions at the first 20 positions of mapped reads compared to a reference sequence. The filtering threshold for predicted accuracy was determined by the Kneedle method59 and we imposed an additional cut-off of transition frequency (minimum 0.01 at the first position) to filter out contigs with weakly damaged reads that could introduce a random noise generated by the inherent error of the Illumina method. Additionally, the same analytical process of authentication was performed for 943 randomly selected contigs from the Metagenomic Gut Viruses (MGV) database31 to compare deamination patterns between modern and ancient viral genomes (Fig. 1c, d).
Viral contigs identification
Three machine learning tools were used to identify viral contigs. The first was Jaeger v.1.1.0, a deep-learning model that identifies phage genome sequences in metagenomes (https://github.com/Yasas1994/Jaeger) based on automatic compositional feature extraction. The second and third were VIBRANT v.1.2.124 and VirSorter2 v.2.2.325, which rely on analysing HMM profiles representing conserved families and/or domains similar to predicted proteins but applying different classifiers and reference databases. Jaeger and VIBRANT were run with default settings. For VirSorter2, we used the positional arguments ‘--include-groups dsDNAphage,NCLDV,ssDNA,lavidaviridae all’. Contigs classified as viral by at least two tools were further analysed.
Bacteriophage lifestyle prediction
Bacteriophage lifestyles were predicted based on the presence of similar prophages in bacterial genomes and lysogeny-associated protein domains. Prophages were defined as BLASTn v.2.13.0+ hits against UHGG collection45 and GTDB44 with minimum 50% coverage of the aMGV (only genomes longer than 5000 nucleotides were used as a query). To predict lifestyle based on domain content we used BACPHLIP v.0.9.660. We classified bacteriophages as temperate, if at least one method indicated this lifestyle, other genomes were considered as virulent.
Genomes quality assessment
CheckV v.1.0.161 was applied to assess the genome quality of ancient viral contigs using the ‘end_to_end’ command. Ancient viral genomes classified as complete, high-quality, medium-quality, or fragments longer than 20 kb and with at least one viral gene were considered in the next steps.
Viral contigs clustering
The ancient MGVs (n = 383) were clustered into 298 species-level viral operational taxonomic units (vOTUs) using scripts, published in the CheckV repository (https://bitbucket.org/berkeleylab/checkv/src/master/). Accordingly, sequences were grouped based on 95% ANI and 85% alignment fraction of the shorter sequence, as recommended in MIUViG (Minimum information about an uncultivated virus genome)26.
Gene-sharing network
We selected 10 modern phage genomes for each aMGV from the IMG/VR (v.4 - high-confidence genomes only) to compare ancient phages with their modern counterparts from different environments. Specifically, we selected genomes with the highest number of shared proteins determined by a DIAMOND v.2.0.1562 search in the ‘blastp’ mode (--very-sensitive) with a minimum of 50% query coverage and 50% sequence identity. We then visualised this collection of aMGVs, selected modern viral genomes, and all prokaryotic DNA viruses with assigned ICTV taxonomy (VMR_20-190822_MSL37.2, created 08/31/2022) using vContact2 v.0.11.328. The network was displayed in Cytoscape v.3.9.0 and refined in Inkscape v.1.2.2.
Comparison of aMGVs to contemporary bacteriophage genomes
The genomic sequences of 298 aMGVs were queried in the BLASTn searches against genomes of contemporary viruses from IMG/VR v.4, GenBank, and RefSeq. For each aMGV, we selected the top 30 contemporary virus genomes with the highest BLASTn alignment score and calculated ANI and AAI between the query aMGV and the selected genomes using VIRIDIC v.1.163 and EzAAI v.1.2.264, respectively. The contemporary phage with the highest ANI was identified as the closest known modern relative to each aMGV.
Host prediction
Four computational tools (BLASTn65, PHIST66, VirHostMatcher-Net67, and RaFAH68) were used to assign hosts to ancient phages and sequences from IMG/VR v.4 (only PHIST). PHIST v.1.1.0 and BLASTn v.2.13.0+ predictions were run against representative genomes of GTDB44 database v.07-RS207 (62,291 bacterial species + 3,412 archaeal species). Prokaryotic species whose genomes obtained the highest similarity score to the virus genome and had an e-value < 10−5 (BLASTn) or p-value < 10−5 (PHIST) were assigned as a putative host. For methods integrating machine learning approaches such as VirHostMatcher-Net v.1.0 or RaFAH v.0.1, we selected host predictions with, at minimum, 0.5 and 0.14 scores, respectively.
Taxonomy assignment, clustering, and phylogenetic analysis
Taxonomic assignment of viral genomes was performed in the geNomad v.1.3.3 tool32 using the ‘annotate’ function. To classify aMGVs at the genus and family level, viral genomes were clustered using a combination of gene sharing and AAI. Initially, we selected aMGVs assessed as high-quality or complete by CheckV and then added prokaryotic viruses from RefSeq (n = 4703; access: 30.01.2023) and IMG/VR sequences (n = 265) to form clusters (VC) with aMGVs (see: Methods, Gene-sharing-network). In this collection, pairwise protein sequence alignments were performed using DIAMOND62 with the options ‘-e-value 1 × 10 − 5–max-target-seqs 10000’. Next, we calculated the percentage of shared genes and AAI between each pair of viruses. Following the criteria from previous studies31,69, we kept connections between viruses with >20% AAI and >10% genes shared for the family level and >50% AAI and >20% genes shared for the genus level. Finally, clustering was performed based on the connections between viral genomes using MCL with the option ‘-I 1.2’ for the family level or ‘-I 2’ for the genus level. All scripts used to perform analyses at this step are available at (https://github.com/snayfach/MGV/blob/master/aai_cluster/README.md). To visualise the phylogenetic relationships of genus- and family-level groups, we generated a proteomic tree of 5017 viral sequences using ViPTreeGen v.1.1.370 and GraPhlAn v.1.1.371.
Analyses of the Mushuvirus mushu genome
We performed a BLASTn search of 298 ancient metagenomic viral sequences against the nr/nt NCBI database. This search revealed that one identified aMGV (NODE_310_length_36983_cov_28.516681) was remarkably similar to present-day Mushuvirus mushu (NC_047913.1). This genome was present in a vContact2 cluster, along with one reference from NCBI (contemporary genome of Mushuvirus mushu) and 10 vOTUs from IMG/VR v.4 (high-confidence genomes only). All 12 sequences from the vContact2 cluster were aligned using MAFFT v.7.308 to differentiate the core of the phage genome (~36,623 bp) from the flanking regions coming from host integration sites (~241 bp and ~119 bp in the ancient contig). Only those core sequences were used in further analyses. To assess the nucleotide variability of modern and ancient Mushuvirus mushu sequences, we calculated the intergenomic similarity within the cluster using the VIRIDIC63 with the following parameters: ‘-word_size 7 -reward 2 -penalty −3 -gapopen 5 -gapextend 2’. A phylogenetic tree (Fig. 4d) was constructed with the obtained similarity matrix using the bioNJ algorithm with default parameters72. To annotate protein-coding genes in the analysed genomes, we used an end-to-end script (https://github.com/Yasas1994/phage_contig_annotator) that annotates phage genes based on the HMMER v.3.3.2 search against Prokaryotic Virus Remote Homologous Groups73. Visualisation and manual curation of the genome were conducted in Geneious Prime v.2023.04 (Fig. 4c). The MAFFT plugin for the same tool was used to generate multiple sequence alignments of entire viral genomes. The identity of protein-coding genes at the amino acid level was calculated based on local alignment performed using EMBOSS v.6.6.0.074. To calculate SNVs in the ancient genome, a Python script was used (https://github.com/pinbo/msa2snp). We detected template and variable regions of DGRs using myDGR38. Finally, we assigned potential hosts by searching for sequences similar to the prophage core and flanking regions (blastn -task megablast) in the UHGG collection45 and GTDB44. We measured the population microdiversity of Mushuvirus mushu in modern hunter gathers’ gut (Fig. 4e) by mapping raw sequences to the complete bacteriophage genomes from samples where Mushuvirus mushu was previously detected37 using Bowtie2 with default settings. Next, we calculated average nucleotide diversity and average divergent sites using InStrain v.1.8.036.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Ancient phage genome sequences and their gene annotations generated in this study have been deposited in the Zenodo database [https://doi.org/10.5281/zenodo.7919433]. The reconstructed ancient genome sequence of Mushuvirus mushu is available from NCBI GenBank under accession BK063464. Supporting data generated in this study are provided in the Supplementary Information, Source Data and Supplementary Data files. Accession numbers of ancient metagenomic samples used in this study are available in the AncientMetagenomeDir [https://github.com/SPAAM-community/AncientMetagenomeDir]. Other databases used in the study include: IMG/VR v.4 [https://genome.jgi.doe.gov/portal/IMG_VR/], GTDB v.07-RS207 [https://data.ace.uq.edu.au/public/gtdb/data/releases/release207/], UHGG v.2.0 [http://ftp.ebi.ac.uk/pub/databases/metagenomics/mgnify_genomes/human-gut/v2.0/], NCBI GenBank release 251 [https://www.ncbi.nlm.nih.gov/genbank/] and NCBI RefSeq release 215 [https://www.ncbi.nlm.nih.gov/refseq/], PHROG v.4 [https://phrogs.lmge.uca.fr], and Virus Metadata Resource release 12/02/2022 from ICTV [https://ictv.global/vmr]. Source data are provided with this paper.
Code availability
Bioinformatic scripts and a guide to the data analysis performed in this study are provided in the GitHub repository (https://github.com/rozwalak/Ancient_gut_phages)75.
References
Hatfull, G. F. & Hendrix, R. W. Bacteriophages and their genomes. Curr. Opin. Virol. 1, 298–303 (2011).
Roux, S., Matthijnssens, J. & Dutilh, B. E. Encyclopedia of Virology. 133–140 (Elsevier, 2021).
Pappas, N. et al. Encyclopedia of Virology. 124–132 (Elsevier, 2021).
Call, L., Nayfach, S. & Kyrpides, N. C. Illuminating the virosphere through global metagenomics. Annu Rev. Biomed. Data Sci. 4, 369–391 (2021).
Edwards, R. A. et al. Global phylogeography and ancient evolution of the widespread human gut virus crAssphage. Nat. Microbiol 4, 1727–1736 (2019).
Minot, S. et al. Rapid evolution of the human gut virome. Proc. Natl Acad. Sci. USA 110, 12450–12455 (2013).
Kupczok, A. et al. Rates of mutation and recombination in siphoviridae phage genome evolution over three decades. Mol. Biol. Evol. 35, 1147–1159 (2018).
Spyrou, M. A., Bos, K. I., Herbig, A. & Krause, J. Ancient pathogen genomics as an emerging tool for infectious disease research. Nat. Rev. Genet. 20, 323–340 (2019).
Barreat, J. G. N. & Katzourakis, A. Paleovirology of the DNA viruses of eukaryotes. Trends Microbiol. 30, 281–292 (2022).
Alempic, J.-M. et al. An update on eukaryotic viruses revived from ancient permafrost. Viruses 15, 564 (2023).
Nishimura, L., Fujito, N., Sugimoto, R. & Inoue, I. Detection of ancient viruses and long-term viral evolution. Viruses 14, 1336 (2022).
Appelt, S. et al. Viruses in a 14th-century coprolite. Appl. Environ. Microbiol. 80, 2648–2655 (2014).
Santiago-Rodriguez, T. M. et al. Natural mummification of the human gut preserves bacteriophage DNA. FEMS Microbiol. Lett. 363, fnv219 (2016).
Eisenhofer, R. & Weyrich, L. Proper authentication of ancient DNA is still essential. Genes (Basel) 9, 122 (2018).
Borry, M., Hübner, A., Rohrlach, A. B. & Warinner, C. PyDamage: automated ancient damage identification and estimation for contigs in ancient DNA de novo assembly. PeerJ 9, e11845 (2021).
Nishimura, L. et al. Identification of ancient viruses from metagenomic data of the Jomon people. J. Hum. Genet. 66, 287–296 (2021).
Wibowo, M. C. et al. Reconstruction of ancient microbial genomes from the human gut. Nature 594, 234–239 (2021).
Klapper, M. et al. Natural products from reconstructed bacterial genomes of the Middle and Upper Paleolithic. Science (1979) 380, 619–624 (2023).
Maixner, F. et al. The 5300-year-old Helicobacter pylori genome of the Iceman. Science (1979) 351, 162–165 (2016).
Tett, A. et al. The prevotella copri complex comprises four distinct clades underrepresented in Westernized populations. Cell Host Microbe 26, 666–679.e7 (2019).
Hagan, R. W. et al. Comparison of extraction methods for recovering ancient microbial DNA from paleofeces. Am. J. Phys. Anthropol. 171, 275–284 (2020).
Borry, M. et al. CoproID predicts the source of coprolites and paleofeces using microbiome composition and host DNA content. PeerJ 8, e9001 (2020).
Maixner, F. et al. Hallstatt miners consumed blue cheese and beer during the Iron Age and retained a non-Westernized gut microbiome until the Baroque period. Curr. Biol. 31, 5149–5162.e6 (2021).
Kieft, K., Zhou, Z. & Anantharaman, K. VIBRANT: automated recovery, annotation and curation of microbial viruses, and evaluation of viral community function from genomic sequences. Microbiome 8, 90 (2020).
Guo, J. et al. VirSorter2: a multi-classifier, expert-guided approach to detect diverse DNA and RNA viruses. Microbiome 9, 37 (2021).
Roux, S. et al. Minimum Information about an Uncultivated Virus Genome (MIUViG). Nat. Biotechnol. 37, 29–37 (2019).
Der Sarkissian, C. et al. Ancient metagenomic studies: considerations for the wider scientific community. mSystems 6, e0131521 (2021).
Bin Jang, H. et al. Taxonomic assignment of uncultivated prokaryotic virus genomes is enabled by gene-sharing networks. Nat. Biotechnol. 37, 632–639 (2019).
Camargo, A. P. et al. IMG/VR v4: an expanded database of uncultivated virus genomes within a framework of extensive functional, taxonomic, and ecological metadata. Nucleic Acids Res. 51, D733–D743 (2023).
Walker, P. J. et al. Recent changes to virus taxonomy ratified by the International Committee on Taxonomy of Viruses (2022). Arch. Virol. 167, 2429–2440 (2022).
Nayfach, S. et al. Metagenomic compendium of 189,680 DNA viruses from the human gut microbiome. Nat. Microbiol 6, 960–970 (2021).
Camargo, A. P. et al. Identification of mobile genetic elements with geNomad. Nat. Biotechnol. https://doi.org/10.1038/s41587-023-01953-y (2023).
Cornuault, J. K. et al. Phages infecting Faecalibacterium prausnitzii belong to novel viral genera that help to decipher intestinal viromes. Microbiome 6, 65 (2018).
Juge, N. Relationship between mucosa-associated gut microbiota and human diseases. Biochem. Soc. Trans. 50, 1225–1236 (2022).
Kupczok, A. & Dagan, T. Rates of molecular evolution in a marine synechococcus phage lineage. Viruses 11, 720 (2019).
Olm, M. R. et al. inStrain profiles population microdiversity from metagenomic data and sensitively detects shared microbial strains. Nat. Biotechnol. 39, 727–736 (2021).
Carter, M. M. et al. Ultra-deep sequencing of Hadza hunter-gatherers recovers vanishing gut microbes. Cell 186, 3111–3124.e13 (2023).
Sharifi, F. & Ye, Y. MyDGR: a server for identification and characterization of diversity-generating retroelements. Nucleic Acids Res. 47, W289–W294 (2019).
Doulatov, S. et al. Tropism switching in Bordetella bacteriophage defines a family of diversity-generating retroelements. Nature 431, 476–481 (2004).
Roux, S. et al. Ecology and molecular targets of hypermutation in the global microbiome. Nat. Commun. 12, 3076 (2021).
Auger, S. et al. Gene co-expression network analysis of the human gut commensal bacterium Faecalibacterium prausnitzii in R-Shiny. PLoS ONE 17, e0271847 (2022).
Barr, J. J. et al. Bacteriophage adhering to mucus provide a non–host-derived immunity. Proc. Natl Acad. Sci. USA 110, 10771–10776 (2013).
Barr, J. J. et al. Subdiffusive motion of bacteriophage in mucosal surfaces increases the frequency of bacterial encounters. Proc. Natl Acad. Sci. USA 112, 13675–13680 (2015).
Parks, D. H. et al. GTDB: an ongoing census of bacterial and archaeal diversity through a phylogenetically consistent, rank normalized and complete genome-based taxonomy. Nucleic Acids Res. 50, D785–D794 (2022).
Almeida, A. et al. A unified catalog of 204,938 reference genomes from the human gut microbiome. Nat. Biotechnol. 39, 105–114 (2021).
Bellas, C. M., Schroeder, D. C., Edwards, A., Barker, G. & Anesio, A. M. Flexible genes establish widespread bacteriophage pan-genomes in cryoconite hole ecosystems. Nat. Commun. 11, 4403 (2020).
Marston, M. F. & Martiny, J. B. H. Genomic diversification of marine cyanophages into stable ecotypes. Environ. Microbiol. 18, 4240–4253 (2016).
Jang, S. & Harshey, R. M. Repair of transposable phage Mu DNA insertions begins only when the E. coli replisome collides with the transpososome. Mol. Microbiol. 97, 746–758 (2015).
Simmonds, P., Aiewsakun, P. & Katzourakis, A. Prisoners of war — host adaptation and its constraints on virus evolution. Nat. Rev. Microbiol. 17, 321–328 (2019).
Wertheim, J. O. & Kosakovsky Pond, S. L. Purifying selection can obscure the ancient age of viral lineages. Mol. Biol. Evol. 28, 3355–3365 (2011).
Weller, C. & Wu, M. A generation-time effect on the rate of molecular evolution in bacteria. Evolution 69, 643–652 (2015).
Fernandez-Guerra, A. et al. A 2-million-year-old microbial and viral communities from the Kap København Formation in North Greenland. bioRxiv https://doi.org/10.1101/2023.06.10.544454 (2023).
Fellows Yates, J. A. et al. Community-curated and standardised metadata of published ancient metagenomic samples with AncientMetagenomeDir. Sci. Data 8, 31 (2021).
Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 17, 10 (2011).
Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data (2010).
Nurk, S., Meleshko, D., Korobeynikov, A. & Pevzner, P. A. metaSPAdes: a new versatile metagenomic assembler. Genome Res. 27, 824–834 (2017).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012).
Li, H. et al. The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Satopaa, V., Albrecht, J., Irwin, D. & Raghavan, B. Finding a ‘Kneedle’ in a Haystack: Detecting Knee Points in System Behavior. In 2011 31st International Conference on Distributed Computing Systems Workshops. 166–171 (IEEE, 2011).
Hockenberry, A. J. & Wilke, C. O. BACPHLIP: predicting bacteriophage lifestyle from conserved protein domains. PeerJ 9, e11396 (2021).
Nayfach, S. et al. CheckV assesses the quality and completeness of metagenome-assembled viral genomes. Nat. Biotechnol. 39, 578–585 (2021).
Buchfink, B., Reuter, K. & Drost, H.-G. Sensitive protein alignments at tree-of-life scale using DIAMOND. Nat. Methods 18, 366–368 (2021).
Moraru, C., Varsani, A. & Kropinski, A. M. VIRIDIC—A Novel Tool to Calculate the Intergenomic Similarities of Prokaryote-Infecting viruses. Viruses 12, 1268 (2020).
Kim, D., Park, S. & Chun, J. Introducing EzAAI: a pipeline for high throughput calculations of prokaryotic average amino acid identity. J. Microbiol. 59, 476–480 (2021).
Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. Basic local alignment search tool. J. Mol. Biol. 215, 403–410 (1990).
Zielezinski, A., Deorowicz, S. & Gudyś, A. PHIST: fast and accurate prediction of prokaryotic hosts from metagenomic viral sequences. Bioinformatics 38, 1447–1449 (2022).
Wang, W. et al. A network-based integrated framework for predicting virus–prokaryote interactions. NAR Genom. Bioinform. 2, 1–19 (2020).
Coutinho, F. H. et al. RaFAH: host prediction for viruses of Bacteria and Archaea based on protein content. Patterns 2, 100274 (2021).
Li, S. et al. A catalog of 48,425 nonredundant viruses from oral metagenomes expands the horizon of the human oral virome. iScience 25, 104418 (2022).
Nishimura, Y. et al. ViPTree: the viral proteomic tree server. Bioinformatics 33, 2379–2380 (2017).
Asnicar, F., Weingart, G., Tickle, T. L., Huttenhower, C. & Segata, N. Compact graphical representation of phylogenetic data and metadata with GraPhlAn. PeerJ 3, e1029 (2015).
Gascuel, O. BIONJ: an improved version of the NJ algorithm based on a simple model of sequence data. Mol. Biol. Evol. 14, 685–695 (1997).
Terzian, P. et al. PHROG: families of prokaryotic virus proteins clustered using remote homology. NAR Genom. Bioinform. 3, lqab067 (2021).
Rice, P., Longden, I. & Bleasby, A. EMBOSS: the european molecular biology open software suite. Trends Genet. 16, 276–277 (2000).
Rozwalak, P., Barylski, J., Wijesekara, Y., Dutilh, B. E. & Zielezinski, A. Ultraconserved bacteriophage genome sequence identified in 1300-year-old human palaeofaeces. GitHub https://doi.org/10.5281/zenodo.10224491 (2023).
O’Leary, N. A. et al. Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 44, D733–D745 (2016).
Acknowledgements
This work was funded by the Polish Ministry of Science and Higher Education under the programme “Perły Nauki”, project number PN/01/0063/2022. The total value of the project 228,448PLN was awarded to P.R.; A.Z is supported by Polish National Science Centre [2018/31/D/NZ2/00108], and B.E.D. is supported by the European Research Council (ERC) Consolidator grant 865694: DiversiPHI, the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany ́s Excellence Strategy – EXC 2051 – Project-ID 390713860, the Alexander von Humboldt Foundation in the context of an Alexander von Humboldt-Professorship founded by the German Federal Ministry of Education and Research. B.E.D and Y.W. are supported by the European Union’s Horizon 2020 research and innovation program, under the Marie Skłodowska-Curie Actions Innovative Training Networks grant agreement no. 955974 (VIROINF). The computations were performed at the PLGrid Infrastructure and the Poznan Supercomputing and Networking Center (grant pl0074-02 and grant pl0243-01).
Author information
Authors and Affiliations
Contributions
P.R. conceived the idea of studying ancient phages; P.R., A.Z., J.B. and B.E.D. designed the research; P.R. and A.Z. selected samples and performed preprocessing, assembly and aDNA authentication; J.B. and Y.W. performed identification of viral contigs; P.R. and A.Z. created a gene-sharing network and performed host prediction; P.R., A.Z. and J.B. performed taxonomy assignment, clustering, phylogenetic studies, and the analyses of the Mushuvirus mushu genome; Y.W. contributed to phage genome annotation; P.R., A.Z., J.B., and B.E.D. analysed the results. P.R. wrote the manuscript with substantial contributions from A.Z., J.B. and B.E.D. All authors reviewed and approved the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Christopher Bellas and the other, anonymous, reviewers for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Rozwalak, P., Barylski, J., Wijesekara, Y. et al. Ultraconserved bacteriophage genome sequence identified in 1300-year-old human palaeofaeces. Nat Commun 15, 495 (2024). https://doi.org/10.1038/s41467-023-44370-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-023-44370-0
- Springer Nature Limited