

Abstract
The axially chiral indole-aryl motifs are present in natural products and biologically active compounds as well as in chiral ligands. Atroposelective indole formation is an efficient method to construct indole-based biaryls. We report herein the result of a chiral phosphoric acid catalyzed asymmetric cycloaddition of 3-alkynylindoles with azonaphthalenes. A class of indole-based biaryls were prepared efficiently with excellent yields and enantioselectivities (up to 98% yield, 99% ee). Control experiment and DFT calculations illustrate a possible mechanism in which the reaction proceeds via a dearomatization of indole to generate an allene-iminium intermediate, followed by an intramolecular aza-Michael addition. This approach provides a convergent synthetic strategy for enantioselective construction of axially chiral heterobiaryl backbones.
Similar content being viewed by others
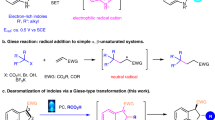
Introduction
Axially chiral compounds are present in a plethora of natural products, biologically active compounds, and privileged chiral ligands and catalysts1,2,3. Consequently, developing protocols for the catalytic enantioselective construction of axially chiral scaffolds has attracted a great deal of interest among researchers in synthetic chemistry4,5. Many strategies have been developed, including desymmetrization or kinetic resolution of prochiral biaryls6,7,8,9,10,11,12, aryl–aryl coupling13,14,15, direct arylation16,17,18,19,20,21,22, chirality conversion23,24,25,26,27,28,29,30,31,32,33, cycloaddition34,35,36,37,38,39, and so on40,41,42,43,44,45,46,47,48. Recently, the catalytic enantioselective construction of chiral atropisomeric heterobiaryl backbones has received increasing attention from the chemistry community. However, there are still some challenges to be overcome in this field of research.
Indole-based biaryls have been found in nature and have been used as ligands for asymmetric synthesis49,50,51. Recently, a few outstanding cases of the enantioselective synthesis of indole–aryl skeletons have been reported22,29,33,52,53,54,55,56,57,58,59,60,61,62,63,64,65,66. Generally, there are two strategies to construct indole-based biaryls enantioselectively. One is asymmetric coupling reaction of indole derivatives with other aryls22,56,57,58,59,60,61,62, the other is in situ construction of indole skeletons atroposelectively63,64,65,66. Interestingly, o-alkynylanilines were usually employed for the synthesis of indoles possessing chiral axis by the latter strategy. For instance, Kitagawa and coworkers reported an elegant Pd(II)-catalyzed enantioselective hydroamination reaction to construct indoles bearing an N-aryl chiral axis53,55. Li and coworkers merged C−H activation with nucleophilic cyclization to realize oxidative coupling of indoles and o-alkynylanilines for the synthesis of biindolyls63. Zhu and coworkers developed a palladium-catalyzed asymmetric Cacchi reaction for the construction of indoles bearing a chiral axis66. Yan and coworkers established an organocatalytic enantioselective construction of chiral indole-aryl skeletons through an asymmetric annulation64. Notably, the intramolecular annulation strategy, a linear synthesis has been adopted to form a single C–N bond to generate the indole skeleton in all these protocols (Fig. 1a). While the intermolecular strategy, a convergent synthesis strategy for the atroposelective construction of indole–aryls remains limited. Therefore, to realize a divergent synthesis of such indole-based biaryls, designing of more efficient strategies is of great significance.

a Construction of indole derivatives via intramolecular cyclization. b CPA-catalyzed atroposelective intermolecular [3 + 2] cycloaddition.
In fact, transition metal catalyzed cyclization reaction of azobenzene derivatives with alkynes to construct indole derivatives without chiral axis has been reported67,68,69. Although organo-catalyzed cycloaddition of alkenes with azonaphthalenes has been well documented22,29,33,70, the organo-catalyzed asymmetric cycloaddition reaction of azo benzene derivatives with alkynes is still lacking. The reason may be ascribed to a relative lower reactivity of alkyne than that of alkene in the cycloaddition reaction with azonaphthalenes. We envisioned that installation of electron-donating groups to alkyne moiety would enhance such reactivity, and further activated by organocatalyst would realize the desired cycloaddition to give the corresponding indole–aryl atropisomers.
In order to achieve this goal, we needed to overcome the following issues: (1) realization of efficient annulation of alkynes with azonaphthalenes; (2) the installation of a suitable group onto the 1-ethynylnaphthene to avoid free rotation around the C–C axis; (3) the choice of a competent chiral catalyst to activate reaction partners and to control the stereoselectivity. Herein, we report a formal [3 + 2] cycloaddition strategy for atroposelective construction of indole-based biaryls via chiral phosphoric acid (CPA)-catalyzed cycloaddition reaction of 3-alkynylindoles with azonaphthalenes (Fig. 1b).
Results
We commenced the reaction of 1-ethynylnaphthol with azonaphthalene to examine the possibility of our strategy. Unfortunately, no reaction occurred when a series of reaction conditions were attempted (for details, see Supplementary Table 1). The reason may be due to that the electron density of the alkyne was still insufficient to perform the nucleophilic addition to azonaphthalenes. Then we changed the naphthols with more electron-rich indoles, 3-alkynylindol derivatives as the substrate. To our delight, the desired cycloaddition products could be obtained smoothly.
As shown in Table 1, chiral phosphoric acid 16a (10 mol%) catalyzed the reaction of 3-alkynylindole 1a with azonaphthalene 8a in toluene at −50 oC for 72 h, the desired cycloaddition product 9a was obtained in moderate yield and enantioselectivity (entry 1). An increase in enantioselectivity was realized when catalyst 16b was used (entry 2). The CPA 16c, bearing a 9-phenanthryl group at the 6,6′-position of SPINOL, provided the desired product 9a with good yield and enantioselectivity (87%, 86% ee, entry 3). Having realized the efficiency of the annulation, we turned our effort to improve the enantioselectivity. First, the installation of suitable protection groups onto the naphthol of 3-alkynylindole derivatives was performed to avoid free rotation around the C–C axis during the reaction. As expected, the enantioselectivity declined to around 40% when methoxymethyl was substituted with a less bulky methyl group (entry 4). On the other hand, the more bulky group benzyl or tert-Bu has an enhancing effect on the enantioselectivity, albeit with the reaction efficiency was dramatically decreased, probably due to the steric reason (entries 5,6 vs entry 4). These results demonstrated that the R group plays an important role in the stereocontrol in this reaction. The use of substrate 5a containing a cyclohexyl group could afford the corresponding product in 65% yield with 87% ee (entry 7). Interestingly, iso-Pr returned the desired product in 95% yield with 90% ee (entry 8). Moreover, 7a bearing a 3-pentyl group was found to be the optimal substrate, affording the desired product 15a in good yield and excellent enantioselectivity (75%, 94% ee, entry 9). Further optimization of the reaction by a variety of solvents (CH2Cl2, CH3CN, CCl4, and CHCl3) was subsequently investigated (entries 10–13), and CHCl3 gave the best result (97% ee, entry 13). In addition, a further decrease of the reaction temperature could not improve the enantioselectivity of the reaction (entry 14). Meanwhile, increasing the temperature to −25 oC or 0 oC has a negative effect on the reaction transformation (entries 15–16). Fortunately, when the catalyst loading was reduced to 5 mol% or 2 mol%, the enantioselectivity almost remained unchanged and excellent yields were still obtained (entries 17–18). Further reducing the amount of the catalyst to 1 mol% resulted in a measurable decrease in the yield (entry 19). The thermal racemization of these atropisomers was conducted to investigate the configurational stability (for details, see Supplementary Information). Compound 15a displays an excellent stability with a barrier to enantiomerization of 173 kJ mol−1, and the half-life time of racemization was determined to be 3.3 × 109 years at 25 oC.
With the optimal reaction conditions in hand, we examined the substrate scope of the protocol (Fig. 2). First, the substitution effect at the indole was examined. Substituents of the phenyl ring of the indole core with halogens at the 5-position, such as F, Cl, and Br, afforded the corresponding axially chiral products with excellent yields and enantioselectivities (15b–d, 90–96%, 97–98% ee). The methyl or methoxy-group substituent at the 5-position of the indole could be well tolerated to give the expected products 15e and 15f, respectively, with excellent results (up to 98% yield, 98% ee). In addition, product 15g with a 5-Ph substituent indole was obtained in 75% yield and 96% ee. The halogen (F, Cl, and Br)-containing substrates at the 6-position of the indole were tolerated under the standard conditions, all proceeding in excellent yields and enantioselectivities (15h–j, 97–98%, 95–97% ee). Substrates with the substituents CH3 and Ph at the 6-position of the indole all resulted in the corresponding desired products with good yields and excellent enantioselectivities (15k, 15l, 97% ee). A 7-chloro-substituted indole substrate 7m returned an 87% yield with 98% ee. Interestingly, when chloro substituent was installed at the 4-position of the indole, the corresponding product 15n bearing two stereogenic axes was obtained in good yield with excellent enantioselectivity and moderate diastereoselectivity. Substrate 7o was also used to construct the compound containing two stereogenic axes, unfortunately, poor stereoselectivity was observed.

aReactions were carried out with 7 (0.24 mmol), 8 (0.20 mmol), and 16c (0.004 mmol) in CHCl3 (2.0 mL) at −50 °C under N2. The yields shown are for isolated products and the ee values were determined by HPLC analysis. bThe reaction temperature is 40 °C.
Next, we examined the substrate scope with regard to the naphthyl ring moiety and other azonaphthalenes. 3-Pentan-3-yloxy at the 3-position of the naphthyl ring resulted in the desired product 15p in 80% yield with 93% ee. Different substituents at the 6-position of the naphthyl ring, such as CH3, Br, and Ph all could afford the desired products in good yields and excellent enantioselectivities (15q–s, 73–98% yields, 97–98% ee). Moreover, both electron-rich and electron-deficient substituents at the 7-position of the naphthyl ring returned the desired axially chiral compounds in up to 97% yield with up to 99% ee (15t–v). On the other hand, the substrates with different substituents at the 6 or 7-position of the azonaphthalene were well tolerated and delivered the corresponding products 15w–z in high yields (63–95%) and excellent enantioselectivities (92–99% ee). Among them, the product 15y was obtained in 63% yield and 93% ee at 40 °C. Finally, azonaphthalene with different N-protecting groups could be employed, delivering the corresponding axially chiral products in moderate-to-good yields (52–83%) and excellent enantioselectivities (15aa–ae, 94–97% ee). Besides naphthyl-substituted alkynylindole, phenyl-substituted alkynylindole 7af was also treated under the optimal reaction conditions, satisfyingly, the desired axially chiral product 15af was obtained in 79% yield and 80% ee. The relatively lower enantioselectivity may be due to its lower configurational stability. Indeed, compound 15af displays a lower stability with a barrier to enantiomerization of 100 kJ mol−1, and the half-life time of racemization was determined to be 4.8 hours at 25 oC (for details, see Supplementary Information). The absolute configuration of the product was assigned on the basis of the X-ray crystallographic structure of 15x.
Interestingly, compound 15ag bearing two chiral axes can be generated with 94% ee by this strategy starting from 7ag, albeit a lower yield (Fig. 3). To test the practicality of this method, a gram-scale reaction of 7a with 8a was carried out under the standard reaction conditions. As expected, compound 15a was obtained successfully with excellent yield and enantioselectivity (Fig. 4a). In addition, reduction of 15a with LiAlH4 provided the secondary amine product 17 in good yield and no changes occurred in the stereoselectivity during this reaction. The 3-pentyl group could also be removed smoothly by using BBr3 in CH2Cl2 for 4 h, giving phenol product 18 in quantitative yield without any loss of enantiopurity, which is a useful building block for further transformation. Trifluoromethylation of 18 afforded product 19 in excellent yield and enantioselectivity (95% yield, 97% ee), which is a potential partner in metal-catalyzed coupling reactions (Fig. 4b). The protecting group on the azonitrogen center could be removed, affording compound 20 in good yield. The axially chiral benzoindolenaphthyl-derived thiourea compound 21 bearing 3,5-bis(trifluoromethyl)phenyl group was prepared smoothly in 90% yield according to the classical approach for the preparation of cinchona-derived thiourea catalyst (Fig. 4c).

The reaction was carried out with 7ag (0.05 mmol), 8a (0.125 mmol), and 16c (0.002 mmol) in CHCl3 (0.5 mL) at −50 °C under N2.

a Gram-scale synthesis of axially chiral compound 15a. b Transformation of compound 15a. c Synthesis of axially chiral organocatalyst.
On the basis of the above transformation, we attempted to transform compound 15a into potential useful phosphine ligands to showcase the practical value of the products. As shown in Fig. 5, treatment of 20 with [Ir[dF(CF3)ppy]2(dtbbpy)PF6] catalyst under 5w blue LED lamp, in CH3OH/CH3CN provided 22. Compound 24 was prepared from 22, and the enantioselectivity was retained even after three steps (98% ee). Finally, compound 24 was transformed into the benzoindolenaphthyl phosphine ligand 25 by reduction with HSiCl3 reagent. A preliminary application of 25 (98% ee) as ligand in the asymmetric allylation reaction of (E)-1,3-diphenylallyl acetate with dimethyl malonate revealed that 25 is efficient at promoting the reaction (99% yield), although the enantioselectivity of product 28 needs to be improved (39% ee).

a Synthesis of chiral phosphine ligand 25. b Asymmetric allylation using 25 as the ligand.
To get mechanistic insights into this reaction, a control experiment was carried out under the optimal conditions. The desired product could not be obtained when the N–H group of 7a was protected with methyl, which demonstrated that the N–H group plays an important role in the process. To better understand the mechanism and origins of stereo- and regioselectivity, density-functional theory (DFT) calculations were conducted using Gaussian 09 program. All the intermediates and transition states were optimized by employing the xc-functional ωB97X-D with dispersion correction (ωB97X-D) and the 6-31G(d) basis set. Normal vibrational mode analysis at the same level of theory confirmed that the optimized structures are minima (zero imaginary frequency) or saddle points (one imaginary frequency). Based on optimized geometries, single-point energies and solvent effects in chloroform were computed with the range-separated functional ωB97X-D and the 6-311G (d,p) basis set using the SMD model. To save computational time, the phenanthryl moieties in the catalyst were replaced with methyls and 3-pentyl in the substrate with methyl. The free-energy profiles (in kcal/mol) of the most favorable pathway for the phosphoric acid-catalyzed cycloaddition of alkynylindoles with azonaphthalenes are shown in Supplementary Fig. 2. First, the chiral phosphoric acid acts as a bifunctional catalyst to simultaneously activate 3-alkynylindoles and azosubstrates through a dual hydrogen-bonding activation mode to generate a complex. Subsequent dearomatization of 3-substituted indole generates the transient allene-iminium intermediate62,71,72,73,74, which further undergoes an intramolecular aza-Michael addition to afford the axially chiral intermediate. Finally, deprotonation and aromatization yields the desired product.
Figure 6 shows the optimized structures and relative free energies of the four competing stereoselectivity-determining allene–iminium transition states TS-1a, TS-1b, TS-1c, and TS-1d, and the structures of their corresponding allene–iminium intermediates. Indeed, the calculated re-face attack TS-1a is found to be 3.7 kcal/mol lower in the relative free energy than the si-face attack in TS-1b. The potential energy of the allene–iminium intermediate INT-2a was also found to be lower than that of the intermediate INT-2b by 0.89 kcal/mol (for details, see Supplementary Information). Transition states TS-1a and TS-1c would generate S- and R-configuration product, respectively. The relative activation-free energy (ΔΔG) for TS-1a is 12.98 kcal/mol more favorable than TS-1c, which corroborated the observed enantioselectivity that the (S)-selective product was obtained. In addition, the TS-1a that ultimately leads to the major conformation product is 6.22 kcal/mol more stable than TS-1d, which is in good agreement with the experimental result.

DFT-optimized chiral phosphoric acid-catalyzed transition states TS-1a–d, and their corresponding allene–iminium intermediates INT-2a–d.
Discussion
In this work, we show the CPA-catalyzed intermolecular [3 + 2] formal cycloaddition of 3-alkynylindoles with azonaphthalene for the enantioselective construction of indole-based atropisomers. A type of axially chiral arylindole derivatives were afforded in high yields and excellent stereoselectivities (up to 98% yield, 99% ee). In addition, this transformation allows lowering of the catalyst loading to 2 mol% without considerable loss of reactivity and enantioselectivity. This reaction not only provides a useful method for constructing enantioenriched indole-based biaryl atropisomer scaffolds, but also advances the chemistry of catalytic asymmetric reactions of alkynylindoles.
Methods
General procedure for the enantioselective synthesis of 15
To a stirred solution of 7 (0.24 mmol), 8 (0.20 mmol) and CPA16c (0.004 mmol) in CHCl3 (2.0 mL) at −50 °C in one portion, the mixture was stirred until TLC revealed the absence of the starting material. The solvent was removed under reduced pressure, the residue was purified by flash-column chromatography (petroleum ether/EtOAc) to yield the corresponding product 15.
Synthesis of 18
To a mixture of 15a (113 mg, 0.2 mmol, 1.0 eq) in CH2Cl2 (2 mL) at −78 °C was added BBr3 (0.2 mL, 1.0 mol/L CH2Cl2, 1.0 eq) dropwise, and the mixture was stirred for 4 hours at room temperature. TLC showed complete consumption of the starting material. The reaction was quenched with 10% NaHCO3 solution and extracted with CH2Cl2 (3 × 5 mL). The combined organics were washed with brine and dried over Na2SO4. The solvent was removed under reduced pressure, the residue was purified by column chromatography on silica gel (20% EtOAc in hexane) to yield 18 as a white solid (98.4 mg, 99% yield).
Synthesis of 19
The reaction was performed using 18 (98.4 mg, 0.2 mmol, 1.0 eq), 4-nitrophenyl trifluoromethanesulfonate (65 mg, 0.24 mmol, 1.2 eq), and K2CO3 (41.5 mg. 0.3 mmol, 1.5 eq) in THF (2.0 mL) at 40 oC. After finishing, the reaction was quenched with H2O (5 mL). The solution was extracted with CH2Cl2 (3 × 5 mL), washed with brine (5.0 mL), and dried over anhydrous Na2SO4. The solvent was evaporated under reduced pressure. The crude product was purified by flash-column chromatography (10% EtOAc in hexane) giving 19 as a white solid (119.5 mg, 95% yield).
Data availability
The X-ray crystallographic coordinates for structures that support the findings of this study have been deposited at the Cambridge Crystallographic Data Centre (CCDC) with the accession code CCDC 2005065 (15x) (www.ccdc.cam.ac.uk/data_request/cif). The authors declare that all other data supporting the findings of this study are available within the article and Supplementary Information files, and also are available from the corresponding author upon request.
References
Tang, W. & Zhang, X. New chiral phosphorus ligands for enantioselective hydrogenation. Chem. Rev. 103, 3029–3070 (2003).
Kozlowski, M. C., Morgan, B. J. & Linton, E. C. Total synthesis of chiral biaryl natural products by asymmetric biaryl coupling. Chem. Soc. Rev. 38, 3193–3207 (2009).
Bringmann, G., Gulder, T., Gulder, T. A. M. & Breuning, M. Atroposelective total synthesis of axially chiral biaryl natural products. Chem. Rev. 111, 563–639 (2011).
Wencel-Delord, J., Panossian, A., Leroux, F. R. & Colobert, F. Recent advances and new concepts for the synthesis of axially stereoenriched biaryls. Chem. Soc. Rev. 44, 3418–3430 (2015).
Wang, Y.-B. & Tan, B. Construction of axially chiral compounds via asymmetric organocatalysis. Acc. Chem. Res. 51, 534–547 (2018).
Mori, K. et al. Enantioselective synthesis of multisubstituted biaryl skeleton by chiral phosphoric acid catalyzed desymmetrization/kinetic resolution sequence. J. Am. Chem. Soc. 135, 3964–3970 (2013).
Armstrong, R. J. & Smith, M. D. Catalytic enantioselective synthesis of atropisomeric biaryls: a cation-directed nucleophilic aromatic substitution reaction. Angew. Chem. Int. Ed. 53, 12822–12826 (2014).
Mori, K., Itakura, T. & Akiyama, T. Enantiodivergent atroposelective synthesis of chiral biaryls by asymmetric transfer hydrogenation: chiral phosphoric acid catalyzed dynamic kinetic resolution. Angew. Chem. Int. Ed. 55, 11642–11646 (2016).
Wang, J., Chen, M.-W., Ji, Y., Hu, S.-B. & Zhou, Y.-G. Kinetic resolution of axially chiral 5- or 8-substituted quinolines via asymmetric transfer hydrogenation. J. Am. Chem. Soc. 138, 10413–10416 (2016).
Yu, C., Huang, H., Li, X., Zhang, Y. & Wang, W. Dynamic kinetic resolution of biaryl lactones via a chiral bifunctional amine thiourea-catalyzed highly atropo-enantioselective transesterification. J. Am. Chem. Soc. 138, 6956–6959 (2016).
Jolliffe, J. D., Armstrong, R. J. & Smith, M. D. Catalytic enantioselective synthesis of atropisomeric biaryls by a cation-directed O-alkylation. Nat. Chem. 9, 558–562 (2017).
Liu, Y., Tse, Y.-L. S., Kwong, F. Y. & Yeung, Y.-Y. Accessing axially chiral biaryls via organocatalytic enantioselective dynamic-kinetic resolution-semipinacol rearrangement. ACS Catal. 7, 4435–4440 (2017).
Cammidge, A. N. & Crépy, K. V. L. The first asymmetric Suzuki cross-coupling reaction. Chem. Commun. 1723-1724 (2000).
Guo, Q.-X. et al. Highly enantioselective oxidative couplings of 2-naphthols catalyzed by chiral bimetallic oxovanadium complexes with either oxygen or air as oxidant. J. Am. Chem. Soc. 129, 13927–13938 (2007).
Yamaguchi, K., Yamaguchi, J., Studer, A. & Itami, K. Hindered biaryls by C–H coupling: bisoxazoline-Pd catalysis leading to enantioselective C–H coupling. Chem. Sci. 3, 2165–2169 (2012).
De, C. K., Pesciaioli, F. & List, B. Catalytic asymmetric benzidine rearrangement. Angew. Chem. Int. Ed. 52, 9293–9295 (2013).
Li, G.-Q. et al. Organocatalytic aryl–aryl bond formation: an atroposelective [3,3]-rearrangement approach to BINAM derivatives. J. Am. Chem. Soc. 135, 7414–7417 (2013).
Pan, C., Zhu, Z., Zhang, M. & Gu, Z. Palladium-catalyzed enantioselective synthesis of 2-aryl cyclohex-2-enone atropisomers: platform molecules for the divergent synthesis of axially chiral biaryl compounds. Angew. Chem. Int. Ed. 56, 4777–4781 (2017).
Bringmann, G. et al. Atroposelective synthesis of axially chiral biaryl compounds. Angew. Chem. Int. Ed. 44, 5384–5427 (2005).
Chen, Y.-H. et al. Atroposelective synthesis of axially chiral biaryldiols via organocatalytic arylation of 2-Naphthols. J. Am. Chem. Soc. 137, 15062–15065 (2015).
Wang, J.-Z. et al. Symmetry in cascade chirality-transfer processes: a catalytic atroposelective direct arylation approach to BINOL derivatives. J. Am. Chem. Soc. 138, 5202–5205 (2016).
Qi, L.-W., Mao, J.-H., Zhang, J. & Tan, B. Organocatalytic asymmetric arylation of indoles enabled by azo groups. Nat. Chem. 10, 58–64 (2018).
Yang, H., Chen, J. & Zhou, L. Construction of axially chiral compounds via central-to-axial chirality conversion. Chem. Asian J. 15, 2939–2951 (2020).
Nguyen, T. T. Traceless point-to-axial chirality exchange in the atropselective synthesis of biaryls/heterobiaryls. Org. Biomol. Chem. 17, 6952–6963 (2019).
Quinonero, O. et al. Combining organocatalysis with central-to-axial chirality conversion: atroposelective Hantzsch-type synthesis of 4-arylpyridines. Angew. Chem. Int. Ed. 55, 1401–1405 (2016).
Raut, V. S. et al. Enantioselective syntheses of furan atropisomers by an oxidative central-to-axial chirality conversion strategy. J. Am. Chem. Soc. 139, 2140–2143 (2017).
Link, A. & Sparr, C. Remote central-to-axial chirality conversion: direct atroposelective ester to biaryl transformation. Angew. Chem. Int. Ed. 57, 7136–7139 (2018).
He, X.-L. et al. Asymmetric Barton–Zard reaction to access 3-pyrrole-containing axially chiral skeletons. ACS Catal. 9, 4374–4381 (2019).
Hu, Y.-L. et al. Conversion of two stereocenters to one or two chiral axes: atroposelective synthesis of 2,3-diarylbenzoindoles. Chem. Sci. 10, 6777–6784 (2019).
Bao, X., Rodriguez, J. & Bonne, D. Bidirectional enantioselective synthesis of bis-benzofuran atropisomeric oligoarenes featuring two distal C–C stereogenic axes. Chem. Sci. 11, 403–408 (2020).
Zhang, D. et al. Diversified transformations of tetrahydroindolizines to construct chiral 3-arylindolizines and dicarbofunctionalized 1,5-diketones. J. Am. Chem. Soc. 142, 15975–15985 (2020).
Wang, S.-J., Wang, Z., Tang, Y., Chen, J. & Zhou, L. Asymmetric synthesis of quinoline-naphthalene atropisomers by central-to-axial chirality conversion. Org. Lett. 22, 8894–8898 (2020).
Xu, W.-L., Zhao, W.-M., Zhang, R.-X., Chen, J. & Zhou, L. Organocatalytic cycloaddition–elimination cascade for atroposelective construction of heterobiaryls. Chem. Sci. 12, 14920–14926 (2021).
Gutnov, A. et al. Cobalt(I)-catalyzed asymmetric [2+2+2] cycloaddition of alkynes and nitriles: synthesis of enantiomerically enriched atropoisomers of 2-arylpyridines. Angew. Chem. Int. Ed. 43, 3795–3797 (2004).
Shibata, T., Fujimoto, T., Yokota, K. & Takagi, K. Iridium complex-catalyzed highly enantio- and diastereoselective [2+2+2] cycloaddition for the synthesis of axially chiral teraryl compounds. J. Am. Chem. Soc. 126, 8382–8383 (2004).
Nishida, G., Noguchi, K., Hirano, M. & Tanaka, K. Asymmetric assembly of aromatic rings to produce tetra-ortho-substituted axially chiral biaryl phosphorus compounds. Angew. Chem. Int. Ed. 46, 3951–3954 (2007).
Kwon, Y., Chinn, A. J., Kim, B. & Miller, S. J. Divergent control of point and axial stereogenicity: catalytic enantioselective C−N bond-forming cross-coupling and catalyst-controlled atroposelective cyclodehydration. Angew. Chem. Int. Ed. 57, 6251–6255 (2018).
Liu, Y. et al. Organocatalytic atroposelective intramolecular [4+2] cycloaddition: synthesis of axially chiral heterobiaryls. Angew. Chem. Int. Ed. 57, 6491–6495 (2018).
Zheng, S.-C., Wang, Q. & Zhu, J. Catalytic atropenantioselective heteroannulation between isocyanoacetates and alkynyl ketones: synthesis of enantioenriched axially chiral 3-arylpyrroles. Angew. Chem. Int. Ed. 58, 1494–1498 (2019).
Lotter, D., Neuburger, M., Rickhaus, M., Häussinger, D. & Sparr, C. Stereoselective arene-forming aldol condensation: synthesis of configurationally stable oligo-1,2-naphthylenes. Angew. Chem. Int. Ed. 55, 2920–2923 (2016).
Zhang, J.-W. et al. Discovery and enantiocontrol of axially chiral urazoles via organocatalytic tyrosine click reaction. Nat. Commun. 7, 10677 (2016).
Yao, Q.-J., Zhang, S., Zhan, B.-B. & Shi, B.-F. Atroposelective synthesis of axially chiral biaryls by palladium-catalyzed asymmetric C−H olefination enabled by a transient chiral auxiliary. Angew. Chem. Int. Ed. 56, 6617–6621 (2017).
Zhang, L., Zhang, J., Ma, J., Cheng, D.-J. & Tan, B. Highly atroposelective synthesis of arylpyrroles by catalytic asymmetric Paal–Knorr reaction. J. Am. Chem. Soc. 139, 1714–1717 (2017).
Yang, T., Guo, X., Yin, Q. & Zhang, X. Intramolecular asymmetric reductive amination: synthesis of enantioenriched dibenz[c,e]azepines. Chem. Sci. 10, 2473–2477 (2019).
Mao, J.-H. et al. Organocatalyst-controlled site-selective arene C–H functionalization. Nat. Chem. 13, 982–991 (2021).
Wang, J.-Y. et al. Atroposelective construction of axially chiral alkene-indole scaffolds via catalytic enantioselective addition reaction of 3-alkynyl-2-indolylmethanols†. Chin. J. Chem. 39, 2163–2171 (2021).
Xu, D. et al. Diversity-oriented enantioselective construction of atropisomeric heterobiaryls and N-aryl indoles via vinylidene ortho-quinone methides. CCS Chem. 3, 2680–2691 (2021).
Mi, R. et al. Rhodium-catalyzed atroposelective access to axially chiral olefins via C−H bond activation and directing group migration. Angew. Chem. Int. Ed. 61, e202111860 (2022).
Bringmann, G. et al. Murrastifoline-F: first total synthesis, atropo-enantiomer resolution, and stereoanalysis of an axially chiral N,C-coupled biaryl alkaloid. J. Am. Chem. Soc. 123, 2703–2711 (2001).
Mino, T. et al. N-Aryl indole-derived C–N bond axially chiral phosphine ligands: synthesis and application in palladium-catalyzed asymmetric allylic alkylation. Tetrahedron. Asymmetry 21, 711–718 (2010).
Mishra, S., Liu, J. & Aponick, A. Enantioselective alkyne conjugate addition enabled by readily tuned atropisomeric P,N-ligands. J. Am. Chem. Soc. 139, 3352–3355 (2017).
Li, T.-Z., Liu, S.-J., Tan, W. & Shi, F. Catalytic asymmetric construction of axially chiral indole-based frameworks: an emerging area. Chem. Eur. J. 26, 15779–15792 (2020).
Ototake, N. et al. Catalytic enantioselective synthesis of atropisomeric indoles with an N-C chiral axis. Chem. Eur. J. 16, 6752–6755 (2010).
Kamikawa, K. et al. Simultaneous induction of axial and planar chirality in arene–chromium complexes by molybdenum-catalyzed enantioselective ring-closing metathesis. Chem. Eur. J. 21, 4954–4957 (2015).
Morimoto, Y. et al. Enantioselective synthesis of N–C axially chiral indoles through chiral palladium-catalyzed 5-endo-hydroaminocyclization. Tetrahedron 72, 5221–5229 (2016).
He, C., Hou, M., Zhu, Z. & Gu, Z. Enantioselective synthesis of indole-based biaryl atropisomers via palladium-catalyzed dynamic kinetic intramolecular C–H cyclization. ACS Catal. 7, 5316–5320 (2017).
Zhang, H.-H. et al. Design and enantioselective construction of axially chiral naphthyl-indole skeletons. Angew. Chem. Int. Ed. 56, 116–121 (2017).
Ma, C. et al. Design and catalytic asymmetric construction of axially chiral 3,3’-bisindole skeletons. Angew. Chem. Int. Ed. 58, 3014–3020 (2019).
Zhu, S. et al. Organocatalytic atroposelective construction of axially chiral arylquinones. Nat. Commun. 10, 4268 (2019).
Xia, W. et al. Chiral phosphoric acid catalyzed atroposelective C−H amination of arenes. Angew. Chem. Int. Ed. 59, 6775–6779 (2020).
Chen, Y.-H. et al. Organocatalytic enantioselective synthesis of atropisomeric aryl-p-quinones: platform molecules for diversity-oriented synthesis of biaryldiols. Angew. Chem. Int. Ed. 59, 11374–11378 (2020).
Wang, C.-S. et al. Axially chiral aryl-alkene-indole framework: a nascent member of the atropisomeric family and its catalytic asymmetric construction. Chin. J. Chem. 38, 543–552 (2020).
Tian, M., Bai, D., Zheng, G., Chang, J. & Li, X. Rh(III)-catalyzed asymmetric synthesis of axially chiral biindolyls by merging C–H activation and nucleophilic cyclization. J. Am. Chem. Soc. 141, 9527–9532 (2019).
Peng, L. et al. Organocatalytic asymmetric annulation of ortho-alkynylanilines: synthesis of axially chiral naphthyl-C2-indoles. Angew. Chem. Int. Ed. 58, 17199–17204 (2019).
Jiang, F. et al. A strategy for synthesizing axially chiral naphthyl-indoles: catalytic asymmetric addition reactions of racemic substrates. Angew. Chem. Int. Ed. 58, 15104–15110 (2019).
He, Y.-P., Wu, H., Wang, Q. & Zhu, J. Palladium-catalyzed enantioselective Cacchi reaction: asymmetric synthesis of axially chiral 2,3-disubstituted indoles. Angew. Chem. Int. Ed. 59, 2105–2109 (2020).
Burger, K. & Rottegger, S. 1,3-Cycloaddition von alkinen an hetero-1,3-diene 1-aminopyrrole aus 1,2-diazabuta-1,3-dienen und 1-diethylaminopropin. Tetrahedron Lett. 25, 4091–4094 (1984).
Dürr, U., Heinemann, F. W. & Kisch, H. Transition metal complexes of diazenes XLI1For XL, see: H. Kisch, Z. Naturforsch. 52B (1997) 994.1: cobalt catalyzed addition of internal alkynes to 1,2-diaryldiazenes: formation of 2,3-dihydrocinnolines, mono- and distilbenylazobenzenes. J. Org. Chem. 558, 91–101 (1998).
Zhang, S., Wang, B., Jia, X. & Yuan, Y. Rhodium-catalyzed cascade annulation reaction via C−H activation of azobenzenes with terminal alkynes: a synthesis of indolo[1,2-b]cinnolines. Adv. Synth. Catal. 361, 451–455 (2019).
Li, C., Xu, D.-N., Ma, C., Mei, G.-J. & Shi, F. Diastereo- and enantioselective construction of dihydrobenzo[e]indole scaffolds via catalytic asymmetric [3 + 2] cycloannulations. J. Org. Chem. 83, 9190–9200 (2018).
Zi, W., Zuo, Z. & Ma, D. Intramolecular dearomative oxidative coupling of indoles: a unified strategy for the total synthesis of indoline alkaloids. Acc. Chem. Res. 48, 702–711 (2015).
Yi, J.-C., Liu, C., Dai, L.-X. & You, S.-L. Synthesis of C3-methyl-substituted pyrroloindolines and furoindolines via cascade dearomatization of indole derivatives with methyl iodide. Chem. Asian J. 12, 2975–2979 (2017).
Zheng, C. & You, S.-L. Catalytic asymmetric dearomatization (CADA) reaction-enabled total synthesis of indole-based natural products. Nat. Prod. Rep. 36, 1589–1605 (2019).
Sheng, F.-T., Wang, J.-Y., Tan, W., Zhang, Y.-C. & Shi, F. Progresses in organocatalytic asymmetric dearomatization reactions of indole derivatives. Org. Chem. Front. 7, 3967–3998 (2020).
Acknowledgements
We thank the National Natural Science Foundation of China (NSFC-21672170, 91954120), Natural Science Basic Research Plan in Shaanxi Province of China (2018JC-020, 2021JZ-40), and China Postdoctoral Science Foundation (2018M643705) for financial support. The calculations were performed at Chemical HPC Center of NWU.
Author information
Authors and Affiliations
Contributions
H.Y. and H.R.S. contributed equally. L.Z. and J.C. conceived and designed the experiments. H.Y., H.R.S., H.W., S.Y., and G.G.Z. performed the experiments and prepared the Supplementary Information. R.Q.H. and L.Y. performed DFT calculations. H.Y, H.R.S., and L.Z. wrote the paper. All authors analyzed and discussed the results and commented on the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yang, H., Sun, HR., He, RQ. et al. Organocatalytic cycloaddition of alkynylindoles with azonaphthalenes for atroposelective construction of indole-based biaryls. Nat Commun 13, 632 (2022). https://doi.org/10.1038/s41467-022-28211-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-022-28211-0
- Springer Nature Limited