

Abstract
Direct asymmetric reductive amination is one of the most efficient methods for the construction of chiral amines, in which the scope of the applicable amine coupling partners remains a significant challenge. In this study we describe primary alkyl amines effectively serve as the N-sources in direct asymmetric reductive amination catalyzed by the iridium precursor and sterically tunable chiral phosphoramidite ligands. The density functional theory studies of the reaction mechanism imply the alkyl amine substrates serve as a ligand of iridium strengthened by a (N)H-O(P) hydrogen-bonding attraction, and the hydride addition occurs via an outer-sphere transition state, in which the Cl-H H-bonding plays an important role. Through this concise procedure, cinacalcet, tecalcet, fendiline and many other related chiral amines have been synthesized in one single step with high yields and excellent enantioselectivity.
Similar content being viewed by others


Introduction
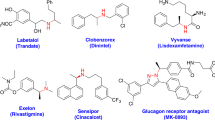
Enantiomerically enriched N-alkyl amines are common structural motifs for active pharmaceutical ingredients (Fig. 1a). The increasing demand has driven the development of novel and efficient methods for their synthesis1,2,3, including two practical and highly efficient routes, asymmetric hydrogenation4,5,6,7,8,9 and reductive amination10,11,12,13,14,15 (Fig. 1b). Direct asymmetric reductive amination (DARA) is one of the most efficient approaches for the construction of chiral amines by enabling the coupling of ketones with amines in a single step rather than taking a circuitous route through imine/enamine preparation, reduction, and/or the following N-deprotection. For the transition-metal-catalyzed DARA, as some progress has been made, the applicable scope of the amine coupling partners are still very limited. Apart from the intramolecular DARAs16,17,18,19,20,21,22,23, most reported N-sources of DARAs generally fall into three categories: inorganic ammonium salts24,25,26,27,28,29/ammonia30 for the synthesis of primary amines; aryl amines31,32,33,34,35, the popularly applied N-sources, albeit often times the N-Ar group in the product is not desired and needs to be removed, which lead to secondary chiral amine products; and one example of secondary amine sources for the construction of tertiary chiral amine products36. Besides, there are some other sporadically reported special amines, including benzyl amines37,38, diphenylmethylamine39,40,41, hydrazine42, and hydrazides43,44.

a Representing N-alkyl pharmaceuticals. b Practical methods for the synthesis N-alkyl amines. c This work: primary alkyl amines as N-sources for DARA.
Previously there was only one example that utilized an alkyl amine, MeNH2, as the N-coupling partners in transition-metal-catalyzed DARAs45, and some instances in which special alkyl amines, such as benzyl amines38,39 diphenylmethylamine39,40,41, and allylamine46, were used. Even the literature for the asymmetric hydrogenation of corresponding N-alkyl imines to directly form the N-alkyl amines are extraordinary scarce47,48,49,50,51,52, of which in two cases the N-alkyl amines were in situ transformed into N-SiH2Ph and N-Boc products in order to decrease the inhibitory effects on the catalysts49,50,51. One of the main reasons for this limitation in DARA research is that the alkyl groups could not coordinate with or interact with the functioning catalytic species via secondary interactions, for example, hydrogen-bonding, π effects, and electrostatic interactions.
The inhibitory effects of nitrogen-containing species, especially the electron-rich N-alkyl amines and imines in this case, on the catalyst is also a major factor. Another difficulty for lagging the effective utility of primary alkyl amines in DARAs is the direct products are secondary amines, which could serve as new coupling partners to continue to react with ketone substrates and form tertiary amines36. We postulate that the application of readily steric tuning chiral ligands, which are capable of efficiently confining the position of the alkyl groups during the catalytic process, along with the accelerations from the additives, may tackle this problem.
Herein we report a highly efficient DARA of various ketones with primary alkyl amines for the synthesis of corresponding chiral secondary N-alkyl amines catalyzed by 0.05 mol% iridium-phosphoramidite complex (Fig. 1c). The evolved phosphoramidite chiral ligands with bulky 3,3′-1-naphthyl substituents successfully manage the enantioselective process of the reaction with the lack of secondary interactions between the alkyl groups and the catalytic complex. Moreover, computational studies have been carried out to provide useful information about the reaction mechanism and reveal an outer-sphere hydride addition pathway, in which two H-bonding attractions, one between (P)O of the phosphoramidite chiral ligand and (N)H and the other between the chlorine on iridium and the imine substrate, play important roles.
Result and discussion
Steric tuning of the chiral monodentate phosphoramidite ligands
The present study was initiated by the direct reductive amination of acetophenone 1 and 3-phenylpropylamine 2, to mimic an alkyl amine, which was catalyzed by the catalyst in situ generated from [Ir(cod)Cl]2 and H8-BINOL-based chiral ligand L1 (Table 1). The monodentate phosphoramidite ligands have found success in numerous catalytic research areas because of their high degree of tunability on both electronic and steric properties, and ease of preparation at low cost53,54. From the brief solvent screening we can see, although multiple solvents led to excellent yields for the reaction, only protic methanol and trifluoroethanol displayed enantioselectivity moderately (Table 1, entries 1–6).
To sufficiently exploit the readily fine-tuning feature of this kind of ligands, we deliberately modified the H8-BINOL back-bone by introducing methyl, phenyl, and 1-naphthyl groups at the 3,3′-positions and synthesized ligands L2, L3, and L4. Compared with the results from L1, the reaction yield and enantioselectivity of L2 decreased by 17% and 11% respectively, indicating that the methyl groups at the 3,3′-positions compromised the proper coordination of the substrate to the transition-metal complex. While the substituents were enlarged into phenyl groups, the ee value was improved significantly to 69% (Table 1, entry 8). To further enlarge the size of imbedded segments to 1-naphthyl groups (L4), the enantioselectivity was elevated to 86% (Table 1, entry 9). The improved stereoselectivity indicates the spacious 1-naphthyl groups on L4 could effectively confine the imine substrate for better steric differentiation (Fig. 2)55

a Structures of developed ligands. b Ligand–substrate interactions.
Additives in DARA reactions have been proven to be perfectly competent in improving both reaction reactivity and selectivity significantly31,32,33,34,35,36,37,38,39,40,41,42,43,44,56,57,58. Titanium isopropoxide is well known to be able to facilitate the formation of the imine intermediates59. Some other common additives, iodine, Brønsted/Lewis acids/bases, and non-coordinate anion were also evaluated (Table 1, entries 10–14). Among them, Lewis acid FeCl3 effectively increased the reaction yield and stereoselectivity (Table 1, entry 14). Further solvent screening revealed the combination of trifluoroethanol and methyl acetate provided a satisfying ee value at 97% with an excellent reaction yield. We speculated that it was Brønsted acid HCl generated from the hydrolysis of Lewis acid FeCl3 in the reaction system that successfully promoted the stereoselectivity. This was proved by the result from the mere addition of 30 mol% hydrochloride salt of 2 (Table 1, entry 17). When the catalyst loading was reduced to 0.02 mol%, the reaction still proceeded smoothly without significant erosion in reactivity and selectivity with 88% yield and 95% ee (Table 1, entry 19).
Substrate scope
Based on the optimal conditions for our iridium-catalyzed DARA, firstly the scope of aromatic ketones was probed with 3-phenylpropylamine 2 as a representative amine partner applying 0.05 mol% the in situ generated iridium catalyst (Fig. 3a). In cases the enantiomers of the products were unable to be separated by available chiral HPLC columns, benzylamine was used instead as the coupling partner of the ketones. At first, we examined the effects of the electronic property and steric hindrance on the reactions for the substituted arylethanones. It appeared the common substituents including small-sized groups at the ortho-position had no significant influence on the reaction yields or stereoselectivity (products 3–21), regardless of their electron-withdrawing or donating properties. It is worth mentioning that for some steric-hindered substrates, the corresponding products were still obtained in excellent ee values (products 22–23, 26–29). To achieve better results for space-demanding products 27–29, modified chiral ligand L5 (Fig. 3) was applied and displayed higher stereoselectivity with the opposite spatial configuration than L4, further demonstrating the versatility of this class of ligands. To our delight, various functional groups, including nitro (-NO2), benzyloxy (-OBn), thio-ether (-SMe), tert-butyl carbamate (-NHBoc), borate ester, and ester, were all well-tolerated during this transformation, in which the corresponding products 30–35 were obtained in good yields and excellent ees. Heteroaromatic ketone 2-acetylthiophene could be employed in this reaction to provide 36 in a high yield and good enantioselectivity.

a Aromatic ketone scope. b Amine scope. Conditions: [Ir]-L4 0.05 mol%; ketone 0.3 mmol, amine 0.95 equiv. (MeNH2 and nPrNH2 were 1.5 equiv.), Ti(OiPr)4 1.2 equiv., FeCl3 30 mol%, solvent (CF3CH2OH/MeOAc 1:1) 1.2 mL, 40 oC, 24 h; Yields were isolated yields; Enantiomeric excesses were determined by chiral HPLC after the products were converted to the corresponding acetamides. aReaction conditions: [Ir]-L5 0.1 mol%, Ti(OiPr)4 1.2 equiv., 1,8-diazabicyclo[5.4.0]undec-7-ene 10 mol%, Et3N·HCl 30 mol%, MeOAc 2 mL, 60 atm H2, 60 oC, 24 h.
Next, the scope of primary aliphatic amines was explored with acetophenone or 1′-acetonaphthone as the representative ketone partners (Fig. 3b). It is important to note that benzylamine was a valuable coupling partner for the DARA with 1 leading to the corresponding N-Bn product, which could be easily deprotected under mild conditions to afford α-phenylethylamine60,61, a versatile building-block and common resolution reagent in organic synthesis. The aromatic amine aniline as a weaker nucleophile was also a suitable N-source for the reductive coupling reaction with acetophenone to provide excellent results. The DARAs of 1 and pure aliphatic amine sources bearing linear or cyclic alkyl groups also proceeded smoothly to afford products 40–46 with excellent enantioselectivity and yields. Importantly, N-sources with heteroaromatic segments, such as furan, thiophene, indole, and pyridine, reductively coupled with acetophenone successfully to provide products 47–50. Besides, the catalytic system effectively tolerated NHBoc, olefin, and ether functional groups (products 51–53).
After having successfully conducted the DARA reactions of aromatic ketones with amines, we were interested in the applicability of this iridium catalysis in alkyl ketones for the synthesis of chiral aliphatic amines, which are difficult to synthesize via asymmetric catalytic methods, due to the lack of aforementioned secondary interactions between substrates and the catalytic complex62,63. To date, the successful DARA examples of alkyl–alkyl ketones are very scarce, in which some special amine partners, anilines, diphenylmethanamine, and benzhydrazide, were utilized to gain additional anchors with the catalysts31,32,33,34,35,40. Accomplishment in the reductive coupling of aromatic ketones prompted us to validate this protocol for more challenging alkyl–alkyl ketone substrates. Fortunately, the iridium-L4 catalytic system is found to have well functioned in this substrate category (Fig. 4a). Accordingly, various aliphatic ketones with the linear, branch, and cyclic segments prosperously reacted with alkyl amines to effectively furnish the chiral aliphatic amines 54–61 in excellent yields and enantioselectivities. Importantly, this procedure was also very effective towards ketones and amines with pure alkyl components to afford the all-alkyl amines 59–61. L6 was utilized in the synthesis of products 60 and 61 for higher ee values, which again displayed the advantage of the readily fine-tuning property of this type of ligand.

a Aliphatic ketone scope. b Applications in drugs and key intermediates synthesis. Conditions: [Ir]-L4 0.05 mol%; ketone 0.3 mmol, amine 0.95 equiv. (MeNH2 was 1.5 equiv.), Ti(OiPr)4 1.2 equiv., FeCl3 30 mol%, solvent (CF3CH2OH/MeOAc 1:1) 1.2 mL, 40 oC, 24 h; Yields were isolated yields; Enantiomeric excesses were determined by chiral HPLC or GC after the products were converted to the corresponding acetamides (for product 59, it was converted to benzamide). For substrates in Fig. 4a, FeCl3 0 mol%, amine 1.1 equiv., solvent (CF3CH2OH/MeOAc 1:5) 1.2 mL. a(R)-L6 was used instead of L4. b(S)-L4 was used instead of L4.
Practical applications
To showcase the practical utility, a 10-gram-scale reaction was carried out (Fig. 4b). Catalyzed by 0.05 mol% of the Ir-L4 complex, 10 grams of acetophenone efficiently reductively coupled with 8.5 grams of benzylamine to afford 16.7 grams of N-benzyl-1-phenylethan-1-amine 38 in 94% yield and 95% ee, which were similar to the results from the small-scale reaction. To further demonstrate potential, this iridium catalysis was applied in the synthesis of a collection of life-science important pharmaceuticals and key intermediates. Utilizing 3-(3-(trifluoromethyl)phenyl) propan-1-amine as the amine source, the calcimimetic agent cinacalcet could be synthesized at gram-scale via this protocol in 96% yield and 94% ee. Similarly, tecalcet and fendiline were prepared in excellent ee’s and high yields. With methyl amine as the coupling partner, the key intermediates of rivastigmine and orvepitant, 62 and 66, were effectively constructed.
Mechanistic studies
To gain insights into the complete reaction features of this iridium catalysis, we performed DFT calculations using Gaussian 09 program64 at the B3LYP-D3 level of theory with the 6–311 G(d,p) basis set for C, P, N, O, F, Cl, H and LANL2DZ for Ir. Since we utilized a mixed solvent in the experiment, we separately carried out the calculation in both solvents and found the energies in CF3CH2OH (see Supplementary Data 1) are commonly lower than in EtOAc (see Supplementary Data 2). Based on the calculation results and literatures on iridium catalysis65,66,67,68, we proposed a possible reaction pathway (Fig. 5a) and summarized the energy profiles (Fig. 5b). L3, acetophenone 1, and methyl amine were employed to simplify the calculation process. Initially methyl amine coordinates to Ir and breaks the dimer I69, in situ generated from [Ir(cod)Cl]2 and L3, to form complex II, which is substantially stabilized by an H-bonding attraction between the (N)-H of methyl amine and (P)-O of the chiral ligand L3 (Fig. 5b, TS3-S) with the H–O distance at 2.0 Å. This step is rather facile and exergonic by 12.8 kcalmol−1, followed by the oxidative addition, in which H2 is activated and adds to Ir(I) via transition state TS1 to afford Ir(III) (intermediate IV). Then the incoming second H2 molecule prefers to coordinate to the cis-position of P than the trans-position (see competing intermediates IV′ and IV″ in Fig. 5) to form complex V. And with the help of the imine substrate, it heterocleaves and the resulting hydride adds to Ir to form a tight ion pair complex67 VI through transition state TS2 with an activation barrier of 9.9 kcalmol−1.

a Catalytic cycle. b Gibbs energy profiles.
The origins of enantioselectivity occur in the hydride addition step. From the optimized transition state TS3-S we can see that the distance between the chloride ion on Ir and the proton on imine is 2.53 Å, indicating there is an H-bonding interaction (Fig. 5b). The hydride adds from the Si-face of the imine substrate (TS3-S) is thermodynamically favored by 1.6 kcalmol−1 than from the Re-face (TS3-R), to afford the amine product and be back to complex IV with the release of 26.4 kcalmol−1 of Gibbs free energy. In the competing transition state (TS3-R’), there exists a similar H-bonding interaction between one of the hydride ions on Ir and the proton on imine (H-H 2.22 Å, Fig. 5b). During the catalytic cycle, the imine substrate does not coordinate directly with the iridium metal center, that is, the hydride addition is an outer-sphere process, in which the bulky aromatic groups at the 3,3′-positions of the chiral BINOL-based phosphoramidite ligands L3 and L4 contribute by forcing the imine substrate to the outer sphere of the catalytic complex.
We also calculated the Gibbs energy for the “inner-sphere” H-addition pathways, in which the oxidation state of iridium changes from +1 to +3 (see Supplementary Data 3). The two different pathways share early intermediates II, III, and IV, and transition state TS1. Compared with the single hydride addition transition state TS3 of the “outer-sphere” pathway, there are two H-addition transition states for the “inner-sphere” one, IS-TS2 and IS-TS3, through which two hydrides add to C and N of the imine substrate subsequently. The Gibbs free energy for the second transition state, IS-TS3, is much higher than that of TS3, thus enabling the “outer-sphere” alternative to occur more likely.
In summary, we have successfully applied primary alkyl amines as the N-sources in direct catalytic asymmetric reductive aminations of a broad range of ketones. A notable feature of the applied chiral phosphoramidite ligands is their tunability for accommodating our specific substrates. Using as low as 0.02 mol% catalyst, the developed procedure is effective towards both aromatic and aliphatic ketones, offering various chiral secondary alkyl amine in excellent enantioselectivity and high yields. A collection of pharmaceuticals and important intermediates were facially synthesized in one single step. The 10-gram-scale experiment further demonstrates the practical utility of this methodology. The DFT studies reveal the amine substrate serves as a ligand of the transition-metal center and the hydride addition occurs through an outer-sphere transition state, in which there are two H-bonding attractions, one between the (N)-H of amine substrate and (P)-O of the chiral ligand and the other between the chloride ion on Ir and the proton on the imine intermediate. Our protocol greatly broadens the scope of the N-coupling partners for current asymmetric reductive amination research, opening the door for the direct and efficient synthesis of related chiral secondary amines.
Methods
General procedure for direct reductive amination
In a nitrogen-filled glovebox, [Ir(cod)Cl]2 (2.0 mg, 3 mmol) and L4 (3.3 mg, 12.6 mmol) were dissolved in anhydrous trifluoroethanol (2 mL) in a 10 mL vial equipped with a stir bar. The above solution was stirred at room temperature for 20 min to in situ generate the Ir-L4 complex. To a 5 mL vial equipped with a stir bar were added ketone (0.3 mmol) and amine (0.29 mmol, 0.95 equiv.) substrates, followed by the addition of anhydrous trifluoroethanol (0.5 mL), Ti(OiPr)4 (0.36 mmol, 1.2 equiv.), FeCl3 (0.09 mmol, 30 mol%), and the solution of the Ir-L4 complex (50 μL, 0.05 mol%). The total amount of solvent was made to 1.2 mL (CF3CH2OH/MeOAc = 1:1). The resulting vial was transferred to an autoclave, which was purged with H2 3 times and then charged with H2 (40 atm), and stirred at 40 oC for 24 h. After the reaction was complete, the hydrogen gas was released slowly and the reaction solution was concentrated to give the crude products, which were purified by column chromatography (silica gel, petroleum ether/EtOAc from 10/1 to 2/1 with 0.5% Et3N) to afford the final product.
Data availability
The authors declare that the data supporting the conclusions of this study are available within the article and its Supplementary Information file.
References
Nugent, T. C. Chiral Amine Synthesis: Methods, Developments and Applications (Wiley-VCH, 2010).
Li, W. & Zhang, X. Stereoselective Formation of Amines. Top. Curr. Chem. 343, 33–73 (2014).
Trowbridge, A., Walton, S. M. & Gaunt, M. J. New strategies for the transition-metal catalyzed synthesis of aliphatic amines. Chem. Rev. 120, 2613–2692 (2020).
Blaser, H. & Federsel, H. Asymmetric Catalysis on Industrial Scale: Challenges, Approaches and Solutions 2nd edn (Wiley-VCH, 2010).
Xie, J.-H., Zhu, S.-F. & Zhou, Q.-L. Transition metal-catalyzed enantioselective hydrogenation of enamines and imines. Chem. Rev. 111, 1713–1760 (2011).
Ager, D. J., de Vries, A. H. M. & de Vries, J. G. Asymmetric homogeneous hydrogenations at scale. Chem. Soc. Rev. 41, 3340–3380 (2012).
Seo, C. S. G. & Morris, R. H. Catalytic homogeneous asymmetric hydrogenation: successes and opportunities. Organometallics 38, 47–65 (2019).
Barrios-Rivera, J., Xu, Y., Wills, M. & Vyas, V. K. A diversity of recently reported methodology for asymmetric imine reduction. Org. Chem. Front. 7, 3312–3342 (2020).
Cabré, A., Verdaguer, X. & Riera, A. Recent advances in the enantioselective synthesis of chiral amines via transition metal-catalyzed asymmetric hydrogenation. Chem. Rev. 122, 269–339 (2022).
Wang, C. & Xiao, J. Asymmetric reductive amination. Top. Curr. Chem. 343, 261–282 (2014).
Afanasyev, O. I., Kuchuk, E., Usanov, D. L. & Chusov, D. Reductive amination in the synthesis of pharmaceuticals. Chem. Rev. 119, 11857–11911 (2019).
Irrgang, T. & Kempe, R. Transition-metal-catalyzed reductive amination employing hydrogen. Chem. Rev. 120, 9583–9674 (2020).
Murugesan, K. et al. Catalytic reductive aminations using molecular hydrogen for synthesis of different kinds of amines. Chem. Soc. Rev. 49, 6273–6328 (2020).
Tian, Y., Hu, L., Wang, Y.-Z., Zhang, X. & Yin, Q. Recent advances on transition-metal-catalysed asymmetric reductive amination. Org. Chem. Front. 8, 2328–2342 (2021).
Reshi, N. U. D., Saptal, V. B., Beller, M. & Bera, J. K. Recent progress in transition-metal-catalyzed asymmetric reductive amination. ACS Catal. 11, 13809–13837 (2021).
Williams, G. D., Pike, R. A., Wade, C. E. & Wills, M. A one-pot process for the enantioselective synthesis of amines via reductive amination under transfer hydrogenation conditions. Org. Lett. 5, 4227–4230 (2003).
Strotman, N. A. et al. Reaction development and mechanistic study of a ruthenium catalyzed intramolecular asymmetric reductive amination en route to the dual orexin inhibitor Suvorexant (MK-4305). J. Am. Chem. Soc. 133, 8362–8371 (2011).
Zhou, H., Liu, Y., Yang, S., Zhou, L. & Chang, M. One-pot N-deprotection and catalytic intramolecular asymmetric reductive amination for the synthesis of tetrahydroisoquinolines. Angew. Chem. Int. Ed. 56, 2725–2729 (2017).
Song, B., Yu, C.-B., Ji, Y., Chen, M.-W. & Zhou, Y.-G. Synthesis of chiral sultams via palladiumcatalyzed intramolecular asymmetric reductive amination. Chem. Commun. 53, 1704–1707 (2017).
Song, B., Chen, M.-W. & Zhou, Y.-G. Facile synthesis of chiral indolines through asymmetric hydrogenation of in situ generated indoles. Org. Chem. Front. 5, 1113–1117 (2018).
Chen, Y., He, Y.-M., Zhang, S., Miao, T. & Fan, Q.-H. Rapid construction of structurally diverse quinolizidines, indolizidines, and their analogues via ruthenium-catalyzed asymmetric cascade hydrogenation/reductive amination. Angew. Chem. Int. Ed. 58, 3809–3813 (2019).
Yang, T., Guo, X., Yin, Q. & Zhang, X. Intramolecular asymmetric reductive amination: synthesis of enantioenriched dibenz[c,e]azepines. Chem. Sci. 10, 2473–2477 (2019).
Wang, L.-R. et al. Highly enantioselective ruthenium-catalyzed cascade double reduction strategy: construction of structurally diverse julolidines and their analogues. Org. Lett. 22, 2251–2255 (2020).
Kadyrov, R. & Riermeier, T. H. Highly enantioselective hydrogen-transfer reductive amination: catalytic asymmetric synthesis of primary amines. Angew. Chem. Int. Ed. 42, 5472–5474 (2003).
Steinhuebel, D., Sun, Y., Matsumura, K., Sayo, N. & Saito, T. Direct asymmetric reductive amination. J. Am. Chem. Soc. 131, 11316–11317 (2009).
Lou, Y. et al. Dynamic kinetic asymmetric reductive amination: synthesis of chiral primary β-amino lactams. Angew. Chem. Int. Ed. 57, 14193–14197 (2018).
Tan, X. et al. Asymmetric synthesis of chiral primary amines by ruthenium-catalyzed direct reductive amination of alkyl aryl ketones with ammonium salts and molecular H2. J. Am. Chem. Soc. 140, 2024–2027 (2018).
Hu, L., Zhang, Y., Zhang, Q.-W., Yin, Q. & Zhang, X. Ruthenium-catalyzed direct asymmetric reductive amination of diaryl and sterically hindered ketones with ammonium salts and H2. Angew. Chem. Int. Ed. 59, 5321–5325 (2020).
Gao, Z., Liu, J., Huang, H., Geng, H. & Chang, M. An iridium catalytic system compatible with inorganic and organic nitrogen sources for dual asymmetric reductive amination reactions. Angew. Chem. Int. Ed. 60, 27307–27311 (2021).
Gallardo-Donaire, J. et al. Direct asymmetric ruthenium-catalyzed reductive amination of alkyl−aryl ketones with ammonia and hydrogen. J. Am. Chem. Soc. 140, 355–361 (2018).
Blaser, H.-U., Buser, H.-P., Jalett, H.-P., Pugin, B. & Spindler, F. Iridium ferrocenyl diphosphine catalyzed enantioselective reductive alkylation of a hindered aniline. Synlett. 1999, 867−868 (1999).
Rubio-Pérez, L., Pérez-Flores, F. J., Sharma, P., Velasco, L. & Cabrera, A. Stable preformed chiral palladium catalysts for the one-pot asymmetric reductive amination of ketones. Org. Lett. 11, 265–268 (2009).
Li, C., Villa-Marcos, B. & Xiao, J. Metal-Brønsted acid cooperative catalysis for asymmetric reductive amination. J. Am. Chem. Soc. 131, 6967–6969 (2009).
Yang, P., Lim, L., Chuanprasit, P., Hirao, H. & Zhou, J. Nickel-catalyzed enantioselective reductive amination of ketones with both arylamines and benzhydrazide. Angew. Chem. Int. Ed. 55, 12083–12087 (2016).
Liu, R. et al. Iridium-catalyzed enantioselective reductive amination of aromatic ketones. Catal. Sci. Technol. 10, 5448–5452 (2020).
Wu, Z. et al. Secondary amines as coupling partners in direct catalytic asymmetric reductive amination. Chem. Sci. 10, 4509–4514 (2019).
Tararov, V. I., Kadyrov, R., Riermeier, T. H. & Borner, A. On the reductive amination of aldehydes and ketones catalyzed by homogeneous Rh(i) complexes. Chem. Commun. 1867−1868 (2000).
Kadyrov, R., Riermeier, T. H., Dingerdissen, U., Tararov, V. & Borner, A. The first highly enantioselective homogeneously catalyzed asymmetric reductive amination: synthesis of α-N-benzylamino acids. J. Org. Chem. 68, 4067–4070 (2003).
Huang, H., Liu, X., Zhou, L., Chang, M. & Zhang, X. Direct asymmetric reductive amination for the synthesis of chiral β-arylamines. Angew. Chem. Int. Ed. 55, 5309–5312 (2016).
Huang, H., Zhao, Y., Yang, Y., Zhou, L. & Chang, M. Direct catalytic asymmetric reductive amination of aliphatic ketones utilizing diphenylmethanamine as coupling partner. Org. Lett. 19, 1942–1945 (2017).
Huang, H., Wu, Z., Gao, G., Zhou, L. & Chang, M. Iridium-catalyzed direct asymmetric reductive amination of aromatic ketones. Org. Chem. Front. 4, 1976–1980 (2017).
Chen, Z.-P., Hu, S.-B., Zhou, J. & Zhou, Y.-G. Synthesis of chiral trifluoromethyl-substituted hydrazines via Pd-catalyzed asymmetric hydrogenation and reductive amination. ACS Catal. 5, 6086–6089 (2015).
Chang, M., Liu, S. & Zhang, X. Direct catalytic asymmetric reductive amination of simple aromatic ketones. Org. Lett. 15, 4354–4357 (2013).
Chen, Z.-P., Hu, S.-B., Chen, M.-W. & Zhou, Y.-G. Synthesis of chiral fluorinated hydrazines via Pd-catalyzed asymmetric hydrogenation. Org. Lett. 18, 2676–2679 (2016).
Verzijl, G. K. M., Schuster, C., Dax, T., de Vries, A. H. M. & Lefort, L. Asymmetric synthesis of a key intermediate for Tofacitinib via a dynamic kinetic resolution-reductive amination protocol. Org. Process Res. Dev. 22, 1817–1822 (2018).
Ros, A. et al. Transfer hydrogenation of α-branched ketimines: enantioselective synthesis of cycloalkylamines via dynamic kinetic resolution. Adv. Synth. Catal. 347, 1917–1920 (2005).
Han, Z., Wang, Z., Zhang, X. & Ding, K. Spiro[4,4]-1,6-nonadiene-based phosphine–oxazoline ligands for iridium-catalyzed enantioselective hydrogenation of ketimines. Angew. Chem. Int. Ed. 48, 5345–5349 (2009).
Salomo, E. et al. Direct asymmetric hydrogenation of N-methyl and N-alkyl imines with an Ir(III)H catalyst. J. Am. Chem. Soc. 140, 16967–16970 (2018).
Verdaguer, X., Lange, U. E. W., Reding, M. T. & Buchwald, S. L. Highly enantioselective imine hydrosilylation using (S,S)-ethylenebis(η5-tetrahydroindenyl)titanium difluoride. J. Am. Chem. Soc. 118, 6784–6785 (1996).
Chen, F. et al. Asymmetric hydrogenation of N-alkyl ketimines with phosphine-free, chiral, cationic Ru-MsDPEN catalysts. Chem. Eur. J. 17, 1109–1113 (2011).
Wakchaure, V. N., Kaib, P. S. J., Leutzsch, M. & List, B. Disulfonimide-catalyzed asymmetric reduction of N-alkyl imines. Angew. Chem. Int. Ed. 54, 11852–11856 (2015).
Blasius, C. K., Heinrich, N. F., Vasilenko, V. & Gade, L. H. Tackling N-alkyl imines with 3d metal catalysis: highly enantioselective iron-catalyzed synthesis of α-chiral amines. Angew. Chem. Int. Ed. 59, 15974–15977 (2020).
Minnaard, A. J., Feringa, B. L., Lefort, L. & de Vries, J. G. Asymmetric hydrogenation using monodentate phosphoramidite ligands. Acc. Chem. Res. 40, 1267–1277 (2007).
Fu, W. & Tang, W. Chiral monophosphorus ligands for asymmetric catalytic reactions. ACS Catal. 6, 4814–4858 (2016).
Mitschke, B., Turberg, M. & List, B. Confinement as a unifying element in selective catalysis. Chem 6, 2515–2532 (2020).
Vogl, E. M., Gröger, H. & Shibasaki, M. Towards perfect asymmetric catalysis: additives and cocatalysts. Angew. Chem. Int. Ed. 38, 1570–1577 (1999).
Yu, Z., Jin, W. & Jiang, Q. Brønsted acid activation strategy in transition-metal catalyzed asymmetric hydrogenation of N-unprotected imines, enamines, and N-heteroaromatic compounds. Angew. Chem. Int. Ed. 51, 6060–6072 (2012).
Hong, L., Sun, W., Yang, D., Li, G. & Wang, R. Additive effects on asymmetric catalysis. Chem. Rev. 116, 4006–4123 (2016).
Mattson, R. J., Pham, K. M., Leuck, D. J. & Cowen, K. A. An improved method for reductive alkylation of amines using titanium(IV) isopropoxide and sodium cyanoborohydride. J. Org. Chem. 55, 2552–2554 (1990).
Martínez-Montero, L., Díaz-Rodríguez, A., Gotor, V., Gotor-Fernández, V. & Lavandera, I. Broadening the chemical scope of laccases: selective deprotection of N-benzyl groups. Green. Chem. 17, 2794–2798 (2015).
Wakchaure, V. N., Nicoletti, M., Ratjen, L. & List, B. Towards a practical Brønsted acid catalyzed and hantzsch ester mediated asymmetric reductive amination of ketones with benzylamine. Synlett 18, 2708–2710 (2010).
Breuer, M. et al. Industrial methods for the production of optically active intermediates. Angew. Chem. Int. Ed. 43, 788–824 (2004).
Liu, T.-L., Wang, C.-J. & Zhang, X. Synthesis of chiral aliphatic amines through asymmetric hydrogenation. Angew. Chem. Int. Ed. 52, 8416–8419 (2013).
Frisch. M. J. et al. Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2013.
Dobereiner, G. E. et al. Iridium-catalyzed hydrogenation of N-heterocyclic compounds under mild conditions by an outer-sphere pathway. J. Am. Chem. Soc. 133, 7547–7562 (2011).
Li, M.-L. et al. Mechanism studies of Ir-catalyzed asymmetric hydrogenation of unsaturated carboxylic acids. J. Am. Chem. Soc. 139, 541–547 (2017).
Qu, B. et al. Enantioselective synthesis of α-(hetero)aryl piperidines through asymmetric hydrogenation of pyridinium salts and its mechanistic insights. Org. Lett. 20, 1333–1337 (2018).
Cui, C.-X. et al. Mechanism of Ir-catalyzed hydrogenation: a theoretical view. Coord. Chem. Rev. 412, 213–251 (2020).
Nagano, T. et al. Additive effects of amines on asymmetric hydrogenation of quinoxalines catalyzed by chiral iridium complexes. Chem. Eur. J. 18, 11578–11592 (2012).
Acknowledgements
Financial support from the National Natural Science Foundation of China (21772155, M.C.) and the Scientific Fund of Northwest A&F is gratefully acknowledged. We greatly acknowledge the HPC of Northwest A&F University for the DFT calculations carried out in this work.
Author information
Authors and Affiliations
Contributions
Z.W. and H.G. established the reaction conditions. W.W. performed the DFT calculations. Z.W., G.G., and H.H. expanded the substrate scope. M.C. conceived and supervised the project and wrote the manuscript. All the authors discussed the results and commented on the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wu, Z., Wang, W., Guo, H. et al. Iridium-catalyzed direct asymmetric reductive amination utilizing primary alkyl amines as the N-sources. Nat Commun 13, 3344 (2022). https://doi.org/10.1038/s41467-022-31045-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-022-31045-5
- Springer Nature Limited
This article is cited by
-
Effect of Cu − hydrazine complex formation on HyBRID decontamination reactions for magnetite, nickel ferrite, and chromite
Carbon Letters (2024)
-
Nickel-catalyzed enantioselective reductive amination of benzylic ketones in alcohols
Science China Chemistry (2024)
-
Practical N-alkylation via homogeneous iridium-catalyzed direct reductive amination
Science China Chemistry (2023)