

Abstract
The demands for cost-effective solar fuels have triggered extensive research in artificial photosynthesis, yet the efforts in designing high-performance particulate photocatalysts are largely impeded by inefficient charge separation. Because charge separation in a particulate photocatalyst is driven by asymmetric interfacial energetics between its reduction and oxidation sites, enhancing this process demands nanoscale tuning of interfacial energetics on the prerequisite of not impairing the kinetics and selectivity for surface reactions. In this study, we realize this target with a general strategy involving the application of a core/shell type cocatalyst that is demonstrated on various photocatalytic systems. The promising H2O2 generation efficiency validate our perspective on tuning interfacial energetics for enhanced charge separation and photosynthesis performance. Particularly, this strategy is highlighted on a BiVO4 system for overall H2O2 photosynthesis with a solar-to-H2O2 conversion of 0.73%.
Similar content being viewed by others


Introduction
Solar energy is deemed as a key solution to the increasing global demands on clean energy and related climate issues1,2,3. To tackle the spatiotemporal fluctuations of solar radiation, a promising strategy is to capture solar energy in storable and transportable solar fuels (e.g., H2 and H2O2) by artificial photosynthesis4,5. Among primary photosynthetic systems, the particulate photocatalyst is considered as the most cost-effective one because of its high simplicity and scalability, yet its energy conversion efficiency still needs improving for viable applications6,7.
In pursuit of high-performance particulate photocatalysts, a key challenge is to improve charge separation8. Enhancing charge separation demands exquisitely tuning interfacial energetics between reduction and oxidation sites, and in the meantime not impairing the kinetics and selectivity for surface reactions. For instance, most particulate photocatalysts are n-type semiconductors with a Fermi level close to their conduction band, and the photocatalyst/cocatalyst interfaces typically form Schottky junctions, with upward band bending on both the reduction and oxidation sites (Fig. S1a)9. The Schottky junction at the oxidation sites facilitates hole migration to the surface reaction sites, favoring charge separation. In contrast, the Schottky junction at the reduction sites impedes electron migration and traps holes, undermining charge separation. Further, it reduces the band offset potential between the reduction and oxidation sites (ΔV), the fundamental driving force for charge separation (Fig. S1a)10,11. Such detrimental effects of a high Schottky barrier at the reduction sites have been verified in the previous studies (e.g., Pd-loaded BiVO4 for H2O2 generation12, and Pt-loaded GaN or LaTiO2N for H2 evolution13,14). Therefore, the Schottky barrier at reduction sites is a major hurdle limiting charge separation and further artificial photosynthesis performance. Yet, how to lower the Schottky barrier at reduction sites remains a critical challenge for particulate photocatalysts (Fig. S1b).
To tune interfacial energetics while preserving the kinetics and selectivity of surface reactions, a possible strategy is to apply a binary cocatalyst composed of two parts for the respective function. Such a strategy has been applied in the photoelectrochemical water splitting systems15,16. For example, Esposito et al. applied a Ti/Pt double-layer cocatalyst on a p-Si photocathode for enhancing H2 evolution. In this system, the Ti layer formed a desirable junction with p-type Si for charge separation, while the Pt layer on the Ti layer efficiently catalyzed H2 evolution16. However, applying this strategy on a particulate photocatalyst is challenged by its complicated configuration where reduction and oxidation sites are assembled on the same surface with a nanoscale distance. Under such an intricate condition, the energetics at reduction sites need to be tuned selectively and locally without affecting those at the oxidation sites, otherwise the overall charge separation will be affected. These challenges have impeded the practice of interfacial energetics tuning for enhancing charge separation in a particulate photocatalyst.
Here, we report the interfacial energetics tuning in a particulate photocatalyst for photosynthesis of H2O2, an emerging liquid solar fuel substitute for gaseous H217. Organic semiconductors have been developed for photocatalytic H2O2 generation with reported solar-to-H2O2 conversion (STH) up to 1.5%18,19,20,21. Yet, organic materials have a potential stability concern because photocatalytic H2O2 generation is inevitably accompanied by hydroxyl radical (•OH) generation (H2O2 + hv → 2•OH or H2O2 + e− + H+ → •OH + H2O), which can damage organic structures22,23. Our group recently developed an inorganic BiVO4 photosynthesis system with a STH of 0.29%. The apparent quantum yield (AQY) of the system is 5.8% at 420 nm, indicating that majority of the charge carriers recombined during reaction and charge separation needs further tuned for higher photocatalytic performance12. In this study, we constructed a Ag/Pd binary cocatalyst with core/shell structure on the {010} reduction facets of BiVO4. BiVO4 is a visible-light-responsive photocatalyst promising for long-term H2O2 generation because it is resistive to •OH (a potent oxidant inevitably formed during H2O2 generation) compared to the organic counterparts24,25,26. The Pd shell served to steer the oxygen-reduction pathway to two-electron process for H2O2 synthesis27,28,29, while the Ag core served to lower the Schottky barrier between BiVO4 and Pd. In-depth time-resolved spectroscopic investigations and electrical numerical simulations on photogenerated charge carriers demonstrate that Ag core significantly lowered the Schottky barrier at the {110} reduction facets and enhanced charge separation in BiVO4. Without using any sacrificial agent, the rationally designed BiVO4 photocatalyst achieved a new record among inorganic photocatalysts (Table S1). Even comparing with the sacrificial system, CoOx/BiVO4/(Ag/Pd) has a comparable H2O2 production yield (Table S1). The feasibility of core/shell cocatalyst construction for interfacial-energetics-tuning was further demonstrated on C3N4 and TiO2, demonstrating its high generality for enhancing charge separation and performance in photosynthesis systems.
Results and discussion
Interfacial energetics tuning through applying core/shell type cocatalyst
We first synthesized faceted BiVO4 particles using a solid-liquid-reaction method. The X-ray diffraction (XRD) pattern of BiVO4 particles matched well with that of monoclinic BiVO4, with {010} and {110} facet peaks located at 30.6°and 18.7°, respectively (Fig. S2). Scanning electron microscope (SEM) images (Fig. S3) show that the BiVO4 particles exhibit a decahedron structure with clear facets. Following previous studies12,30, the top/bottom and side facets are assigned to {010} reduction facets and {110} oxidation facets, respectively.
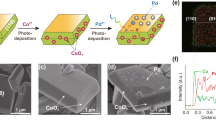
We then loaded reductive and oxidative cocatalysts onto the corresponding facets of BiVO4 via stepwise photodeposition. CoOx served to enhance water oxidation and was deposited onto {110} facets via photooxidation of Co2+ ions (CoOx/BiVO4, Fig. 1a). SEM image shows that CoOx particles are loaded on the surface of BiVO4 (Fig. S4a). Energy-dispersive X-ray spectroscopy (EDS) elemental mapping and line profile indicate that Co signal on {110} facets is much higher than that on {110} facets, confirming the selective loading of CoOx on the {110} facets (Fig. 1b). The particles exhibit prominent Co 2p X-ray photoelectron spectroscopy (XPS) peaks (Fig. S5). The Co 2p3/2 peak can be deconvoluted to a Co2+ peak at 781.6 eV and a Co3+ peak at 780.6 eV, suggesting that the valence of Co was in-between +2 and +3 and therefore it can be denoted as CoOx.

a Stepwise and facet-selective photodeposition of Co, Ag, and Pd on BiVO4. b Energy-dispersive X-ray spectroscopy (EDS) elemental mapping and line profile along with the white arrow of CoOx/BiVO4/(Ag/Pd). We increased Co (2 wt%), Ag (1 wt%), and Pd (1 wt%) loadings for more clear observation. c, d Scanning transmission electron microscopy (STEM)-EDS elemental mapping of Ag/Pd particles loaded on BiVO4. We increased Ag (1 wt%) and Pd (1 wt%) loadings for more clear observation. e Ultraviolet photoelectron spectroscopy (UPS) spectra of BiVO4, BiVO4/Ag, BiVO4/Pd, and BiVO4/(Ag/Pd). f Schematic illustration of {010} reduction facet interfacial energetics tuning through Ag/Pd core/shell cocatalyst construction on BiVO4.
Selective deposition of Ag and Pd on the {010} facets of BiVO4 was performed via stepwise photoreduction of Ag+ and PdCl42− to form a core/shell structure (CoOx/BiVO4/(Ag/Pd), Fig. 1a). SEM images clearly show Ag (Fig. S4b) and Ag/Pd nanoparticles on the {010} facets (Fig. S4c, d). Prominent Ag 3d and Pd 3d XPS peaks demonstrate the successful loading of metallic Ag/Pd cocatalysts, with the Ag 3d peaks at 368.1 eV and 374.2 eV assigned to the Ag 3d5/2 and Ag 3d3/2, respectively (Fig. S6), and the Pd 3d peaks at 335.1 eV and 340.6 eV assigned to Pd 3d5/2 and Pd 3d3/2, respectively (Fig. S7). The minor Pd2+ peak at 337.0 eV is attributed to PdO due to the partial oxidation of Pd (Fig. S7).
Consistent with elemental mapping observations, line profile confirms the facet-selective loading of cocatalysts, with 3.0-fold stronger Co signals on the {110} facets compared to those on the {010} facets, as well as 6.1- and 3.3-fold stronger Ag and Pd signals on the {010} facets compared to those on the {110} facets (Fig. 1b). Notably, the Ag and Pd signals in line profile overlap in distance (Fig. 1b), indicating the co-loading of Ag and Pd nanoparticles on the same sites. The facet-selective loading of Co, Ag and Pd was further confirmed by STEM-EDS mapping from top view and side view of CoOx/BiVO4/Ag (Fig. S8) and CoOx/BiVO4/Pd (Fig. S9).
We further analyzed the structure of the Ag/Pd nanoparticles by scanning transmission electron microscopy-energy dispersive spectrometer (STEM-EDS). Top view element mapping images show a great overlap between Ag and Pd, with the distribution area of Pd slightly larger than Ag, indicating the formation of core (Ag)/shell (Pd) structure (Fig. 1c). Such a structure is further displayed by a distinct Pd shell slightly larger than Ag core as shown in the side view images (Fig. 1d). These results demonstrate the core/shell structure of the Ag/Pd cocatalyst, formed via selective deposition of Pd on Ag nanoparticles where photoexcited electrons accumulated (Fig. 1a).
The effectiveness of loading Ag on tuning interfacial energetics was investigated by comparing the overall Schottky barrier heights of BiVO4, BiVO4/Pd, BiVO4/Ag, and BiVO4/(Ag/Pd) using ultraviolet photoelectron spectroscopy (UPS). Here, the overall Schottky barrier heights are quantified by the distance between valence band and the Fermi level position, (Ev − Ef)31. The (Ev − Ef) of BiVO4, BiVO4/Ag, BiVO4/Pd, and BiVO4/(Ag/Pd) were determined to be 2.22, 2.35, 2.25 and 2.36 eV, respectively (Fig. 1e). Since Pd has a high work function (5.6 eV), BiVO4/Pd built up a high Schottky barrier. In comparison, BiVO4/Ag exhibits a lower Schottky barrier attributed to the low work function of Ag (4.3 eV). Such a low Schottky barrier was maintained on BiVO4/(Ag/Pd) with a direct contact between Ag and BiVO4 (Fig. 1f). Consistent with UPS results, the binding energies of Bi and V in XPS spectra follow the order of BiVO4/Ag <BiVO4/Pd <BiVO4 (Fig. S10), indicating that Ag on {010} facets reduced excitation energy of BiVO4 valence electrons by lowering the Schottky barrier. Notably, both the UPS and XPS analyses are based on overall energetics of BiVO4 rather than the local one at the {010} facets. Therefore, the impact of Ag on the Schottky barrier at {010} facets should be even higher than UPS observations (confirmed by electrical simulation results, see below). While it is desirable to directly compare the local Schottky barriers at {010} facets with recently developed potential-sensing electrochemical atomic force microscopy in Boettcher group, its application on a particulate photocatalyst is still under progress32,33.
Enhanced photocatalytic H2O2 generation with interfacial energetics tuning
We first compare the photocatalytic H2O2 generation performance of different particulate photocatalysts under visible light irradiation (λ > 400 nm) in pure water. The performance of CoOx/BiVO4/(Ag/Pd) was optimized by adjusting the loading amount of Co, Ag, and Pd to 0.3 wt%, 0.08 wt%, 0.4 wt%, respectively (Fig. S11–13). Pd loading (i.e., BiVO4/Pd) enhanced H2O2 generation by 7.9-fold compared to bare BiVO4 (Fig. 2a), attributing to promoted selectivity (Fig. 2b). In the meanwhile, loading Co onto BiVO4/Pd (i.e., CoOx/BiVO4/Pd) improved water oxidation efficiency, resulting in a 3.9-fold enhancement of 20-min H2O2 generation (245 μM). Most importantly, with successful tuning of interfacial energetics, CoOx/BiVO4/(Ag/Pd) enhanced the H2O2 generation by 2.1-fold (520 μM, Fig. 2a). Interestingly, when the core/shell structure was reversed (i.e., Pd core and Ag shell, denoted as CoOx/BiVO4/(Pd/Ag)), the photocatalytic H2O2 generation rate was much decreased, even lower than that of CoOx/BiVO4/Pd (Fig. 2a). This result demonstrates that forming a BiVO4/Ag junction at {010} reduction sites instead of random Ag loading is key to improve photocatalytic H2O2 generation.

a Time courses of photocatalytic H2O2 generation. Reaction conditions: photocatalyst, 1 mg/mL; 50 ml DI water saturated with O2; light source, LED visible light, 300 mW cm−2, λ > 400 nm. b Selectivity of H2O2 production for BiVO4, BiVO4/Ag, BiVO4/Pd, and BiVO4/(Ag/Pd). Reaction conditions: photocatalyst, 1 mg/ml; 50 mL DI water with 0.1 M H3BO3 and 0.075 M ScCl3 saturated with O2 (pH 6.8), 10 v/v% methanol as electron donor; light source, LED visible light, 100 mW/cm2, λ > 400 nm. H2O2 selectivity is defined as the ratio of electrons utilized for H2O2 synthesis to the total number of electrons consumed (i.e., electrons donated by methanol). c Preparation of various core/shell cocatalyst and correlation between H2O2 photosynthesis performance and core junction metal work function. Reaction conditions: photocatalyst, 1 mg/ml; 50 mL DI water saturated with O2 (pH 6.8); light source, LED visible light, 300 mW/cm2, λ > 400 nm; reaction time, 20 min. d Decay in H2O2 photosynthesis activity of CoOx/BiVO4/(Ag/Pd) and C3N4/Pd under •OH-rich conditions. Aging conditions: photocatalyst, 1 mg/mL; 50 ml DI water with 25 mM H2O2; light source, 254 nm ultraviolet radiation light; •OH was generated via the reaction H2O2 + hν → 2•OH. e Repetitive use of CoOx/BiVO4/(Ag/Pd) for H2O2 photosynthesis. Reaction conditions: photocatalyst amount, 1 mg/ml; 50 ml DI water saturated with O2; light source, LED visible light, 300 mW cm−2, λ > 400 nm. f Time courses of photocatalytic H2O2 generation over CoOx/Mo:BiVO4/(Ag/Pd) and the corresponding STH efficiency. Reaction conditions: photocatalyst, 10 mg; photocatalyst, 1 mg/ml; 10 mL DI water with 0.1 M H3BO3 and 0.075 M ScCl3 saturated with O2 (pH 6.8); light source, xenon lamp solar simulator, 100 mW cm−2, AM 1.5 G; irradiation area, 4.5 cm−2. g Apparent quantum yield (AQY) of H2O2 photosynthesis over CoOx/Mo:BiVO4/(Ag/Pd) as a function of the incident light wavelength. Reaction conditions: photocatalyst, 10 mg; photocatalyst, 1 mg/ml; 10 mL DI water with 0.1 M H3BO3 and 0.075 M ScCl3 saturated with O2 (pH 6.8); light source, monochromatic LED light.
The construction of BiVO4/Ag junction did not impair the surface reaction selectivity. For instance, as compared to the low selectivity of BiVO4/Ag towards H2O2 generation (13 %), the H2O2 generation selectivity of BiVO4/(Ag/Pd) (81 %) was similar to that of BiVO4/Pd (85 %, Fig. 2b), suggesting that the Pd shell fully covered Ag core and acted as the dominant H2O2 generation sites. H2O2 production on CoOx/BiVO4/(Pd/Ag) proceeded via a two-electron oxygen-reduction path, as demonstrated by halted H2O2 production in the presence of IO3¯ as electron scavenger or under N2-purged condition and enhanced H2O2 production when methanol as hole scavenger was added (Fig. S14).
To further show the effectiveness of tuning interfacial energetics on improving photosynthesis performance, we constructed Au/Pd and Rh/Pd core/shell structure cocatalysts and examined their H2O2 generation activity with BiVO4 (Fig. 2c). The work functions of Au (5.1 eV) and Rh (5.0 eV) were higher than Ag (4.3 eV), yet lower than Pd (5.6 eV). Therefore, Au and Rh in theory should also be capable of lowering the Schottky barrier on {010} facets, yet to a lower extent than Ag. The photocatalytic H2O2 generation shows a distinct negative correlation with metal work function (Fig. 2c), further demonstrating the validity of lowering Schottky barrier for enhanced charge separation and photosynthesis performance.
CoOx/BiVO4/(Ag/Pd) exhibits high chemical stability during H2O2 generation. After 24-h incubation in •OH-rich condition, CoOx/BiVO4/(Ag/Pd) displayed nominal decay in H2O2 production performance (Fig. 2d). In stark contrast, C3N4/Pd as a representative organic photocatalyst was highly susceptible to •OH, with ~80% decay in H2O2 production performance after 24-h incubation (Fig. 2d). Because photocatalytic H2O2 generation is inevitably accompanied by •OH generation (H2O2 + hν → 2 •OH or H2O2 + e¯ + H+ → •OH + H2O), such resistance to •OH as shown by CoOx/BiVO4/(Ag/Pd) is pivotal for long-term H2O2 photosynthesis. The high stability of CoOx/BiVO4/(Ag/Pd) was further demonstrated by its stable catalytic performance over repetitive use up to five cycles (Fig. 2e). Notably, the photocatalytic H2O2 generation over CoOx/BiVO4/(Ag/Pd) decayed over time in each cycle (Fig. 2e). This issue has been noticed in the previous studies on photocatalytic H2O2 generation12, which is caused by H2O2 self-decomposition instead of photocatalyst inactivation and can be solved by adding H2O2 stabilizers such as Sc3+ (see data below)34,35.
With exquisite tuning of interfacial energetics and surface reactions, BiVO4 exhibits a high overall H2O2 photosynthesis efficiency. Here we applied Mo-doped BiVO4 (Mo:BiVO4) for further photocatalytic performance evaluation because doping the V5+ sites of BiVO4 with Mo6+ can increase its bulk conductivity36,37. Without any sacrificial reagent, CoOx/Mo:BiVO4/(Ag/Pd) produced 9.7 mM H2O2 in one hour, corresponding to an AQY of 3.0% and a STH of 0.73% at full spectrum (Fig. 2f). Such an efficiency leveraged the record for inorganic semiconductors by 252% and is comparable with the efficiencies of most organic semiconductors (Table S1). This H2O2 production performance was confirmed to be reproducible with three batches of CoOx/Mo:BiVO4/(Ag/Pd) (Fig. S15). The wavelength-dependent AQYs measured by light-emitting diode (LED) light irradiation agree well with the absorption spectrum (Fig. 2g), suggesting that H2O2 was generated following the bandgap excitation of BiVO4. The AQY at 420 nm was determined to be 13.1%, the highest reported for inorganic semiconductors to the best of our knowledge (Table S1).
Time-resolved-spectroscopic analyses revealing enhanced charge separation through interfacial energetics tuning
To investigate the impact of surface energetics tuning on charge separation, charge-carrier dynamics in CoOx/BiVO4/(Ag/Pd) and CoOx/BiVO4/Pd were thoroughly studied by transient absorption spectroscopy (TAS). The transient kinetics of photogenerated charge carriers in BiVO4 were monitored upon band-gap excitation using 470 nm (~2.64 eV) laser pulses, with photogenerated surface trapped holes probed at 505 nm and photogenerated free/shallowly trapped electrons probed at 2000 nm38,39,40.
As shown in Fig. 3a, CoOx/BiVO4/(Ag/Pd) exhibits a longer electron lifetime than CoOx/BiVO4/Pd, indicating an enhanced charge-separation process in the former case with a higher ΔV. Yet, considering that CoOx/BiVO4/(Ag/Pd) was about three times more active than CoOx/BiVO4/Pd for H2O2 generation, their difference on electron decay is smaller than expectation. We therefore infer that a higher ΔV not only enhances charge separation by preserving charge carriers from recombination, but also by driving these charge carriers to the corresponding cocatalysts (Fig. 3e). As a result, electron accumulation in BiVO4 is competed by the rapid electron transfer to Ag/Pd.

a, b Transient profiles of a free/shallowly trapped electrons probed at 2000 nm and b trapped holes probed at 505 nm. Photoexcitation of the samples was performed using 470 nm laser pulses (duration: 6 ns, fluence: 3 mJ/pulse, frequency: 1 Hz). Measurements were carried out in vacuum (base pressure: ~ 10−5 Torr). c, d The transient profiles of trapped holes probed at 505 nm for c CoOx/BiVO4/Pd and d CoOx/BiVO4/(Ag/Pd) in vacuum and in the presence of HCOOH vapor. Photoexcitation of the samples was performed using 470 nm laser pulses (duration: 6 ns, fluence: 3 mJ/pulse, frequency: 1 Hz). Measurements were carried out in vacuum or in the presence of 20 Torr HCOOH. e Schematic illustration of the charge-separation process enhanced by surface energetics tuning.
To verify whether a higher ΔV in CoOx/BiVO4/(Ag/Pd) can enhance charge separation by driving charge carriers to cocatalysts, we further investigated the hole decay of CoOx/BiVO4/Pd and CoOx/BiVO4/(Ag/Pd). Since photogenerated holes exhibit comparably strong signal and long lifetime in the microsecond-millisecond timescales39,40, we also show the hole kinetics of bare BiVO4 and BiVO4/Pd for a detailed comparison. As shown in Fig. 3b, BiVO4/Pd exhibits a longer hole lifetime than bare BiVO4 due to electron transfer to Pd. Loading CoOx on BiVO4/Pd (i.e., CoOx/BiVO4/Pd) further prolonged the hole lifetime, even though the sluggish decay of long-lived holes was partially offset by hole transfer to CoOx. We attribute the prolonged hole lifetime to (i) synergistically enhanced charge separation by Pd and CoOx coloading, and (ii) more efficient electron transfer to Pd than hole transfer to CoOx, which have been confirmed in our previous study12.
CoOx/BiVO4/(Ag/Pd) shows a shorter hole lifetime than CoOx/BiVO4/Pd, proving that the elevated ΔV enhanced migration of holes from bulk BiVO4 to CoOx. Such enhanced hole migration can lead to higher hole accumulation in the CoOx of CoOx/BiVO4/(Ag/Pd) and accelerate surface reactions (Fig. 3e). As a proof, we examined the reactivity of holes in CoOx of CoOx/BiVO4/(Ag/Pd) and CoOx/BiVO4/Pd towards oxidation of formic acid (HCOOH), a representative hole scavenger41,42. For both samples, HCOOH as a hole scavenger accelerated the hole decays (Figs. 3c and 3d), where the acceleration was more significant in CoOx/BiVO4/(Ag/Pd) than in CoOx/BiVO4/Pd. For instance, at 1 ms, the hole signal intensity for CoOx/BiVO4/Pd decreased by 69%, while that for CoOx/BiVO4/(Ag/Pd) decreased by 87%. This indicates that 31% of holes remained in BiVO4 for CoOx/BiVO4/Pd, whereas only 13% for CoOx/BiVO4/(Ag/Pd). The corresponding percentage decrease of the TA signal represents the population of holes utilized for HCOOH oxidation.
This is a notable result of elevated ΔV that enhanced the charge separation by driving higher extent of holes transferred to CoOx for HCOOH oxidation. Collectively, the TAS results indicate that lowering the Schottky barrier at {010} facets by constructing BiVO4/Ag junction effectively enhances charge separation by surviving charge carriers as well as driving these charge carriers to corresponding cocatalysts for surface reactions (Fig. 3e).
Electrical simulation revealing impacts of surface energetics on charge separation
To quantitatively study the effects of surface energetics on charge separation, COMSOL Multiphysics was applied to simulate the photocatalytic H2O2 generation reactions on CoOx/BiVO4/Pd and CoOx/BiVO4/(Ag/Pd) particles. A three-dimensional (3D) model for BiVO4 particles (Fig. S16) was used in the simulation to accurately represent experimental conditions. The optoelectronic parameters used in the COMSOL simulation were obtained from our previous studies and literature as summarized in Table S2. The charge-carrier generation in the particle model was detailed in Section S1, Fig. S17–19. The experimental photoatalytic H2O2 generation rates were converted to photocurrent densities (Section S2) in order to quantitatively compare with the photocurrent densities obtained by comsol simulation.
We dissected the operating cocatalyst-loaded BiVO4 as a combination of a solar cell and an electrocatalyst cell (Section S3, Fig. S20)43. The solar cell was simulated by a 3D particle model with reduction (top/bottom) and oxidation (side) facets as the cathode and anode, respectively (Fig. 4a, b). The barrier height at the oxidation facets was fixed at 1.23 V, while that at the reduction facets was varied to obtain the corresponding light-responsive J–V curves. The dark J–V curve of the electrocatalyst, H2O2 generation on cathode and O2 evolution on anode, was experimentally measured (Fig. S21). The intersect of the light-responsive J–V curve from the simulated solar cell and J–V curve from the electrocatalyst cell represents the photocurrent density of cocatalyst-loaded BiVO4 particles (Fig. S22).

a, b Schematic model and band diagram of BiVO4 as a solar cell. c Current density vs. applied potential with barrier heights at cathodic sites (Φ010) varied from 0 to 0.4 V (gray dotted arrow). The barrier height of the anodic side was fixed at 1.23 V. Two dash lines labeled with 0.0081 and 0.0257 mA cm-2 were used to indicate the photocurrent densities converted from experimental H2O2 generation rates of CoOx/BiVO4/Pd and CoOx/BiVO4/(Ag/Pd), respectively. The operating conditions of CoOx/BiVO4/Pd and CoOx/BiVO4/(Ag/Pd) were marked with two void circles. Their detailed optoelectronic properties are presented in the following figures. d–i 2D cross-sectional plots of the optoelectronic properties of a BiVO4 particle, including conduction band energy (eV) (d, e), electron concentration (f, g), and hole concentration (h, i). j–m 1D plots of energy band diagram (j, k) and mobile charge carrier density (l, m).
The photocurrent densities of cocatalyst-loaded BiVO4 particles with various barrier heights at the reduction facets were shown in Fig. 4c. The photocurrent densities for CoOx/BiVO4/Pd and CoOx/BiVO4/(Ag/Pd) were determined to be 0.0081 and 0.0257 mA cm−2, corresponding to a barrier height at the reduction facets of 0.32 and 0.09 V, respectively. This result indicates that Ag between BiVO4 and Pd decreased the Schottky barrier at the reduction facets of BiVO4 by 0.23 V. Notably, this value is larger than that (0.13 V) obtained by UPS (Fig. 1e), which is reasonable because the UPS results reflected the energetics of not only reduction facets but also the oxidation ones.
After extracting the barrier heights of CoOx/BiVO4/Pd and CoOx/BiVO4/(Ag/Pd) at their reduction facets, we further compared their optoelectronic properties including band energy diagram and mobile charge carrier density (Fig. 4d–m). With the barrier height at the cathodic site even slightly higher than that of the anodic site (Fig. 4d, j), the asymmetry in energetics of CoOx/BiVO4/Pd is relatively small. This results in the back-flow and poor accumulation of charge carriers on the surface of BiVO4 (Fig. 4f, h, l). In comparison, with Schottky barrier lowered by Ag, CoOx/BiVO4/(Ag/Pd) particle exhibits a larger asymmetry in energetics (Fig. 4e, k), which efficiently drive electrons and holes to the cathodic and anodic sites, respectively (Fig. 4g, i, m). Overall, the simulations on photocarrier dynamics in BiVO4 particles demonstrate that constructing a BiVO4/Ag junction effectively lowered the Schottky barrier at reduction sites, creating a high ΔV for improving charge separation and photosynthesis performance.
Generality of interfacial-energetics-tuning strategy for enhancing artificial photosynthesis
Our surface-energetic-tuning strategy was further validated on C3N4 and TiO2, two widely studied photocatalysts for artificial photosynthesis (e.g, H2, H2O2)44,45. C3N4/(Ag/Pd) and TiO2/(Ag/Pd) was 1.7 and 1.4 times more active than C3N4/Pd and TiO2/Pd on H2O2 photosynthesis, respectively (Figs. 5a and 5b). Such results clearly manifest the generality of our strategy on enhancing photocatalysis. Notably, the enhancement on C3N4 and TiO2 is weaker than that on faceted BiVO4. This is reasonable because without spatially separating the energetics on different facets, deposited Ag may disturb the energetics of the nearby oxidation sites and thus degrade the overall charge-separation process in photocatalysts.

Time courses of photocatalytic H2O2 generation by a C3N4/Pd and C3N4/(Ag/Pd) and b TiO2/Pd and TiO2/(Ag/Pd). Reaction conditions: photocatalyst, 1 mg/mL; 50 ml DI water saturated with O2; light source, LED visible light (300 mW cm−2, λ > 400 nm) for C3N4 and UVA light (λ = 365 nm) for TiO2. c A general approach for effective interfacial-energetics-tuning and enhanced artificial photosynthesis.
Based on above results, we propose a general approach for effective interfacial-energetics-tuning (Fig. 5c): (i) apply facet engineering to spatially separate the surface energetics for electron and hole accumulation, (ii) selectively deposite metal/metal oxide species with desirable work functions on the required facets to tune the surface energetics for charge separation; and (iii) selectively deposite catalysts on the metal/metal oxide species to tune surface reactions.
Our study validated the tuning of interfacial energetics as a general and effective approach to enhance charge separation, a key challenge in designing high-performance photocatalysts. The construction of core/shell Ag/Pd cocatalyst lowered the Schottky barrier at the {010} facets of BiVO4 without impairing the surface reactions, leading to enhanced overall asymmetry in energetics and charge separation. With successful tuning of interfacial energetics, BiVO4 generated H2O2 with an AQY of 3.0% and a STH efficiency of 0.73% at full spectrum, a new record for inorganic semiconductor-based systems. As first highlighted by BiVO4 for H2O2 generation, such a strategy of tuning interfacial energetics can be generally applied towards other photosynthetic systems for promoting solar fuel production such as water splitting and CO2 reduction.
Methods
Catalyst preparation
Single crystal BiVO4 was prepared by heating the mixture of K2CO3 (1.047 g) and V2O5 (2.272 g) in a ceramic crucible at a heating rate of 1.5 °C/min to 450 °C and annealing for 5 h in a muffle furnace. The obtained K3V5O14 (2 g) was mixed with Bi(NO3)3•5H2O (0.326 g) and dispersed in 50 mL water under ultrasonication for 30 min. The mixture was heated at 70 °C for 10 h under ultrasonication, separated by centrifugation, washed with deionized water, and dried at 70 °C for 8 h.
Cocatalyst deposition
As-prepared BiVO4 (0.2 g) was dispersed in 100 ml water, followed by addition of 0.1 mol NaIO3 and 0.35 mL Co(NO3)2 solution (1.5 g/L). The mixture was irradiated for 3 h using a xenon lamp solar simulator (model 300 DUV; Perfect Light, Inc., light intensity = 0.1 W cm−2, λ > 420 nm), filtered, washed with deionized water, and dried at 60 °C for 8 h. As-prepared CoOx/BiVO4 (0.15 g) was dispersed in 100 ml pure water, followed by addition of 0.36 ml AgNO3 solution (0.33 g/L). The mixture pH was adjusted to 8.5 and irradiated at λ > 420 nm for 20 h. As-prepared CoOx/BiVO4/Ag was dispersed in 100 ml pure water, followed by addition of 0.182 mL Na2PdCl4 solution (3.3 g/L). The mixture was irradiated at λ > 420 nm for 3 h. As-prepared CoOx/BiVO4/(Ag/Pd) was filtered, washed with deionized water, and dried at 60 °C for 8 h. Same photodeposition method was also applied in preparing BiVO4/Ag, BiVO4/Pd, CoOx/BiVO4/Ag, CoOx/BiVO4/Pd, CoOx/BiVO4/(Rh/Pd), CoOx/BiVO4/(Au/Pd), and CoOx/BiVO4/(Pd/Ag) using respective cocatalyst precursors. Similarly, C3N4/Pd, C3N4/(Ag/Pd), TiO2/Pd, and TiO2/(Ag/Pd) were prepared.
Photocatalyst characterizations
XPS measurements were performed with a Thermo Scientific 250Xi system with monochromatic Al Kα as the excitation source. XRD patterns were recorded with a Bruker D8 Advance X-ray diffractometer with Cu Kα radiation (λ = 1.5406 Å) operated at 40 kV and 40 mA. SEM images were taken using a Hitachi SU-8010 microscope equipped with EDS at 30 kV. TEM images were taken using a Hitachi 7650 microscope operated at 100 kV.
Photocatalytic activity tests
Batch assessements of photocatalytic H2O2 production by different photocatalyst were conducted using a high throughput reactor (model slight; Perfect Light Inc., Fig. S23) equipped with LED visible light. The LED light spectrum is shown in Fig. S24. Photocatalyst (50 mg) was dispersed in 50 mL deionized water in a custom-made reactor by ultrasonication for 10 min and purged with O2 for 20 min. H2O2 production was assessed under water bath (12 ± 0.5 °C) with an input light intensity of 300 mW/cm2 (λ > 400 nm; irradiation area = 7.1 cm2). The photocatalytic H2O2 production performance was measured using a xenon lamp (model 300 DUV; Perfect Light Inc., Fig. S25). The spectrum of xenon lamp and the standard AM1.5 G (ASTMG 173) are shown in Fig. S26. Photocatalyst (10 mg) was dispersed in 10 mL deionized water containing 0.1 M H3BO3 and 0.075 M ScCl3 by ultrasonication for 10 min and purged with O2 for 20 min. H2O2 production was assessed under water bath (12 ± 0.5 °C) with an input light intensity of 100 mW cm−2 (AM 1.5 G; irradiation area = 4.5 cm2).
At designated time points, 50 μL suspension was taken for analysis of H2O2 productions and diluted with phosphate buffer (pH=7.4) to a H2O2 concentration (1–15 µM) that is most suitable for accurate H2O2 quantification, followed by centrifugation. Afterwards, 50 μL supernatant was taken and mixed with 50 μL solutions containing phosphate buffer (50 mM, pH = 7.4), ampliflu red (100 µM), and horseradish peroxidase (0.05 U/mL). Ampliflu red selectively reacted with H2O2 in the presence of horseradish peroxidase and formed the product resorufin. Resorufin in the mixture solution was quantified using an Agilent high-performance liquid chromatography coupled to a photo-diode array detector (detection at 560 nm). The calibration curve in Fig. S27 was used to quantitatively analyze H2O2 production. HPLC analysis was carried out in a C18 column at 20 °C with an isocratic mobile phase of 55% sodium citrate buffer (with 10% methanol (v/v), pH 7.4) and 45% methanol (v/v) at a flow rate of 0.5 mL/min.
The photocatalytic H2O2 production selectivity was assessed using methanol as electron donor. H2O2 selectivity is defined as the ratio of electrons utilized for H2O2 synthesis to the total number of electrons consumed (i.e., electrons donated by methanol)46. The consumption amount of methanol was assessed by analyzing its oxidation product formaldehyde. Experimental suspensions contained 1 g/L photocatalyst and 10% methanol (v/v). The suspension was O2 saturated by continuous purging during the irradiation. Nash’s reagent was used for quantification of formaldehyde. Nash’s reagent (0.5 mL) containing 2 M ammonia acetate, 30 mM acetyl acetone, and 35 mM acetic acid was mixed with the sample suspension at 1:1 ratio and heated in oven at 60 °C for 1 h. The absorption spectra of products were measured using UV-vis spectrometer at 415 nm. Formaldehyde standard solution was used to calibrate the absorption in relation to formaldehyde concentration.
TAS measurements
Microsecond-millisecond TAS measurements were carried out using Nd:YAG lasers (Continuum, Surelite I) and custom-built spectrometers14,47. Briefly, the TAS signals for the photoinduced photocarrier were monitored after band-gap excitation using 470 nm laser pulses (duration: 6 ns, fluence: 3 mJ pulse−1). To probe the photoinduced electrons, the IR beam from the MoSi2 coil was irradiated and focused on the film sample. The transmitted IR beam entered a monochromatic grating spectrometer and was detected by a mercury cadmium telluride (MCT) detector (Kolmar). The photoexcited electrons was monitored at 2000 nm. To probe the photogenerated holes, the white continuum beam emitted by a halogen lamp was focused on the sample. The reflected beam from the sample entered the monochromatic grating spectrometer and was detected by Si photodetector. The photogenerated holes was monitored at 505 nm (~2.45 eV). The output electric signal was then amplified with an AC-coupled amplifier (Stanford Research Systems, SR560, 1 MHz) that can monitor responses from 1 microsecond to several milliseconds. Ten to one thousand responses were averaged to obtain the transient decay profile at the probe wavelength. The time resolution of the spectrometer was limited to 1 μs by the bandwidth of the amplifier. The experiments were carried out in vacuum (base pressure ~ 10−5 Torr) and at room temperature. For the measurement with formic acid, the pressure inside the reaction cell was at 20 Torr. For sample preparation, photocatalyst was dispersed in isopropanol, drop-casted on a CaF2 substrate, and dried in air to obtain a powder film with a density of ~ 1.3 mg/cm2.
Numerical simulation
Charge carrier dynamics in BiVO4 was simulated using COMSOL Multiphysics. Detailed information on the charge-carrier distribution, and performance evaluation were elaborated in Section S1–S3.
Data availability
Source data are provided with this paper. The data that support the findings of this study are available from the corresponding author upon reasonable request. Source data are provided with this paper.
Code availability
Source data are provided with this paper. The code used for the electrical simulations are available from the corresponding author upon reasonable request.
References
Ma, J. J. et al. Photochemical intermolecular dearomative cycloaddition of bicyclic azaarenes with alkenes. Science 371, 1338–1345 (2021).
Romero, E., Novoderezhkin, V. I. & van Grondelle, R. Quantum design of photosynthesis for bio-inspired solar-energy conversion. Nature 543, 355–365 (2017).
Chen, S. S., Qi, Y., Li, C., Domen, K. & Zhang, F. X. Surface strategies for particulate photocatalysts toward artificial photosynthesis. Joule 2, 2260–2288 (2018).
Wang, X. C. et al. A metal-free polymeric photocatalyst for hydrogen production from water under visible light. Nat. Mater. 8, 76–80 (2009).
Karamoschos, N. & Tasis, D. Photocatalytic evolution of hydrogen peroxide: a minireview. Energies 15, 6202 (2022).
Mase, K., Yoneda, M., Yamada, Y. & Fukuzumi, S. Seawater usable for production and consumption of hydrogen peroxide as a solar fuel. Nat. Commun. 7, 11470 (2016).
Gopakumar, A. et al. Lignin-supported heterogeneous photocatalyst for the direct generation of H2O2 from seawater. J. Am. Chem. Soc. 144, 2603–2613 (2022).
Yanagi, R., Zhao, T. S., Solanki, D., Pan, Z. H. & Hu, S. Charge separation in photocatalysts: mechanisms, physical parameters, and design principles. ACS Energy Lett. 7, 432–452 (2022).
Nozik, A. J. Photoelectrochemistry: applications to solar energy conversion. Ann. Rev. Phys. Chem. 29, 189–222 (1978).
Pan, Z. H. et al. Elucidating charge separation in particulate photocatalysts using nearly intrinsic semiconductors with small asymmetric band bending. Sustain. Energ. Fuels 3, 850–864 (2019).
Xu, Y. et al. Strategies for efficient charge separation and transfer in artificial photosynthesis of solar fuels. ChemSusChem 10, 4277–4305 (2017).
Liu, T. et al. Overall photosynthesis of H2O2 by an inorganic semiconductor. Nat. Commun. 13, 1034 (2022).
Yoshida, M. et al. ATR-SEIRAS investigation of the fermi level of Pt cocatalyst on a GaN photocatalyst for hydrogen evolution under irradiation. J. Am. Chem. Soc. 131, 13218–13219 (2009).
Yamakata, A. et al. Behavior and energy states of photogenerated charge carriers on Pt- or CoOx-loaded LaTiO2N photocatalysts: time-resolved visible to mid-infrared absorption study. J. Phys. Chem. C 118, 23897–23906 (2014).
Kenney, M. J. et al. High performance silicon photoanodes passivated with ultrathin nickel films for water oxidation. Science 342, 836–840 (2013).
Esposito, D. V., Levin, I., Moffat, T. P. & Talin, A. A. H2 evolution at Si-based metal-insulator-semiconductor photoelectrodes enhanced by inversion channel charge collection and H spillover. Nat. Mater. 12, 562–568 (2013).
Hou, H. L., Zeng, X. K. & Zhang, X. W. Production of hydrogen peroxide by photocatalytic processes. Angew. Chem. Int. Ed. 59, 17356–17376 (2020).
Kofuji, Y. et al. Carbon nitride-aromatic diimide-graphene nanohybrids: metal-free photocatalysts for solar-to-hydrogen peroxide energy conversion with 0.2% efficiency. J. Am. Chem. Soc. 138, 10019–10025 (2016).
Isaka, Y. et al. Photocatalytic production of hydrogen peroxide through selective two-electron reduction of dioxygen utilizing amine-functionalized MIL-125 deposited with nickel oxide nanoparticles. Chem. Commun. 54, 9270–9273 (2018).
Shiraishi, Y. et al. Resorcinol-formaldehyde resins as metal-free semiconductor photocatalysts for solar-to-hydrogen peroxide energy conversion. Nat. Mater. 18, 985–993 (2019).
Shiraishi, Y., Matsumoto, M., Ichikawa, S., Tanaka, S. & Hirai, T. Polythiophene-doped resorcinol-formaldehyde resin photocatalysts for solar-to-hydrogen peroxide energy conversion. J. Am. Chem. Soc. 143, 12590–12599 (2021).
Xiao, J. D. et al. Is C3N4 chemically stable toward reactive oxygen species in sunlight-driven water treatment? Environ. Sci. Technol. 51, 13380–13387 (2017).
Liu, L. J. et al. Linear conjugated polymers for solar-driven hydrogen peroxide production: the importance of catalyst stability. J. Am. Chem. Soc. 143, 19287–19293 (2021).
Zhao, W. et al. Accelerated synthesis and discovery of covalent organic framework photocatalysts for hydrogen peroxide production. J. Am. Chem. Soc. 144, 9902–9909 (2022).
Tian, Q. et al. Nanospatial charge modulation of monodispersed polymeric microsphere photocatalysts for exceptional hydrogen peroxide production. Small 17, 2103224 (2021).
Sun, M. et al. Bifunctional PdOx center at the liquid–solid–gas triphase interface for H2O2 photosynthesis. Acs Catal. 12, 2138–2149 (2022).
Fuku, K. et al. Photocatalytic H2O2 production from O2 under visible light irradiation over phosphate ion-coated Pd nanoparticles-supported BiVO4. Appl. Catal. B-Environ. 272, 119003 (2020).
Shi, H. Y. et al. Mass-transfer control for selective deposition of well-dispersed AuPd cocatalysts to boost photocatalytic H2O2 production of BiVO4. Chem. Eng. J. 443, 136429 (2022).
Wang, K. Y. et al. Optimizing oxygen and intermediate HOO* adsorption of Cu-Pd alloy cocatalyst for boosting photocatalytic H2O2 production of BiVO4. Adv. Sustain. Syst. 6, 2200144 (2022).
Li, R. G. et al. Spatial separation of photogenerated electrons and holes among {010} and {110} crystal facets of BiVO4. Nat. Commun. 4, 1432 (2013).
Chun, W. J. et al. Conduction and valence band positions of Ta2O5, TaON, and Ta3N5 by UPS and electrochemical methods. J. Phys. Chem. B 107, 1798–1803 (2003).
Nellist, M. R. et al. Potential sensing electrochemical atomic force microscopy for in operando analysis of water-splitting catalysts and interfaces. Nat. Energy 3, 46–52 (2018).
Laskowski, F. A. L. et al. Nanoscale semiconductor/catalyst interfaces in photoelectrochemistry. Nat. Mater. 19, 69–78 (2020).
Kato, S., Jung, J., Suenobu, T. & Fukuzumi, S. Production of hydrogen peroxide as a sustainable solar fuel from water and dioxygen. Energy Environ. Sci. 6, 3756 (2013).
Shiraishi, Y., Ueda, Y., Soramoto, A., Hinokuma, S. & Hirai, T. Photocatalytic hydrogen peroxide splitting on metal-free powders assisted by phosphoric acid as a stabilizer. Nat. Commun. 11, 3386 (2020).
Ye, K. H. et al. Enhancing photoelectrochemical water splitting by combining work function tuning and heterojunction engineering. Nat. Commun. 10, 3687 (2019).
Pattengale, B. & Huang, J. The effect of Mo doping on the charge separation dynamics and photocurrent performance of BiVO4 photoanodes. Phys. Chem. Chem. Phys. 18, 32820–32825 (2016).
Ma, Y. M., Pendlebury, S. R., Reynal, A., Le Formal, F. & Durrant, J. R. Dynamics of photogenerated holes in undoped BiVO4 photoanodes for solar water oxidation. Chem. Sci. 5, 2964–2973 (2014).
Yamakata, A., Ranasinghe, C. S. K., Hayashi, N., Kato, K. & Vequizo, J. J. M. Identification of individual electron- and hole-transfer kinetics at CoOx/BiVO4/SnO2 double heterojunctions. ACS Appl. Energy Mater. 3, 1207–1214 (2020).
Suzuki, Y. et al. Rational interpretation of correlated kinetics of mobile and trapped charge carriers: analysis of ultrafast carrier dynamics in BiVO4. J. Phys. Chem. C 121, 19044–19052 (2017).
Ji, Y. F. & Luo, Y. Structure-dependent photocatalytic decomposition of formic acid on the anatase TiO2 {101} surface and strategies to increase its reaction rate. J. Power Sources 306, 208–212 (2016).
Lee, G. D. & Falconer, J. L. Transient measurements of lattice oxygen in photocatalytic decomposition of formic acid on TiO2. Catal. Lett. 70, 145–148 (2000).
Takanabe, K. Photocatalytic water splitting: quantitative approaches toward photocatalyst by design. ACS Catal. 7, 8006–8022 (2017).
Ong, W. J., Tan, L. L., Ng, Y. H., Yong, S. T. & Chai, S. P. Graphitic carbon nitride (g-C3N4)-based photocatalysts for artificial photosynthesis and environmental remediation: Are we a step closer to achieving sustainability? Chem. Rev. 116, 7159–7329 (2016).
Li, R. G. et al. Achieving overall water splitting using titanium dioxide-based photocatalysts of different phases. Energ. Environ. Sci. 8, 2377–2382 (2015).
Chu, C. H. et al. Spatially separating redox centers on 2D carbon nitride with cobalt single atom for photocatalytic H2O2 production. Proc. Natl Acad. Sci. USA 117, 6376–6382 (2020).
Vequizo, J. J. M. et al. Trapping-induced enhancement of photocatalytic activity on brookite TiO2 powders: comparison with anatase and rutile TiO2 powders. ACS Catal. 7, 2644–2651 (2017).
Acknowledgements
This work was supported by National Natural Science Foundation of China (NSFC, No. 22136004, 22006129) and JSPS KAKENHI (No. JP20K22556). We are grateful to Sudan Shen (State Key Laboratory of Chemical Engineering at Zhejiang University) and analysis center of agrobiology and environmental sciences for help in STEM and SEM measurements, respectively. Shu Hu and Rito Yanagi are supported by a grant from the Thistledown Foundation through the Research Corporation for Science Advancement Negative Emissions Science program.
Author information
Authors and Affiliations
Contributions
C.C. and Z.P. designed research. T.L., Z.P., and X.Z. synthesized the catalysts. T.L. and X.Z. conducted photocatalytic performance tests. T.L. conducted material characterization; Ko.K., J.J.M.V., and A.Y. performed TAS measurements and analysis; Y.R., Z.P., and S.H. conducted numerical simulation. T.L., C.C., Z.P., W.Y., B.C., and Ke.K. analyzed data; T.L., C.C., and Z.P. wrote the paper. All authors discussed the results and commented on the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interest.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Liu, T., Pan, Z., Kato, K. et al. A general interfacial-energetics-tuning strategy for enhanced artificial photosynthesis. Nat Commun 13, 7783 (2022). https://doi.org/10.1038/s41467-022-35502-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-022-35502-z
- Springer Nature Limited
This article is cited by
-
Absolute band-edge energies are over-emphasized in the design of photoelectrochemical materials
Nature Catalysis (2024)
-
Efficient and stable visible-light-driven Z-scheme overall water splitting using an oxysulfide H2 evolution photocatalyst
Nature Communications (2024)
-
Advancing wastewater treatment: chitin and derivatives for PPCP contaminant mitigation
Polymer Bulletin (2024)
-
Photocatalytic production of H2O2 and its in-situ environmental applications
Research on Chemical Intermediates (2024)