

Abstract
Tissues are dynamically shaped by bidirectional communication between resident cells and the extracellular matrix (ECM) through cell-matrix interactions and ECM remodelling. Tumours leverage ECM remodelling to create a microenvironment that promotes tumourigenesis and metastasis. In this review, we focus on how tumour and tumour-associated stromal cells deposit, biochemically and biophysically modify, and degrade tumour-associated ECM. These tumour-driven changes support tumour growth, increase migration of tumour cells, and remodel the ECM in distant organs to allow for metastatic progression. A better understanding of the underlying mechanisms of tumourigenic ECM remodelling is crucial for developing therapeutic treatments for patients.
Similar content being viewed by others

Introduction
Cellular phenotypes and molecular functions are fundamentally dependent on signals from outside the cell such as the interactions with the extracellular matrix (ECM). The core matrisome is composed of ~300 unique matrix macromolecules and can be classified into collagens, proteoglycans (such as heparan sulphate proteoglycans, versican and hyaluronan) and glycoproteins (such as laminins, elastin, fibronectin and tenascins)1. These ECM components are modified post-translationally by an array of secreted remodelling enzymes, such as oxidases and proteases. In addition, the ECM binds soluble factors, such as growth factors and other ECM-associated proteins. Cell surface receptors interact with ECM components and ECM-bound factors to mediate cell adhesion and cell signalling thereby regulating processes as diverse as proliferation, differentiation, migration and apoptosis2. ECM can also demonstrate very different mechanical and topographical properties, which, importantly, can influence cell fate and function via different mechanosignalling routes3.
The ECM has two main forms, which differ in function, composition and location. The interstitial matrix forms porous three-dimensional networks around cells that interconnect cells in the stroma and can connect to the basement membrane, which is the other form of ECM structure. The interstitial matrix guarantees the structural integrity of tissues and organs but also modulates processes such as cell differentiation and migration. The protein composition of the interstitial matrix mainly includes collagens I, III, V, etc., fibronectin and elastin. Abundance and composition of the interstitial matrix vary between tissue types, between microenvironments within the same tissue and can be remodelled in response to force stress or trauma such as wound repair or tissue regeneration4. In cancer, remodelling of the interstitial ECM induces a broad range of biophysical and biochemical changes affecting cell signalling, ECM stiffness, cell migration and tumour progression5. In contrast, the basement membrane is a more stable, sheet-like, dense structure that lines the basal surface of, for example, epithelial and endothelial cells, surrounds muscle cells and adipocytes6, and separates tissues into different, well-organised compartments. The basement membrane consists mainly of collagen IV and laminins, which are interconnected through different network-bridging proteins such as nidogen and heparan sulphate proteoglycans (HSPGs)7. Binding of cells to the basement membrane is essential for establishing epithelial cell polarity and is crucial for many developmental processes and maintenance of tissue homoeostasis8. Remodelling of the basement membrane is required for cancer cells to invade stromal tissue and become a malignant tumour9.
Complex ECM remodelling processes, involving over 700 proteins1, change overall abundance, concentration, structure and organisation of individual ECM components, thereby affecting the three-dimensional spatial topology of the matrix around cells, its biochemical and biophysical properties and consequently its effect on cell fate. ECM remodelling is an essential and tightly regulated physiological process in development and in restoring tissue homoeostasis during wound repair10. However, it is not surprising that cells dysregulate this process in pathologic conditions such as inflammatory diseases, tissue fibrosis, and cancer11. Recent research highlights the importance of the tumour-mediated systemic aberrations of the ECM for the establishment of metastasis. In this review, we discuss remodelling mechanisms of extracellular matrices and the implications of these mechanisms during cancer development, and describe recent concepts of ECM remodelling shaping tissues for tumour cells to metastasise. Increasing understanding of these processes opens up the possibilities of therapeutic approaches to target the aberrant ECM and/or the underlying pathologic mechanisms of its remodelling and prevent malignancy.
Mechanisms of tumourigenic ECM remodelling
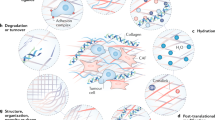
Changes in the ECM are a result of different remodelling mechanisms that can be divided into four main processes: (1) ECM deposition, which changes the abundance and composition of ECM components, thereby affecting biochemical and mechanical ECM properties; (2) chemical modification at the post-translational level, which alters the biochemical properties and structural characteristics of the ECM (Fig. 1a); (3) proteolytic degradation, which releases bioactive ECM fragments and ECM-bound factors and may be required for the liberation of cellular constraints, such as migratory barriers (Fig. 1b); and (4) force-mediated physical remodelling, which affects ECM organisation by aligning ECM fibres and opening-up passages for cell migration (Fig. 1c).

a ECM deposition and modification: using collagen as an example, pre-procollagen is synthesised and translocated to the Golgi, where it becomes a procollagen α-chain. This procollagen molecule undergoes several post-translational modifications (PTMs) to modify its properties. The PTMs include glycosylation, pro-peptide alignment, disulphide bond formation and hydroxylation. Lysine hydroxylation of the procollagen chains by PLODs allows for spontaneous triple helix formation within the cell and secretion into the extracellular space. Here, the pro-peptides on the C- and N-terminal are cleaved by proteases creating collagen fibrils. For further collagen fibre assembly, collagens fibrils are cross-linked by LOX. b ECM degradation: proteases including MMPs cleave the ECM proteins, which releases matrix-bound growth factors (GFs) and cytokines, and ECM fragments, including matrikines and also remove barriers for cell migration. c Force-mediated ECM remodelling: integrin binding to the ECM molecules applies forces to ECM molecules. This can change the conformation of the ECM molecule, thereby exposing binding sites to support self-assembly into fibrils that induces fibre alignment. The mechanical force applied by the integrins in this modification process can also cause non-proteolytic breaching of the basement membrane that will allow cancer cell invasion.
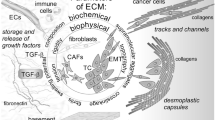
Tissue homoeostasis is dependent on the tight regulation between ECM deposition, modification, degradation and organisation that give rise to the biochemical and biophysical ECM properties. Changing single elements can turn over the delicate balance of ECM remodelling events. ECM alterations have implications on complex cellular signalling networks since ECM components serve as ligands for various cell surface receptors such as integrins, syndecans and receptor tyrosine kinases. Thus, it is not surprising that cancer cells and tumour-associated stromal cells modify all four ECM remodelling mechanisms, creating a cancer-supporting matrix that actively contributes to the pathology of the tumour (Fig. 2)12.

a, b Tumour-derived factors activate stromal cells which differentiate into cancer-associated fibroblasts (CAFs) leading to the secretion and deposition of large amounts of ECM components along with the cancer cells. c ECM-modifying enzymes such as LOX expressed by tumour cells and CAFs cross-link and align collagen fibres, which increases matrix stiffness around the tumour, and e the formation of a physical barrier to evade immune surveillance by T-cells. d Increased matrix stiffness promotes the interaction between ECM components and cell-surface receptors on tumour cells that triggers mechanosignalling mediated by integrins. f To sustain a tumourigenic microenvironment, tumour cells and resident immune cells secrete cytokines, chemokines and growth factors (GFs), which differentiate and recruit bone marrow-derived cells (BMDCs). g The BMDCs, CAFs and tumour cells secrete ECM-degrading proteases, including MMPs, which are cell surface-bound (e.g., MT1-MMP) or secreted (e.g., MMP-9). h Proteolytic ECM degradation generates bioactive matrikines and i releases matrix-bound GFs. These factors induce pro-tumourigenic ECM signalling that promotes tumour proliferation, migration, invasion and angiogenesis. j These combined changes to the ECM create a hypoxic environment. Neutrophils secrete potent MMP-9 that degrades ECM and releases matrix-bound VEGF that forms a concentration gradient for new angiogenic sprouting. k Stimulated by dense ECM, the tumour cells may gain endothelial-like functions and mimic the vasculature that connects to blood vessels.
Cellular deposition of tumourigenic ECM
In principle, ECM components can be produced and deposited by any cell. Tumour cells themselves show altered expression of ECM components, such as collagens I and III and ECM-modifying enzymes, such as lysyl oxidases (LOX) and LOX-like proteins (LOXL) (Fig. 2a)12,13,14,15. However, both in healthy tissue and tumours, the major producers of ECM in the interstitial matrix are fibroblasts, especially when activated and transformed to myofibroblasts; chondrocytes and osteoblasts are also significant, depending on tissue and context16. Myofibroblasts have a combined phenotypic characteristic of fibroblasts and smooth muscle cells, whereby they secrete ECM and exert contractile functions enabling mechanical alterations of the topology of the ECM, respectively. They can originate from different cell types and are mostly characterised by de novo expression of α-smooth muscle actin (α-SMA), although there are other markers of myofibroblasts in different conditions (reviewed in refs. 16,17). Activation of myofibroblasts is triggered by various pro-inflammatory factors18, most importantly transforming growth factor β (TGF-β)19. ECM remodelling by myofibroblasts is an essential physiologic process, for example in tissue regeneration and wound repair, which includes their inactivation after restoring homoeostasis. However, sustained inflammatory stimuli and subsequent TGF-β secretion by immune cells such as macrophages and tumour cells can generate deregulated, hyperproliferative and overactive myofibroblasts that are the major culprits in pathologic conditions such as fibrosis and cancer20.
Tumour cells primarily orchestrate the recruitment and activation of stromal cells that are the major depositors of ECM components in the tumour microenvironment (Fig. 2b) by the secretion of various pro-fibrotic growth factors and inflammatory factors such as TGF-α, TGF-β, fibroblast growth factor (FGF)-2, platelet-derived growth factor (PDGF) and epidermal growth factor (EGF)21. Recent evidence indicates that several of these stromal activation signals can be secreted in exosomes, which may impact the activation and differentiation to a myofibroblast phenotype, as exosomal TGF-β elevates FGF-2 production in fibroblasts compared to non-exosomal TGF-β22.
The tumour-derived activation factors induce the differentiation of stromal cells towards so-called cancer-associated fibroblasts (CAFs), which function as myofibroblasts and remodel the ECM to support tumour growth17,20,23,24. These activated stromal cells are of multiple origins, prominently including tissue-resident or bone marrow-derived fibroblasts25, but also, for example, adipocytes26. Interestingly, stress-induced adrenergic signalling in ovarian cancer cells can also activate CAFs to deposit elevated amounts of collagen that can be inhibited by β-blockers27. Furthermore, in aged tissue, stromal cells show alterations in ECM remodelling properties such as changes in the expression of matrix metalloproteinases (MMPs) and chemokines that support tumour development and metastasis28. In melanoma, aged fibroblasts express lower amounts of the ECM-modifying protein hyaluronan and proteoglycan link protein 1 (HAPLN1) that leads to reduced integrity of lymphatic vasculature and increased metastasis29. Changes in the tumour ECM can also further activate stromal cells in a feed-forward mechanism30,31.
ECM deposition by CAFs may depend on their spatial arrangement in the tumour. In pancreatic ductal adenocarcinoma (PDAC), CAFs in direct interaction with adjacent tumour cells show active TGF-β signalling and deposit collagen, whereas CAFs at a greater distance are also activated by the tumour cells, but are unresponsive to TGF-β, deposit HA and establish a tumourigenic, pro-inflammatory environment through the expression of cytokines such as IL-6, or antigen presentation32,33,34; this fosters the recruitment and activation of immune cells, which, as discussed below, contribute to the establishment of the cancer niche.
It is important to note that subpopulations of CAFs additionally support tumour growth through mechanisms independent of ECM remodelling, such as promoting cancer stemness or preventing cancer cell recognition by T-cells35,36. CAFs also may have tumour-suppressive functions such as restraining tumour vasculature or supporting immune surveillance, since the ablation of CAFs in murine models of PDAC led to a more aggressive tumour phenotype, which also correlates with patient data37,38. Hence, the heterogeneity, spatial distribution and potential plastic nature of CAFs need to be considered carefully in the context of targeting the stromal compartment of tumours.
Changes in ECM composition in cancer
ECM components possess both tumour-suppressing and tumour-promoting properties. For example, depending on its molecular weight hyaluronan (HA) functions as a tumour suppressor or a tumour promoter (reviewed in Bohaumilitzky et al.39). The tumour-resistance of the longest-lived rodent, the naked mole-rat, involves the expression of a unique HA with high molecular mass (HMM-HA) as a major ECM component. HMM-HA signalling through the CD44 receptor activates the expression of key tumour suppressor genes40, leading to a hypersensitive cell-cycle arrest, a common mechanism of tumour suppression41. In contrast, high levels of HA, and in particular small HA oligosaccharides (LMM-HA), are associated with poor prognosis in several tumours such as colorectal, breast and prostate cancer42,43,44,45,46. Here, dysregulated HA synthetase and HA-degrading hyaluronidase lead to the accumulation of LMM-HA42,47. LMM-HA directly interacts with cell surface receptors regulating pro-tumourigenic signalling cascades48, including glycolysis, the main source of energy in tumours, and promotes migration49. LMM-HA signalling through CD44 also increases the resistance to cellular stress and thereby may promote tumour development50.
The most common tumourigenic alteration of ECM homoeostasis is an increased deposition of fibrillar collagen13,51,52, which has direct tumour-promoting properties. Additionally, increased deposition of major ECM components including fibronectin, HA and tenascin C into the interstitial matrix results in a fibrotic phenotype, termed desmoplasia, which is similar to the alterations observed during organ fibrosis. Desmoplasia is a key characteristic of various cancers such as breast cancer and PDAC and is associated with poor prognosis53,54,55. For example, in a murine breast cancer model, the increased deposition of collagen I results in increased tumour formation and development of metastasis, directly linking ECM remodelling with aggressive tumour progression in vivo56. Furthermore, collagen V has been associated with altered mitogenic signalling in breast cancer. It can enhance the co-receptor ability of the surface proteoglycan, glypican-1 (GPC1), to modify FGF-2 signalling leading to increased cell proliferation56,57.
Analysing the complex global changes in the ECM may be beneficial for early cancer detection58. Unique ECM signatures that are characteristic of some tumours change during tumour progression and are predictive of clinical outcomes59,60,61. The combined pathologic alterations of the ECM provide a protein fingerprint by the release of cancer-specific ECM fragments into the circulation that may have diagnostic implications as it was shown for lung, ovarian, breast and colorectal cancer62,63,64. Moreover, even before tumour development, increased deposition of collagen I and proteoglycans lead to increased breast density, which is the greatest independent risk factor for the development of breast cancer (Box 1)65,66,67.

Changes in ECM modification and organisation in cancer
Once synthesised, the ECM proteins undergo post-translational modifications (PTMs) inside and outside the cell, which enhances their complexity and three-dimensional organisation (Table 1, reviewed by Yuzhalin et al.68). Post-translational modifications of ECM components affect matrix interactions with other molecules and cellular receptors, localisation within the tissue and ECM degradation69. We elaborate below how several key post-translational modifications are altered in a cancerous environment.
For example, collagen chains are synthesised as long precursors (procollagens) that are further modified by various post-translational modifications, for example, glycosylation of their lysine and hydroxylysine residues by addition of galactose and glucose; and hydroxylation of lysine residues through lysyl hydroxylases (encoded by procollagen-lysine 2-oxoglutarate 5-dioxygenase (PLOD) genes)70,71,72. These modified procollagens form triple helices and are further processed extracellularly by proteases to create collagen fibrils. Additionally, collagen fibrils are covalently cross-linked by extracellular LOX and LOXLs, which is essential for correct collagen fibre assembly and increases tensile strength and stiffness (Fig. 2c)73,74,75 (reviewed by Mouw and Weaver4). Tissue transglutaminase 2 (TG2) cross-links ECM molecules such as fibronectin, HSPG, fibrinogen and collagen IV68 by the transamidation of glutamine residues to the amino group of a lysine residue of another protein chain. This transamidation process results in the formation of covalent N-γ-glutaminyl-ɛ-lysyl-isopeptide bonds, which are resistant to proteolytic degradation69.
Alterations of these post-translational modifications in tumourigenesis cause morphological changes in the ECM architecture that are indicative of tumour progression and influence tumour cell motility76,77. In normal soft tissue, collagen fibres in the interstitial matrix are curly and oriented parallel to the layer of epithelium5. A high deposition of these curly collagen I fibres in non-tumourous breast tissue can be protective against cancer development through the activation of tumour-suppressive phenotypes such as upregulation of genes encoding components of cell–cell junctions, and downregulation of mesenchymal-specific and metalloproteinase-encoding genes78. However, in breast cancer, collagen fibres in proximity to the tumour boundary are linearised, perpendicularly oriented and supportive of invasive tumour growth15,79. Further, PLODs, LOXs and TG2 are frequently overexpressed in cancer68 that, together with force-mediated ECM remodelling events, cause increased cross-linking and linearization of ECM molecules.
The majority of extracellular and transmembrane proteins are glycosylated. Transmembrane glycoproteins function as receptors for ECM molecules; for example, CD44 is heavily glycosylated and its ECM ligand HA is a glycosaminoglycan. Other glycosylated ECM molecules include HSPGs that bind growth factors and modify receptor tyrosine kinase signalling80. Membrane-anchored glycoproteins, together with glycolipids and polysaccharides, form a complex and heterogenous structure on the cell surface, termed the glycocalyx, that interacts with the ECM81. The glycocalyx mediates cell–ECM, cell–cell interactions (for example, during immune cell surveillance82,83), binding of growth factors, and mechanoresponses84. Interestingly, glycosylation patterns of surface proteins frequently change in cancer and may foster pathological cell behaviours84,85,86. Tumour cells upregulate bulky glycoproteins on the cell surface. The resulting bulky glycocalyx can apply tension to ECM-bound integrins, which are important receptors for ECM components, causing integrin clustering and thereby increasing pro-tumourigenic integrin signalling84.
Modifications of ECM components also affect their capacity of binding to growth factors. Extracellular HSPGs regulate the activity of various receptor tyrosine kinase signalling pathways by their ability to store various growth factors, the release of which is regulated by extracellular modification enzymes such as endosulphatases (Sulfs). Sulfs, which are dysregulated in many cancers, modify the sulphation pattern of HSPGs and affect the binding of growth factors. Sulf1 can act as a tumour suppressor, whereas Sulf2 activates pro-tumourigenic signalling pathways87,88,89. Sulf1 is downregulated in ovarian cancer and a subset of hepatocellular carcinoma, which prevents Sulf1-mediated inactivation of receptor tyrosine kinase signalling90,91. However, Sulf2 supports Wnt and PDGFRα signalling in vitro92,93. Since Sulfs act extracellularly, it is likely that stromal cells contribute to Sulf levels in the tumour ECM. Although both Sulf isotypes show similar substrate specificity, conflicting studies, in particular for Sulf1, suggest that the functional consequences of HSPG modification may depend on experimental conditions (in vitro vs. in vivo), context and tumour or tumour subtype.
Degradation of the tumourigenic ECM
The ECM is cleaved and degraded by target-specific proteases such as MMPs, disintegrin and metalloproteinases (ADAMs), disintegrin and metalloproteinases with thrombospondin motifs (ADAMTS), and proteases that specifically cleave at serine, cysteine and threonine residues (reviewed in refs. 10,94). These proteases are secreted primarily by stromal cells, including recruited bone marrow-derived cells, as well as by cancer cells (Fig. 2f–g). Several proteases are regulated by an activation cascade by which different proteases that are secreted as inactive pro-forms or bound to the ECM are released and activated by other proteases9. The activity of proteases is counterbalanced by protease inhibitors (e.g., tissue inhibitor of metalloproteinases, TIMPs)95. The substrates of the different proteases are still not completely understood. Recent advances in the sensitivity of proteomics techniques allow targeted degradomics studies to identify precise proteolytic cleavage targets from protease-substrate interactions96,97. These studies revealed that proteases cleave different substrates when they are bound to the cell membrane compared to their soluble form. ADAM10 and 17 are membrane-anchored and their ectodomains can be released by ADAM8. The soluble ectodomains show different target specificity98.
Proteolytic degradation of ECM components can be both pro-tumourigenic73,94 and anti-tumourigenic99,100. MMP inhibitors, designed to block ECM remodelling and decrease invasion, have thus far underperformed in clinical trials, likely due to the pleiotropic activities of MMPs99. As an example, MMP-8 has pro-tumour functions, where overexpression of MMP-8 in cancer cells is correlated with decreased survival in patients with ovarian cancer101 and with hepatocellular carcinoma102. However, MMP-8 also has anti-tumour activity, as MMP-8 decreased cancer cell invasion in vitro and low expression of MMP-8 was correlated with decreased survival in patients with oral tongue squamous cell carcinoma103. Further, germline deletion of MMP-8 in mice increased susceptibility to chemically induced skin tumours, and bone marrow transplants of MMP-8-expressing neutrophils restored tumour protection104. These conflicting results highlight that context-dependent effects of ECM-degrading enzymes on tumour growth and invasion.
Tumour and stromal cells express increased levels of ECM-degrading proteases that have multiple functions during tumour progression9. First, proteolytic degradation of ECM components allows progressive destruction of the normal ECM and its replacement with tumour-derived ECM. Second, ECM degradation is an important driver of cancer cell motility (see below). Third, ECM-binding of soluble signalling molecules such as growth factors makes them insoluble and inactive and action of proteases liberate them. Fibronectin, for example, binds insulin-like growth factor binding-protein-3, FGF-2 and VEGF-A with high affinity105. The enhanced protease activity in tumours causes subsequent ECM degradation that releases the ECM-bound growth factors and thereby increases their bioavailability (Fig. 2i).
Last, the cleavage of long ECM components produces bioactive, shorter fragments with distinct functions that can be pro- or anti-tumourigenic compared to the full-length ECM component. These fragments often contain structures similar to chemokines or cytokines and are therefore termed matrikines (Fig. 2h, reviewed in refs. 106,107). Matrikines play an important role in angiogenesis and balance the angiogenic switch through promoting and inhibiting properties (reviewed in ref. 108). Anti-angiogenic functions are described for several NC1 domains of basement membrane collagens109,110, such as the matrikine endostatin108,111. A specific domain of the α3 chain of type IV collagen has anti-tumour effects through the inhibition of the expression of membrane-type 1-matrix metalloproteinase (MT1-MMP or MMP14) and integrin β3, which leads to impaired migratory abilities of tumour cells112. On the other hand, various elastin-derived matrikines, such as Val-Gly-Val-Ala-Pro-Gly (VGVAPG) or Ala-Gly-Val-Pro-Gly-Leu-Gly-Val-Gly (AGVPGLGVG) promote tumour progression113. These ECM fragments are products of the degradation of elastin through different proteolytic enzymes (elastases)114 and MMPs115. These matrikines can, in turn, also induce MMP expression and activation, including MT1-MMP and MMP-2, which would explain their tumour-promoting properties116.
Overall, the combined pro-tumourigenic functions of ECM degradation highlight the importance of proteases for tumour progression. Protease activity can be measured in real-time using Proteolytic Activity Matrix Analysis (PrAMA). Cleavage patterns of FRET-based polypeptides thereby reflect specific enzyme activities when compared to purified proteases as standard. This method allows the detection of enhanced protease activity in patient samples such as cerebrospinal fluid, which is indicative of neoplastic meningitis and metastasis to the brain117.
It is important to note that, in addition to proteolysis of ECM, MMPs have significant non-proteolytic functions that can alter tumour progression. For example, MMP-3 binds and inhibits noncanonical Wnt5b, resulting in increased canonical Wnt signalling9, and MT1-MMP promotes basal extrusion during epithelial-to-mesenchymal transition (EMT) and basal mitosis noncatalytically118.
Impact of tumourigenic ECM remodelling
Remodelling of the tumour ECM impacts every stage of tumourigenesis. In this section we describe these effects on the development of a primary tumour, including how a tumourigenic ECM induces pro-tumour signalling in cancer cells, helps to create an immunosuppressive environment and further supports the progression of the tumour to grow, invade stromal tissue and promote vascularisation.
Tumourigenic ECM remodelling rewires signalling
ECM components bind to integrins which are heterodimeric transmembrane receptors with bidirectional signalling that couple them to the actin cytoskeleton within the cell. Force-mediated ECM-integrin interaction induces various downstream signalling events, known as mechanosignalling (Fig. 2d). Repeated adhesion and de-adhesion of cells to the ECM substrate via integrins and the alteration of actin cytoskeletal reorganisation mediates cellular processes such as cell migration119,120. Syndecans are cell surface-bound HSPGs that interact with integrins to support focal adhesion formation and migration80. Integrin-mediated signalling includes pro-survival and pro-apoptotic pathways and their crosstalk with growth factor receptors mediates cellular signalling (reviewed in refs. 121,122,123).
As a result of the alterations in the ECM, tumours often are stiffer than normal tissue, which can be applied in clinical diagnosis124,125. The stiff matrix of the tumour leads to an altered mechanosignalling. High ECM deposition and increased stiffness combined with integrin overexpression in various cancers126 trigger tumour promotion (reviewed in Desgrosellier and Cheresh121).
As mentioned above, tumour-secreted TGF-β activates the differentiation of stromal cells into ECM-remodelling CAFs. Additionally, TGF-β secreted by CAFs can induce STAT3 signalling in tumour cells127. However, in PDAC, TGF-β loss in tumour cells is associated with increased desmoplasia and enhanced matrix tension. The pronounced desmoplasia triggers β1-integrin mechanosignalling that promotes tumour progression through STAT3 activation128. These observations show the complexity of TGF-β signalling in tumour and stromal cells, which can have pro- and anti-tumourigenic functions depending on the context.
Collagens also bind to unique transmembrane receptor tyrosine kinases, known as Discoidin Domain Receptors (DDR1 and DDR2). They induce cellular signalling in conjunction with other receptors such as integrins and Notch129. The high abundance of collagen in the tumour microenvironment triggers the activation of the receptor tyrosine kinase DDR receptors. DDR1 expressed by gastric cancer cells supports invasion and tumour metastasis130. In breast cancer, the interaction of collagen I with DDR1 expressed by tumour cells leads to the induction of stemness-like signalling through the activation of STAT3, which is crucial to mediate the metastatic outgrowth131. Surprisingly, DDR1 deletion in a breast cancer mouse model increases ECM deposition and promotes tumour aggressiveness and metastasis132. DDR2 deficiency in hepatic stellate cells triggers differentiation of CAFs and fosters the establishment of a metastatic niche133. These studies suggest that DDR1 and DDR2 are expressed by tumour and stromal cells and depending on receptor isotype, cell type and context can either support or suppress tumour development129,132.
Tumour-immune cell crosstalk in ECM remodelling
The high ECM remodelling activity mediated by tumour cells and CAFs contributes to an inflammatory tumour environment134. Components of the ECM can function directly as inflammatory stimuli, known as danger-associated molecular patterns (DAMPs)135 that induce immune responses through the interactions with pattern recognition receptors (PRRs) expressed by immune cells136. ECM-degrading proteases liberate these pro-inflammatory ECM components such as LMM-HA and biglycan, as well as matrix-bound growth factors and cytokines136,137. Biglycan, for example, activates PRRs including Toll-like receptor 4 (TLR4) and TLR2 expressed by macrophages that drive the expression of TNF-α and macrophage inflammatory protein–2 (MIP-2/CXCL2)138. Matrikines may also serve as DAMPs in tumours. Versican is degraded by ADAMTS proteases to reveal a bioactive fragment called versikine139. Increased intratumoural levels of versikine leads to increased tumour infiltration of CD8+ T-cells in colorectal cancer140 and myeloma141. Increased CD8+ T-cell infiltration results from versikine-induced expression of the transcription factor Interferon Regulatory Factor 8 (IRF8) in macrophages and enhances generation of CD103+ CD11chiMHCIIhi conventional dendritic cells (cDC) in a mouse model140.
Bone marrow-derived cells, such as tumour-associated macrophages (TAMs) and tumour-associated neutrophils (TANs), are important sources for ECM remodelling proteases in the primary tumour environment (Fig. 2g) and metastatic sites and promote tumour angiogenesis. Neutrophils are recruited to the tumour site by hypoxic conditions142 and constitute the major source of angiogenesis-inducing MMP-9 (Fig. 2j)143. This neutrophil-derived MMP-9 is particularly potent because it is not complexed with the MMP inhibitor, TIMP-1, unlike MMP-9 molecules secreted by other cell types144. TAMs sustain a pro-tumourigenic environment, predominately when polarised to a M2-like phenotype. M2-like TAMs induce proteolytic clearance of interstitial collagen through upregulated MMP expression, including pro-angiogenic TIMP-1-free MMP-9 similar to neutrophils, accompanied by increased endocytosis and lysosomal degradation of collagen145,146. In addition to ECM degradation, in a colorectal cancer model, TAMs contribute to ECM deposition. Interestingly, this study found that here TAMs are the major cell type to upregulate synthesis and assembly of collagens, specifically collagen types I, VI and XIV, and induce deposition, cross-linking and linearization of these collagen fibres near invasive tumour cells147. These data add to growing evidence that immune cells contribute to ECM deposition, as macrophages also deposit the glycoprotein osteonectin, which promotes stromal invasion in a mouse model of breast cancer148.
ECM remodelling supports tumour cell migration and invasion
The altered physical properties of the stiff tumour ECM promote cell migration and invasion into the stromal tissue (reviewed in Kai et al.12). LOX-mediated collagen cross-linking stiffens the ECM in a murine breast cancer model, which triggers an integrin-dependent invasive phenotype (Fig. 2c)15. Additionally, collagen cross-linking mediated by PLOD2 expressed by CAFs in a murine lung adenocarcinoma model, increases tumour invasiveness149. However, the organisation and composition of the ECM, rather than density and stiffness alone, is crucial for tumour cell motility and tumour progression. For example, non-invasive breast tumour cells cultured in Matrigel, which contains non-fibrillar collagen IV, gain the potential for collective invasion when their culture matrix is changed to fibrillar collagen I, potentially due to fewer interactions with cell adhesion receptors, which anchor the cells to collagen IV-rich basement membranes150,151.
During early solid tumour development, at the stage of carcinoma in situ, neoplastic cells are separated from the surrounding stromal cells by a basement membrane creating a physical barrier that prevents tumour cell dissemination (Fig. 3a). The tumour becomes malignant when the tumour cells break through the basement membrane into the interstitial space and invade the surrounding tissues – processes that are accompanied by various ECM remodelling events41.

a In carcinoma in situ, tumour cells are restricted from migrating and invading into the surrounding tissue by the intact basement membrane. Tumour cells and stroma cells may be physically connected through the basement membrane using integrins. ECM in the interstitial matrix is curly. b Basement membrane breakage can be achieved through proteolytic ECM degradation by proteases secreted by tumour cells and activated stroma cells (top) and through non-proteolytic, force-mediated ECM remodelling (lower box). Integrins expressed on invadopodia, an invasive actin-rich cell structure on tumour and cancer-associated fibroblasts (CAFs), bind to ECM molecules and couple them intracellularly to contractile structures. This process pulls the ECM molecules apart and applies force to the basement membrane facilitating non-proteolytic basement membrane breaching. c In invasive tumours, the basement membrane is mostly degraded. CAFs and tumour cells secrete LOX to cross-link collagen fibres. Increased cross-linking and force-mediated ECM remodelling creates linearised ECM in the tumour-surrounding interstitial matrix. Tumour cells migrate along regions with dense, aligned collagen fibres forming migratory tracks for efficient cell migration. (lower box) Membrane-bound proteases expressed on tumour cells and CAFs, such as MT1-MMP localised on invadopodia, degrades the collagen migration barriers. Exosomes contain additional proteases to clear the ECM for tumour cell migration and are released into the interstitial matrix. Integrins on the surface of secreted exosomes bind to fibronectin, which functions as a bridging molecule connecting integrins expressed on tumour cells thereby promoting the formation of invadopodia and tumour cell migration. CAFs can function as leader cells for directed tumour cell migration and are connected to tumour cells through E-cadherin/N-cadherin adhesions.
Proteolytic degradation is important for breaching the basement membrane, although invasion through the basement membrane involves both proteolysis-dependent and -independent mechanisms (Fig. 3b)152,153,154. In vivo, breast tumour cells break through the basement membrane, mediated by membrane-anchored proteases, predominantly MT1-MMP155,156,157. Tumour cells and CAFs may be physically connected bridged by the layer of the basement membrane. Using their pronounced contractile properties CAFs can apply physical forces on the basement membrane and widen the pre-existing holes thus promoting tumour cell invasion in a MMP-independent manner158.
Proteolysis of ECM components is crucial for the active movement of cells through the interstitial matrix as it opens migratory tracks and reduces mechanical stress on the migrating cells76,159,160. T-cells and dendritic cells migrate towards chemoattractants along collagen type I fibres, which are enriched in the tumour environment160,161. However, as T-cell infiltration is non-proteolytic, they cannot migrate through tumour-associated dense collagen and fibronectin regions, and hence are excluded from the tumour and accumulate in the stroma (Fig. 2e)162,163. Also, age-related ECM alterations, such as specific cross-linking events, prevent sufficient T-cell movement164,165. Immunodeficiency due to decreased T-cell infiltration and consequent T-cell death may be attributed to the mechanical stress on the cytoplasm and nucleus of these T-cells during their passage through stiff ECM (reviewed by Moreau et al.164). Although under debate, these ECM migration barriers largely impair immune surveillance of tumours and have been associated with poor clinical outcome163,166.
In regions of dense collagen fibres in the tumour environment, pericellular proteolysis of the interstitial matrix is important for active cell migration and controlled by MMPs, particularly MT1-MMP on tumour cells and CAFs, which degrade the collagen migration barriers167,168,169. MT1-MMP is usually localised on invasive actin-rich cell structures called invadopodia170. There is now substantial evidence that these processes are supported by exosomes. Exosomes have been shown to promote maturation of invadopodia and ECM degradation via the extracellular delivery of MT1-MMP and other proteinases171. Additionally, integrins on the surface of secreted exosomes bind to fibronectin which thereby act as bridging molecules connecting integrins expressed on tumour cells. The local secretion of exosomes may, therefore, induce cellular integrin clustering that triggers adhesion assembly and result in more effective and directed cell migration172.
Owing to their high ECM-remodelling capacity, CAFs may serve as leader cells for tumour cell migration (Fig. 3c)20. They are positioned in front of invading cancer cells and clear the interstitial matrix through proteolytic- and force-mediated remodelling processes. CAFs interact with collagen-rich ECM through the integrins α3 and α5, and Rho-mediated regulation of myosin light chain activity. These interactions apply force to the ECM thereby aligning the collagen fibres173,174. Additionally, fibronectin binding to integrins, such as integrins αvβ3 and α5β1, on CAFs allows receptor clustering, which applies force on fibronectin dimers changing their conformation and exposing binding sites to support self-assembly into fibrils4. The force-mediated fibronectin assembly creates anisotropic fibre orientation and can open gaps in the matrix that allow cell migration175. Collagen linearization can also be mediated by tumour-secreted factors such as the WNT1 inducible signalling pathway gene (WISP1)176, which is triggered by TGF-β.
These processes create stable migratory tracks that are used for the efficient, proteolysis-independent collective invasion of the tumour cells that follow. Tumour cell migration is also actively guided by CAFs through force triggered E-cadherin/N-cadherin adhesion177. The specific architecture of these collagen tracks guides tumour cells to travel longer distances along the aligned collagen fibres by forming fewer but longer protrusions or to use amoeboid blebbing for optimal migration. The aligned matrix thereby supports, in particular, the directed migration of small, plastic tumour cells with stem-like characteristics178,179,180. This may explain why the signature of highly aligned collagen fibres is associated with poor prognosis79.
Overall, the physical properties of the tumour ECM together with its composition, density and the specific architecture of aligned fibres provide optimal migratory conditions for effective tumour cell dissemination and promote aggressive tumour phenotypes.
ECM remodelling is fueled by hypoxia and promotes vascularisation
During tumour progression, rapid growth leads to a tissue architecture with tumour cells located distant from blood vessels with diffusion-limited oxygen and nutrient supply (reviewed in Semenza181). Hypoxia triggers cellular signalling mediated by the hypoxia-inducible transcription factor (HIF‐1). HIF-1 also promotes ECM remodelling through the regulation of various MMPs, for example, an upregulation of MMP-2, MMP-9 and MT1-MMP182,183, and activates collagen fibre alignment via the induction of various collagen-modifying enzymes, like prolyl-4-hydroxylases (P4HA1 and P4HA2), PLOD1 and PLOD2, LOX and LOX-like proteins (LOXL1, LOXL2 and LOXL4)184,185,186 in CAFs (reviewed in Gilkes et al.70), and additionally upregulates integrins, thereby contributing to enhanced pro-tumourigenic mechanosignalling187. Deposition of collagen I by mesothelial cells is also enriched by a hypoxic tumour-mesothelial niche, which promotes the metastasis of ovarian cancer188.
The state of hypoxia is a key driver for angiogenesis. HIF-1 expression in hypoxic tumour cells induces the angiogenic switch through the upregulation and activation of pro-angiogenic proteins such as VEGF to recruit endothelial cells and build new vessels189. To form functional new vessels endothelial tip cells rely on an upregulation of collagens and ECM-modifying enzymes such as LOX and PLOD proteins which was shown across different human tumour types190. VEGF secreted by cancer and stromal cells is bound to the ECM191 but is released through proteolytic cleavage192,193. HIF-1 expression induces immune cell recruitment, which significantly contributes to high concentrations of active MMP-9 in the ECM (Fig. 2j)142. The simultaneous HIF-1-induced upregulation of VEGF and increased MMP-9 levels lead to a high concentration of soluble VEGF in the tumour tissue driving angiogenesis and invasiveness. However, different ECM components promote and/or inhibit angiogenesis, which results in a disrupted tumour vasculature (reviewed in Sottile108). For example, periostin has pro-angiogenic functions194; tenascin-C and fibronectin have both pro- and anti-angiogenic functions depending on context195 (reviewed in Obberghen-Schilling196), and thrombospondin-1 inhibits MMP-9 activation and angiogenesis and thereby suppresses spontaneous tumour development197 (reviewed in Lawler and Lawler198).
The architecture of dense collagen in tumours also triggers the formation of interconnected networks of cancer cells. These tumour cells form ECM-rich vasculature-like structures, in a process called vasculogenic mimicry, that supply the tumour with blood without inducing angiogenesis (Fig. 2k). A specific, dense collagen architecture with small pores and short fibres causes an up-regulation of integrin-β1 and the differentiation of tumour cells towards an endothelial phenotype characterised by the expression of collagen type IV α1 chain and serine peptidase inhibitor Serpin E2, which have anticoagulant properties and are important for blood perfusion150,199. Vasculogenic mimicry of melanoma cells requires matrix remodelling through the expression of MMP-2, MT1-MMP and the deposition and cleavage of a specific laminin chain (γ2 chain of laminin 332)200. Interestingly, tumour-associated macrophages can also form vascular-like networks201. Besides providing an adequate blood supply for the tumour, vasculogenic mimicry is associated with an aggressive tumour phenotype and promotes metastasis199. Further research is needed to elucidate the underlying mechanisms of how the biochemical and biophysical properties of the ECM induce phenotypical changes in tumour cells and lead to formation of vascular-like structures.
Taken together, the characteristics of a tumour microenvironment maintains and promotes itself through complex interdependent pathways that lead to enhanced angiogenesis and invasiveness with the ultimate goal of metastasising to a secondary organ.
ECM remodelling during metastasis
Crucial for cancer therapy is the prevention and elimination of cancer metastasis, which accounts for the majority of cancer-related deaths. Extensive research during the last decade has unravelled the significance of ECM remodelling at each stage of metastasis development, from surviving in circulation, to forming the pre-metastatic and metastatic niches (Fig. 4).

a Angiogenesis and high MMP activity at the primary tumour site lead to a disrupted vasculature that allows tumour cells to intravasate and enter the circulation. Circulating tumour cells (CTCs) may secrete ECM that protects them from immune surveillance. b CTCs may connect via matrix-like interactions with neutrophil extracellular traps (NETs) and NETotic neutrophils using integrins that are expressed on CTCs and neutrophils. c Endothelial cells (EC) deposit and assemble fibrillar fibronectin that promotes the attachment of CTCs to the endothelial wall at distant organs. Increased MMP activity creates a leaky vasculature allowing CTCs to extravasate into the surrounding tissue. ECs also remodel the tissue in distant organs and deposit ECM contributing to the establishment of a pre-metastatic niche. d Various factors derived from the primary tumour such as growth factors, MMPs, LOX, ECM proteins like fibronectin, and exosomes, create a pre-metastatic niche at a distant site to prime the new tissue for metastasis. Stromal cells in the pre-metastatic niche are activated by tumour-derived factors and myofibroblasts remodel the ECM, for example, by the deposition of fibronectin, tenascin C, osteopontin, and versican depending on tissue context. Bone marrow-derived cells (BMDCs) are recruited to the pre-metastatic niche, attach to the remodelled ECM via integrins and contribute to further ECM remodelling in preparation for the arrival of disseminated tumour cells. e CTCs that extravasate through the disrupted vasculature into the distant tissue may become dormant. Proteases expressed in NETs including neutrophil elastase and MMP-9 cleave laminin, generating a specific matrikine that can awaken dormant tumour cells. Together, these ECM remodelling processes support the formation of metastasis.
ECM remodelling in circulation
As discussed above, various ECM remodelling events support the migration and invasion of tumour cells that eventually will be able to enter circulation. Surprisingly, circulating tumour cells (CTCs) in the blood upregulate expression of common stroma-derived ECM proteins, such as collagens (Col1a2, Col3a1), TIMP-2, the proteoglycan decorin (Dcn), the glycoprotein osteonectin (Sparc), and fibronectin, as revealed by single-cell RNA-sequencing of pancreatic CTCs (Fig. 4a)202,203. Yet, it needs to be determined if CTCs actively secrete ECM and which role these ECM components may play in supporting CTCs. Potentially, CTC-secreted ECM could increase autocrine survival signalling of CTCs, protect them from immune cell clearance, similar to platelets surrounding CTCs204 or promote the formation of CTC clusters for efficient metastatic colonisation205. A recent study showed that metastasis is increased by CTCs when captured by neutrophil extracellular traps (NETs) through interaction with integrin-β1, expressed by both CTCs and NETs (Fig. 4b)206. The CTC-secreted ECM components thereby may serve as bridging molecules between NETs and CTCs.
ECM remodelling in the pre-metastatic niche
Potential sites of metastasis possess organ-specific microenvironments, which are very different from that of the primary tumour. The primary tumour triggers the formation of a favourable microenvironment at the distant tissue, which includes ECM remodelling, even before the presence of metastatic tumour cells, creating a so-called pre-metastatic niche207. Of note, at the metastatic site, ECM composition and remodelling processes are distinct from both the primary tumour and the healthy tissue208,209,210 (reviewed in Haye and Erler211). The most common alteration of the ECM in the primary tumour environment is increased collagen deposition. In contrast, mainly fibronectin is involved in the formation of the pre-metastatic niche along with glycoproteins and proteoglycans such as tenascin C, osteopontin and versican207,211.
Primary tumour-derived factors activate stromal cells in the pre-metastatic site to secrete new ECM molecules or remodel and modify the ECM directly (Fig. 4d). Citrullination, which is the conversion of arginine residues into the non-coding amino acid citrulline, affects the electrostatic charge and folding conformation of fibronectin and collagens and thereby their cell-adhesive properties. Citrullination is mediated by peptidyl-arginine deiminases (PADs), that may be released into the extracellular space on neutrophil extracellular traps during inflammation212,213,214 and potentially also during pre-metastatic niche establishment. However, PAD4 secreted by colorectal cancer cells induces citrullination of collagen I in the liver215, which promotes the adhesion of disseminated tumour cells to the liver tissue. In the bone, tumour-derived factors create either an osteolytic (bone-degrading) or osteoplastic (bone-forming) pre-metastatic environment. The more common osteolytic metastasis occurs often in breast cancers as well as other cancers types, where bone homoeostasis is modulated by promoting the activity of bone-resorbing osteoclasts by factors such as IL-6, EGF-like growth factors, TGF-α, NF-κB ligand (RANKL) but also through tumour-derived LOX216,217,218,219. Moreover, ECM degradation is essential for releasing these soluble factors, such as the liberation of EGF-like growth factors and TGF-α in the primary tumour is mediated by MMP-1 and ADAMTS1219. Osteolytic bone metastasis includes the degradation of collagen I, the most common ECM component in the bone. Collagens can also undergo spontaneous non-enzymatic isomerization. Newly synthesised collagen is non-isomerised and is increased in bone metastasis to compensate for the high ECM turnover. Thus, the non-isomerised C-telopeptide of collagen I (α-CTX-I) can be used as a biomarker for bone metastasis from breast and prostate cancers instead of the isomerised form (β-CTX-I)220. In contrast, growth factors produced by prostate cancer cells trigger osteoblast differentiation, creating an osteoblastic lesion in the bone for tumour colonisation221,222. However, osteolytic events may also occur in osteoblastic bone metastasis since bone matrix-bound growth factors, which are released and activated upon ECM degradation, may be important for metastatic cell proliferation.
Signals from the primary tumour also stimulate the recruitment and adhesion of specific bone marrow-derived cells (e.g., macrophages, neutrophils and myeloid progenitor cells) to the pre-metastatic niche (Fig. 4d)223,224. For example, hypoxia-induced LOX expressed by the primary tumour acts at the metastatic site, where it cross-links collagen IV, which provides the adhesion for bone marrow-derived cells, as shown in breast cancer225. With their potent ECM remodelling capabilities, bone marrow-derived cells, in turn, promote colonisation of the metastatic tumour cells226,227. In another example, colorectal cancer metastasises preferentially to the liver, which correlates with high systemic levels of the MMP inhibitor, TIMP-1. Although TIMP-1 inhibits ECM turnover, it has been implicated in the recruitment of neutrophils to the liver, which then remodel the tissue establishing a tumour-permissive niche228. CTCs may contribute to upregulated TIMP-1 levels229.
Cancer cell-derived MMPs, such as MMP-3 and MMP-10, induce vascular disruption at the pre-metastatic site supporting the extravasation of CTCs and enhance ECM turnover, which opens up the way for infiltrating bone marrow-derived cells and tumour cells230. Endothelial cells also deposit ECM components at the metastatic tissue. Mature endothelial cells can suppress breast cancer growth and mediate their quiescence through the secretion of thrombospondin-1. However, an MMP-mediated disrupted vasculature at the metastatic site230 results in the induction of newly sprouting endothelial tip cells that produce TGF-β1 and ECM molecules involved in pre-metastatic niche development, such as periostin, tenascin, versican and fibronectin231.
Likely, many of the tumour-derived factors acting in distant organs to prime the metastatic site for tumour cell colonisation may be transported in the form of exosomes. In their lumens, exosomes transport various proteins, such as proteases, and nucleic acids, mainly miRNAs, which are effective at the target site. The composition of exosomes is heterogeneous depending on cell type, and functional or developmental state of the cell and subject of intense research efforts232,233.
There is now mounting evidence that metastases of certain cancers show a tissue specificity234, suggesting that organ-specific tissue environments including their particular ECM may influence the growth of metastasis235. Surprisingly, tumour-derived exosomes may mediate the non-random pattern of metastasis and promote pre-metastatic niche development. Specific groups of integrins expressed on tumour-derived exosomes dictate their adhesion in a particular organ and thereby may determine the site of the future metastasis. Integrins α6β4 and α6β1 guide exosomes to the lung to target fibroblasts and epithelial cells, while exosomal integrin αvβ5 determines their transport to the liver for the uptake by Kupffer cells236. To date, it is unclear to what extent this role depends on ECM remodelling in the target tissue and which specific alterations in the ECM cause the organotropic exosome distribution. However, these tumour-derived exosomes create pro-inflammatory and fibrotic environments that recruit bone marrow-derived cells, further promoting ECM remodelling to host metastatic cells. Pancreatic cancer-derived exosomes contain macrophage migration inhibitory factor (MIF) that induces TGF-β secretion by Kupffer cells in the liver, which stimulates an enhanced fibronectin deposition by neighbouring hepatic stellate cells. The resulting fibrotic environment recruits bone marrow-derived macrophages237 and ultimately fosters liver metastasis. These studies link the well-known preferred organotropic metastasis pattern of different tumour entities with the novel concept of pre-metastatic niche establishment through organ-specific, exosome-mediated intercellular communication cascades.
ECM remodelling in the metastatic niche
To form metastases, CTCs need to extravasate from the circulation into the new tissue, a process that is dependent on ECM remodelling. Endothelial cells in the liver of colorectal cancer patients deposit and assemble fibrillar fibronectin that allows the attachment of CTCs to the vascular lumen, induces integrin-dependent focal adhesion and eventually the extravasation into the liver tissue (Fig. 4c)238.
After colonisation, stromal and metastatic cells both contribute to create a metastatic niche and actively change the ECM to promote metastastic growth. Interestingly, ECM remodelling of the metastatic niche is different for the same tumour at different sites, indicating tissue tropism. Unique combinations of matrisomal and ECM-associated proteins are deposited by stromal and cancer cells, respectively, at brain, lung, liver and bone metastases in a breast cancer mouse model239. Glycoproteins, such as osteopontin, periostin and tenascin C are deposited either by infiltrating tumour cells or by stromal cells at the metastatic site and are important for the colonisation of tumour cells by inducing stemness-like signalling pathways240,241,242. Breast cancer cells in the lung produce tenascin C, which triggers Notch and Wnt signalling240. Periostin deposition by activated lung fibroblasts also increases Wnt signalling in metastatic breast cancer cells. The expression of osteopontin and tenascin C depends on the c-Jun transcription factor. Interestingly, chemotherapeutic treatment can activate the c-Jun N-terminal kinase (JNK) pathway and the deposition of these glycoproteins, fuelling metastatic progression243. CTCs exhibit mesenchymal characteristics, whereas primary and established metastatic tumour cells show more epithelial phenotypes242. Versican deposition by bone marrow-derived cells promotes metastasis by inducing the transition from mesenchymal to epithelial phenotype in metastatic cells244, suggesting that this proteoglycan may promote the establishment and progression of metastasis242.
Changes in the ECM may also be essential to reactivate disseminated but dormant tumour cells. Recently, matrikines have been associated with awakening dormant tumour cells and promoting metastasis245. In an inflammatory environment, neutrophil extracellular traps bind to extracellular laminin and, owing to the presence of laminin-degrading proteases like neutrophil elastase and MMP-9, induce laminin degradation. This proteolytic processing results in the exposure of a specific laminin epitope. This matrikine triggers an integrin-mediated signalling cascade, which leads to the awakening of dormant cancer cells at sites of metastasis and their subsequent proliferation (Fig. 4e)245. This study shows the importance of the complex interplay of ECM, immune and tumour cells, with direct implications on metastasis formation.
Further detailed research is needed to uncover the drivers of pre-metastatic niche development and to unravel the intrinsic factors that trigger metastatic ECM remodelling, which may lead to promising therapeutic interventions for the prevention and elimination of cancer metastasis.
Conclusions and perspectives
Changes in ECM abundance, composition, architecture and the resulting cell-ECM interactions have fundamental consequences on cell and tissue functions. These processes are spatially and temporally regulated to preserve the homoeostasis of tissues and involve the interplay of different cell types. Aberrations in ECM remodelling play a profound role in the development and progression of cancer. Due to their expression in specific pathologies, these ECM alterations can be used as biomarkers and therapeutic targets, for example, by using nanobody technology, which can selectively detect these alterations in vivo and guide therapeutics to the specific location246. Several recent examples of ECM-targeted immunotherapies have shown promising results in pre-clinical models247,248,249,250. However, detailed ECM compositions in various disease contexts are still unclear as are the specific targets of different proteases. More clinical trials focused on specifically targeting the ECM remodelling events are needed giving its importance in disease (Box 2 and Table 2). Different immune cells significantly contribute to changes in the ECM and are also functionally affected by these changes. In recent years, novel immunotherapies promised wide-ranging improvements of treatment for cancer patients. However, many tumours do not respond to these therapies, which may partly due to the immune infiltration barriers caused by the ECM. Targeting these ECM barriers could result in more effective immunotherapy134.
Progress in investigating cells in their complex intervening three-dimensional networks has resulted in the development of various organoid models that may provide better in vitro tools to investigate the effects of specific matrix perturbations on complex cell–cell and cell–matrix interactions251. However, conclusions drawn from in vitro studies using Matrigel and other matrices should be handled with care since they are highly dependent on experimental conditions and lack the complexity of ECM composition and organisation in vivo. Optimised proteomics analysis now provides tools to study the entire ECM of tissues in various contexts252. An interesting research direction has also been opened up with the popularisation of novel single-cell technologies, which shed light on the cellular context of ECM remodelling and cell–ECM interactions at a single and sub-cellular level. Advanced tissue imaging approaches promise to give high-resolution insights into the spatial and temporal organisation of these cell-ECM interactions. Novel technologies like multiplexed ion beam imaging (MIBI) can provide a quantitative map of the proteomic landscape of single cells in primary tumours and metastases253. With these advanced technologies, biomedical research can take the next step in uncovering the comprehensive interactions of individual cells within their environment, which can lead to the development of more effective ECM-targeted therapies.
References
Naba, A. et al. The extracellular matrix: tools and insights for the “omics” era. Matrix Biol. 49, 10–24 (2016).
Hastings, J. F., Skhinas, J. N., Fey, D., Croucher, D. R. & Cox, T. R. The extracellular matrix as a key regulator of intracellular signalling networks. Br. J. Pharmacol. 176, 82–92 (2019).
Humphrey, J. D., Dufresne, E. R. & Schwartz, M. A. Mechanotransduction and extracellular matrix homeostasis. Nat. Rev. Mol. Cell Biol. 15, 802–812 (2014).
Mouw, J. K., Ou, G. & Weaver, V. M. Extracellular matrix assembly: a multiscale deconstruction. Nat. Rev. Mol. Cell Biol 15, 771–785 (2014).
Egeblad, M., Rasch, M. G. & Weaver, V. M. Dynamic interplay between the collagen scaffold and tumor evolution. Curr. Opin. Cell Biol. 22, 697–706 (2010).
Yurchenco, P. D. Basement membranes: cell scaffoldings and signaling platforms. Cold Spring Harb. Perspect. Biol. 3, a004911 (2011).
Pozzi, A., Yurchenco, P. D. & Iozzo, R. V. The nature and biology of basement membranes. Matrix Biol. 57–58, 1–11 (2017).
Jayadev, R. & Sherwood, D. R. Basement membranes. Curr. Biol. 27, R207–R211 (2017).
Kessenbrock, K., Plaks, V. & Werb, Z. Matrix metalloproteinases: regulators of the tumor microenvironment. Cell 141, 52–67 (2010).
Bonnans, C., Chou, J. & Werb, Z. Remodelling the extracellular matrix in development and disease. Nat. Rev. Mol. Cell Biol. 15, 786–801 (2014).
Cox, T. R. & Erler, J. T. Remodeling and homeostasis of the extracellular matrix: implications for fibrotic diseases and cancer. Dis. Model Mech. 4, 165–178 (2011).
Kai, F. B., Drain, A. P. & Weaver, V. M. The extracellular matrix modulates the metastatic journey. Dev. Cell 49, 332–346 (2019).
Kauppila, S., Stenbäck, F., Risteli, J., Jukkola, A. & Risteli, L. Aberrant type I and type III collagen gene expression in human breast cancer in vivo. J. Pathol. 186, 262–268 (1998).
Poltavets, V., Kochetkova, M., Pitson, S. M. & Samuel, M. S. The role of the extracellular matrix and its molecular and cellular regulators in cancer cell plasticity. Front. Oncol. 8, 431 (2018).
Levental, K. R. et al. Matrix crosslinking forces tumor progression by enhancing integrin signaling. Cell 139, 891–906 (2009). The study describes how ECM stiffness directly contributes to tumour aggressiveness in-vivo.
Hinz, B. et al. Recent developments in myofibroblast biology: paradigms for connective tissue remodeling. Am. J. Pathol. 180, 1340–1355 (2012).
Kalluri, R. The biology and function of fibroblasts in cancer. Nat. Rev. Cancer 16, 582–598 (2016).
Lemoinne, S., Cadoret, A., El Mourabit, H., Thabut, D. & Housset, C. Origins and functions of liver myofibroblasts. Biochim. Biophys. Acta 1832, 948–954 (2013).
Dooley, S., Ten & Dijke, P. TGF-β in progression of liver disease. Cell Tissue Res. 347, 245–256 (2012).
Barbazán, J., Matic & Vignjevic, D. Cancer associated fibroblasts: is the force the path to the dark side? Curr. Opin. Cell Biol. 56, 71–79 (2019).
Heneberg, P. Paracrine tumor signaling induces transdifferentiation of surrounding fibroblasts. Crit. Rev. Oncol. Hematol. 97, 303–311 (2016).
Webber, J., Steadman, R., Mason, M. D., Tabi, Z. & Clayton, A. Cancer exosomes trigger fibroblast to myofibroblast differentiation. Cancer Res. 70, 9621–9630 (2010).
Orimo, A. et al. Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell 121, 335–348 (2005).
Ishii, G., Ochiai, A. & Neri, S. Phenotypic and functional heterogeneity of cancer-associated fibroblast within the tumor microenvironment. Adv. Drug Deliv. Rev. 99, 186–196 (2016).
Raz, Y. et al. Bone marrow-derived fibroblasts are a functionally distinct stromal cell population in breast cancer. J. Exp. Med. 215, 3075–3093 (2018).
Bochet, L. et al. Adipocyte-derived fibroblasts promote tumor progression and contribute to the desmoplastic reaction in breast cancer. Cancer Res. 73, 5657–5668 (2013).
Nagaraja, A. S. et al. Adrenergic-mediated increases in INHBA drive CAF phenotype and collagens. JCI Insight 2, 1–17 (2017).
Mavrogonatou, E., Pratsinis, H., Papadopoulou, A., Karamanos, N. K. & Kletsas, D. Extracellular matrix alterations in senescent cells and their significance in tissue homeostasis. Matrix Biol. 75–76, 27–42 (2019).
Ecker, B. L. et al. Age-related changes in HAPLN1 increase lymphatic permeability and affect routes of melanoma metastasis. Cancer Discov. 9, 82–95 (2019).
Calvo, F. et al. Mechanotransduction and YAP-dependent matrix remodelling is required for the generation and maintenance of cancer-associated fibroblasts. Nat. Cell Biol. 15, 637–646 (2013).
Lachowski, D. et al. Substrate rigidity controls activation and durotaxis in pancreatic stellate cells. Sci. Rep. 7, 2506 (2017).
Öhlund, D. et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J. Exp. Med. 214, 579–596 (2017).
Biffi, G. et al. IL1-induced JAK/STAT signaling is antagonized by TGFβ to shape CAF heterogeneity in pancreatic ductal adenocarcinoma. Cancer Discov. 9, 282–301 (2019).
Elyada, E. et al. Cross-species single-cell analysis of pancreatic ductal adenocarcinoma reveals antigen-presenting cancer-associated fibroblasts. Cancer Discov. 9, 1102–1123 (2019).
Su, S. et al. CD10 + GPR77 + cancer-associated fibroblasts promote cancer formation and chemoresistance by sustaining cancer stemness. Cell 172, 841–856.e816 (2018).
Costa, A. et al. Fibroblast heterogeneity and immunosuppressive environment in human breast cancer. Cancer Cell 33, 463–479.e410 (2018).
Özdemir, B. C. et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell 25, 719–734 (2014). This study describes how CAF depletion and reduction in stromal content leads to more aggressive tumour growth suggesting tumour suppressive functions of CAFs and the tumour ECM.
Rhim, A. D. et al. Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell 25, 735–747 (2014).
Bohaumilitzky, L. et al. A trickster in disguise: hyaluronan’s ambivalent roles in the matrix. Front. Oncol. 7, 242 (2017).
Tian, X. et al. INK4 locus of the tumor-resistant rodent, the naked mole rat, expresses a functional p15/p16 hybrid isoform. Proc. Natl Acad. Sci. USA 112, 1053–1058 (2015).
Hanahan, D. & Weinberg, R. A. Hallmarks of cancer: the next generation. Cell 144, 646–674 (2011).
Schmaus, A., Bauer, J. & Sleeman, J. P. Sugars in the microenvironment: the sticky problem of HA turnover in tumors. Cancer Metast. Rev. 33, 1059–1079 (2014).
Ropponen, K. et al. Tumor cell-associated hyaluronan as an unfavorable prognostic factor in colorectal cancer. Cancer Res. 58, 342–347 (1998).
Anttila, M. A. et al. High levels of stromal hyaluronan predict poor disease outcome in epithelial ovarian cancer. Cancer Res. 60, 150–155 (2000).
Auvinen, P. et al. Hyaluronan in peritumoral stroma and malignant cells associates with breast cancer spreading and predicts survival. Am. J. Pathol. 156, 529–536 (2000).
Lipponen, P. et al. High stromal hyaluronan level is associated with poor differentiation and metastasis in prostate cancer. Eur. J. Cancer 37, 849–856 (2001).
Itano, N. et al. Selective expression and functional characteristics of three mammalian hyaluronan synthases in oncogenic malignant transformation. J. Biol. Chem. 279, 18679–18687 (2004).
Chanmee, T., Ontong, P. & Itano, N. Hyaluronan: a modulator of the tumor microenvironment. Cancer Lett. 375, 20–30 (2016).
Sullivan, W. J. et al. Extracellular matrix remodeling regulates glucose metabolism through TXNIP destabilization. Cell 175, 117–132.e121 (2018).
Schinzel, R. T. et al. The hyaluronidase, TMEM2, promotes ER homeostasis and longevity independent of the UPRER. Cell 179, 1–13 (2019).
Fang, M., Yuan, J., Peng, C. & Li, Y. Collagen as a double-edged sword in tumor progression. Tumour Biol. 35, 2871–2882 (2014).
Zhou, Z.-H. et al. Reorganized collagen in the tumor microenvironment of gastric cancer and its association with prognosis. J. Cancer 8, 1466–1476 (2017).
Esposito, I. et al. Tenascin C and annexin II expression in the process of pancreatic carcinogenesis. J. Pathol. 208, 673–685 (2006).
Whatcott, C. J. et al. Desmoplasia in primary tumors and metastatic lesions of pancreatic cancer. Clin. Cancer Res. 21, 3561–3568 (2015).
Iacobuzio-Donahue, C. A., Argani, P., Hempen, P. M., Jones, J. & Kern, S. E. The desmoplastic response to infiltrating breast carcinoma: gene expression at the site of primary invasion and implications for comparisons between tumor types. Cancer Res. 62, 5351–5357 (2002).
Provenzano, P. P. et al. Collagen density promotes mammary tumor initiation and progression. BMC Med. 6, 11 (2008). Study shows in an elegant in-vivo model the remarkedly influence of increased collagen I deposition on tumour growth and metastasis formation.
Huang, G., Ge, G., Izzi, V. & Greenspan, D. S. α3 Chains of type V collagen regulate breast tumour growth via glypican-1. Nat. Commun. 8, 14351 (2017).
Socovich, A. M. & Naba, A. The cancer matrisome: from comprehensive characterization to biomarker discovery. Semin. Cell Dev. Biol. 89, 157–166 (2018).
Bergamaschi, A. et al. Extracellular matrix signature identifies breast cancer subgroups with different clinical outcome. J. Pathol. 214, 357–367 (2008).
Yuzhalin, A. E., Urbonas, T., Silva, M. A., Muschel, R. J. & Gordon-Weeks, A. N. A core matrisome gene signature predicts cancer outcome. Br. J. Cancer 118, 435–440 (2018).
Naba, A. et al. The matrisome: in silico definition and in vivo characterization by proteomics of normal and tumor extracellular matrices. Mol. Cell Proteom. 11, M111.014647 (2012). Comprehensive study investigating the matrisome of tissues on a global scale acknowledging the complex interplay of ECM proteins.
Willumsen, N. et al. Serum biomarkers reflecting specific tumor tissue remodeling processes are valuable diagnostic tools for lung cancer. Cancer Med. 3, 1136–1145 (2014).
Bager, C. L. et al. Collagen degradation products measured in serum can separate ovarian and breast cancer patients from healthy controls: a preliminary study. Cancer Biomark. 15, 783–788 (2015).
Kehlet, S. N. et al. Excessive collagen turnover products are released during colorectal cancer progression and elevated in serum from metastatic colorectal cancer patients. Sci. Rep. 6, 30599 (2016).
Guo, Y. P. et al. Growth factors and stromal matrix proteins associated with mammographic densities. Cancer Epidemiol. Biomark. Prev. 10, 243–248 (2001).
McCormack, V. A. Breast density and parenchymal patterns as markers of breast cancer risk: a meta-analysis. Cancer Epidemiol. Biomark. Prev. 15, 1159–1169 (2006).
Kerlikowske, K. et al. Automated and clinical breast imaging reporting and data system density measures predict risk for screen-detected and interval cancers: a case-control study. Ann. Intern. Med. 168, 757–765 (2018).
Yuzhalin, A. E., Lim, S. Y., Kutikhin, A. G. & Gordon-Weeks, A. N. Dynamic matrisome: ECM remodeling factors licensing cancer progression and metastasis. Biochim. Biophys. Acta Rev. Cancer 1870, 207–228 (2018).
Karsdal, M. A. et al. Extracellular matrix remodeling: the common denominator in connective tissue diseases. Possibilities for evaluation and current understanding of the matrix as more than a passive architecture, but a key player in tissue failure. Assay. Drug Dev. Technol. 11, 70–92 (2013).
Gilkes, D. M., Semenza, G. L. & Wirtz, D. Hypoxia and the extracellular matrix: drivers of tumour metastasis. Nat. Rev. Cancer 14, 430–439 (2014).
Qi, Y. & Xu, R. Roles of PLODs in collagen synthesis and cancer progression. Front. Cell Dev. Biol. 6, 66 (2018).
Sipilä, K. H. et al. Proline hydroxylation in collagen supports integrin binding by two distinct mechanisms. J. Biol. Chem. 293, 7645–7658 (2018).
Ford, A. J. & Rajagopalan, P. Extracellular matrix remodeling in 3D: implications in tissue homeostasis and disease progression. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 10, e1503 (2018).
Myllyharju, J. & Kivirikko, K. I. Collagens, modifying enzymes and their mutations in humans, flies and worms. Trends Genet. 20, 33–43 (2004).
Canty, E. G. & Kadler, K. E. Procollagen trafficking, processing and fibrillogenesis. J. Cell Sci. 118, 1341–1353 (2005).
Wolf, K. & Friedl, P. Extracellular matrix determinants of proteolytic and non-proteolytic cell migration. Trends Cell Biol. 21, 736–744 (2011).
Nguyen-Ngoc, K.-V. et al. ECM microenvironment regulates collective migration and local dissemination in normal and malignant mammary epithelium. Proc. Natl Acad. Sci. USA 109, E2595–2604 (2012).
Maller, O. et al. Collagen architecture in pregnancy-induced protection from breast cancer. J. Cell Sci. 126, 4108–4110 (2013).
Conklin, M. W. et al. Aligned collagen is a prognostic signature for survival in human breast carcinoma. Am. J. Pathol. 178, 1221–1232 (2011).
Kneln, E. H., Nee, J. C. & Blobe, G. C. Heparan sulfate signaling in cancer. Trends Biochem. Sci. 39, 277–288 (2014).
Fuster, M. M. & Esko, J. D. The sweet and sour of cancer: glycans as novel therapeutic targets. Nat. Rev. Cancer 5, 526–542 (2005).
Hudak, J. E., Canham, S. M. & Bertozzi, C. R. Glycocalyx engineering reveals a Siglec-based mechanism for NK cell immunoevasion. Nat. Chem. Biol. 10, 69–75 (2014).
Jandus, C. et al. Interactions between Siglec-7/9 receptors and ligands influence NK cell-dependent tumor immunosurveillance. J. Clin. Invest. 124, 1810–1820 (2014).
Paszek, M. J. et al. The cancer glycocalyx mechanically primes integrin-mediated growth and survival. Nature 511, 319–325 (2014).
Shurer, C. R. et al. Physical principles of membrane shape regulation by the glycocalyx. Cell 177, 1757–1770.e1721 (2019).
Möckl, L. et al. Quantitative super-resolution microscopy of the mammalian glycocalyx. Dev. Cell 50, 57–72.e56 (2019).
Lanzi, C., Zaffaroni, N. & Cassinelli, G. Targeting heparan sulfate proteoglycans and their modifying enzymes to enhance anticancer chemotherapy efficacy and overcome drug resistance. Curr. Med. Chem. 24, 2860–2886 (2017).
Vivès, R. R., Seffouh, A. & Lortat-Jacob, H. Post-synthetic regulation of HS structure: the yin and yang of the sulfs in cancer. Front. Oncol. 3, 331 (2014).
Rosen, S. D. & Lemjabbar-Alaoui, H. Sulf-2: an extracellular modulator of cell signaling and a cancer target candidate. Expert Opin. Ther. Targets 14, 935–949 (2010).
Lai, J. et al. Loss of HSulf-1 up-regulates heparin-binding growth factor signaling in cancer. J. Biol. Chem. 278, 23107–23117 (2003).
Lai, J. P. et al. hSulf1 Sulfatase promotes apoptosis of hepatocellular cancer cells by decreasing heparin-binding growth factor signaling. Gastroenterology 126, 231–248 (2004).
Phillips, J. J. et al. Heparan sulfate sulfatase SULF2 regulates PDGFRα signaling and growth in human and mouse malignant glioma. J. Clin. Invest. 122, 911–922 (2012).
Nawroth, R. et al. Extracellular sulfatases, elements of the Wnt signaling pathway, positively regulate growth and tumorigenicity of human pancreatic cancer cells. PLoS ONE 2, e392 (2007).
Lu P., Takai K., Weaver V. M., Werb Z. Extracellular matrix degradation and remodeling in development and disease. Cold Spring Harb. Perspect. Biol. 3, a005058 (2011).
Saw, S. et al. Metalloprotease inhibitor TIMP proteins control FGF-2 bioavailability and regulate skeletal growth. J. Cell Biol. 218, 3134–3152 (2019).
Savickas, S. Auf dem Keller U. Targeted degradomics in protein terminomics and protease substrate discovery. Biol. Chem. 399, 47–54 (2017).
Overall, C. M. & Dean, R. A. Degradomics: systems biology of the protease web. Pleiotropic roles of MMPs in cancer. Cancer Metast. Rev. 25, 69–75 (2006).
Scharfenberg, F. et al. Degradome of soluble ADAM10 and ADAM17 metalloproteases. Cell Mol. Life Sci. 77, 331–350 (2019).
Juurikka, K., Butler, G. S., Salo, T., Nyberg, P. & Astrom, P. The role of MMP8 in cancer: a systematic review. Int. J. Mol. Sci. 20, 4506 (2019).
Lopez-Otin, C., Palavalli, L. H. & Samuels, Y. Protective roles of matrix metalloproteinases: from mouse models to human cancer. Cell Cycle 8, 3657–3662 (2009).
Stadlmann, S. et al. Cytokine-regulated expression of collagenase-2 (MMP-8) is involved in the progression of ovarian cancer. Eur. J. Cancer 39, 2499–2505 (2003).
Qin, G. et al. Reciprocal activation between MMP-8 and TGF-beta1 stimulates EMT and malignant progression of hepatocellular carcinoma. Cancer Lett. 374, 85–95 (2016).
Astrom, P. et al. The interplay of matrix metalloproteinase-8, transforming growth factor-beta1 and vascular endothelial growth factor-C cooperatively contributes to the aggressiveness of oral tongue squamous cell carcinoma. Br. J. Cancer 117, 1007–1016 (2017).
Balbin, M. et al. Loss of collagenase-2 confers increased skin tumor susceptibility to male mice. Nat. Genet. 35, 252–257 (2003).
Martino, M. M. & Hubbell, J. A. The 12th-14th type III repeats of fibronectin function as a highly promiscuous growth factor-binding domain. FASEB J. 24, 4711–4721 (2010).
Wells, J. M., Gaggar, A. & Blalock, J. E. MMP generated matrikines. Matrix Biol. 44–46, 122–129 (2015).
Mott, J. D. & Werb, Z. Regulation of matrix biology by matrix metalloproteinases. Curr. Opin. Cell Biol. 16, 558–564 (2004).
Sottile, J. Regulation of angiogenesis by extracellular matrix. Biochim. Biophys. Acta 1654, 13–22 (2004).
Brassart-Pasco, S. et al. Tetrastatin, the NC1 domain of the α4(IV) collagen chain: a novel potent anti-tumor matrikine. PLoS ONE 7, e29587 (2012).
Ramont, L. et al. The NC1 domain of type XIX collagen inhibits in vivo melanoma growth. Mol. Cancer Ther. 6, 506–514 (2007).
O’Reilly, M. S. et al. Endostatin: an endogenous inhibitor of angiogenesis and tumor growth. Cell 88, 277–285 (1997).
Pasco, S. et al. A specific sequence of the noncollagenous domain of the alpha3(IV) chain of type IV collagen inhibits expression and activation of matrix metalloproteinases by tumor cells. Cancer Res. 60, 467–473 (2000).
Da Silva, J. et al. Structural characterization and pro-tumor properties of a highly conserved matrikine. Oncotarget 9, 17839–17857 (2018).
Werb, Z., Banda, M. J., McKerrow, J. H. & Sandhaus, R. A. Elastases and elastin degradation. J. Invest. Dermatol. 79, 154s–159s (1982).
Mecham, R. P. et al. Elastin degradation by matrix metalloproteinases. J. Biol. Chem. 272, 18071–18076 (1997).
Brassart, B., Randoux, A., Hornebeck, W. & Emonard, H. Regulation of matrix metalloproteinase-2 (gelatinase A, MMP-2), membrane-type matrix metalloproteinase-1 (MT1-MMP) and tissue inhibitor of metalloproteinases-2 (TIMP-2) expression by elastin-derived peptides in human HT-1080 fibrosarcoma cell line. Clin. Exp. Metastasis 16, 489–500 (1998).
Conrad, C. et al. Profiling of metalloprotease activities in cerebrospinal fluids of patients with neoplastic meningitis. Fluids Barriers CNS 14, 1–9 (2017).
Andrieu, C. et al. MMP14 is required for delamination of chick neural crest cells independently of its catalytic activity. Development 147, dev183954 (2020).
Sun, Z., Guo, S. S. & Fässler, R. Integrin-mediated mechanotransduction. J. Cell Biol. 215, 445–456 (2016).
Schmidt, S. & Friedl, P. Interstitial cell migration: integrin-dependent and alternative adhesion mechanisms. Cell Tissue Res. 339, 83–92 (2010).
Desgrosellier, J. S. & Cheresh, D. A. Integrins in cancer: biological implications and therapeutic opportunities. Nat. Rev. Cancer 10, 9–22 (2010).
Hamidi, H., Pietilä, M. & Ivaska, J. The complexity of integrins in cancer and new scopes for therapeutic targeting. Br. J. Cancer 115, 1017–1023 (2016).
Ivaska, J. & Heino, J. Cooperation between integrins and growth factor receptors in signaling and endocytosis. Annu. Rev. Cell Dev. Biol. 27, 291–320 (2011).
Sigrist, R. M. S., Liau, J., Kaffas, A. E., Chammas, M. C. & Willmann, J. K. Ultrasound elastography: Review of techniques and clinical applications. Theranostics 7, 1303–1329 (2017).
Butcher, D. T., Alliston, T. & Weaver, V. M. A tense situation: forcing tumour progression. Nat. Rev. Cancer 9, 108–122 (2009).
Winkler, J. et al. Cellular apoptosis susceptibility (CAS) is linked to integrin β1 and required for tumor cell migration and invasion in hepatocellular carcinoma (HCC). Oncotarget 7, 22883–22892 (2016).
Ligorio, M. et al. Stromal microenvironment shapes the intratumoral architecture of pancreatic cancer. Cell 178, 160–175.e127 (2019).
Laklai, H. et al. Genotype tunes pancreatic ductal adenocarcinoma tissue tension to induce matricellular fibrosis and tumor progression. Nat. Med. 22, 497–505 (2016).
Valiathan, R. R., Marco, M., Leitinger, B., Kleer, C. G. & Fridman, R. Discoidin domain receptor tyrosine kinases: new players in cancer progression. Cancer Metastasis Rev. 31, 295–321 (2012).
Yuge, R. et al. Silencing of discoidin domain receptor-1 (DDR1) concurrently inhibits multiple steps of metastasis cascade in gastric cancer. Transl. Oncol. 11, 575–584 (2018).
Gao, H. et al. Multi-organ site metastatic reactivation mediated by non-canonical discoidin domain receptor 1 signaling. Cell 166, 47–62 (2016).
Takai, K. et al. Discoidin domain receptor 1 (DDR1) ablation promotes tissue fibrosis and hypoxia to induce aggressive basal-like breast cancers. Genes Dev. 32, 244–257 (2018).
Badiola, I., Olaso, E., Crende, O., Friedman, S. L. & Vidal-Vanaclocha, F. Discoidin domain receptor 2 deficiency predisposes hepatic tissue to colon carcinoma metastasis. Gut 61, 1465–1472 (2012).
Mushtaq, M. U. et al. Tumor matrix remodeling and novel immunotherapies: the promise of matrix-derived immune biomarkers. J. Immunother. Cancer 6, 65 (2018).
Frevert, C. W., Felgenhauer, J., Wygrecka, M., Nastase, M. V. & Schaefer, L. Danger-associated molecular patterns derived from the extracellular matrix provide temporal control of innate immunity. J. Histochem. Cytochem. 66, 213–227 (2018).
Ouyang, X., Ghani, A. & Mehal, W. Z. Inflammasome biology in fibrogenesis. Biochim. Biophys. Acta 1832, 979–988 (2013).
Babelova, A. et al. Biglycan, a danger signal that activates the NLRP3 inflammasome via toll-like and P2X receptors. J. Biol. Chem. 284, 24035–24048 (2009).
Schaefer, L. et al. The matrix component biglycan is proinflammatory and signals through Toll-like receptors 4 and 2 in macrophages. J. Clin. Invest. 115, 2223–2233 (2005).
Hope, C. et al. Immunoregulatory roles of versican proteolysis in the myeloma microenvironment. Blood 128, 680–685 (2016).
Hope, C. et al. Versican-derived matrikines regulate batf3-dendritic cell differentiation and promote T cell infiltration in colorectal cancer. J. Immunol. 199, 1933–1941 (2017).
Dhakal, B. et al. Versican proteolysis predicts immune effector infiltration and post-transplant survival in myeloma. Leuk. Lymphoma 60, 2558–2562 (2019).
Du, R. et al. HIF1α induces the recruitment of bone marrow-derived vascular modulatory cells to regulate tumor angiogenesis and invasion. Cancer Cell 13, 206–220 (2008).
Deryugina, E. I. et al. Tissue-infiltrating neutrophils constitute the major in vivo source of angiogenesis-inducing MMP-9 in the tumor microenvironment. Neoplasia 16, 771–788 (2014).
Ardi, V. C., Kupriyanova, T. A., Deryugina, E. I. & Quigley, J. P. Human neutrophils uniquely release TIMP-free MMP-9 to provide a potent catalytic stimulator of angiogenesis. Proc. Natl Acad. Sci. USA 104, 20262–20267 (2007).
Madsen, D. H. et al. Tumor-associated macrophages derived from circulating inflammatory monocytes degrade collagen through cellular uptake. Cell Rep. 21, 3662–3671 (2017).
Zajac, E. et al. Angiogenic capacity of M1- and M2-polarized macrophages is determined by the levels of TIMP-1 complexed with their secreted proMMP-9. Blood 122, 4054–4067 (2013).
Afik, R. et al. Tumor macrophages are pivotal constructors of tumor collagenous matrix. J. Exp. Med. 213, 2315–2331 (2016).
Sangaletti, S. et al. Macrophage-derived SPARC bridges tumor cell-extracellular matrix interactions toward metastasis. Cancer Res. 68, 9050–9059 (2008).
Pankova, D. et al. Cancer-associated fibroblasts induce a collagen cross-link switch in tumor stroma. Mol. Cancer Res. 14, 287–295 (2016).
Velez, D. O. et al. 3D collagen architecture induces a conserved migratory and transcriptional response linked to vasculogenic mimicry. Nat. Commun. 8, 1651 (2017).
Cheung, K. J., Gabrielson, E., Werb, Z. & Ewald, A. J. Collective invasion in breast cancer requires a conserved basal epithelial program. Cell 155, 1639–1651 (2013).
Kelley, L. C. et al. Adaptive F-actin polymerization and localized ATP production drive basement membrane invasion in the absence of MMPs. Dev. Cell 48, 313–328.e318 (2019).
Chang, J. & Chaudhuri, O. Beyond proteases: basement membrane mechanics and cancer invasion. J. Cell Biol. 218, 2456–2469 (2019).
Charras, G. & Sahai, E. Physical influences of the extracellular environment on cell migration. Nat. Rev. Mol. Cell Biol. 15, 813–824 (2014).
Castro-Castro, A. et al. Cellular and molecular mechanisms of MT1-MMP-dependent cancer cell invasion. Annu. Rev. Cell Dev. Biol. 32, 555–576 (2016).
Lodillinsky, C. et al. p63/MT1-MMP axis is required for in situ to invasive transition in basal-like breast cancer. Oncogene 35, 344–357 (2016).
Hotary, K., Li, X. Y., Allen, E., Stevens, S. L. & Weiss, S. J. A cancer cell metalloprotease triad regulates the basement membrane transmigration program. Genes Dev. 20, 2673–2686 (2006).
Glentis, A. et al. Cancer-associated fibroblasts induce metalloprotease-independent cancer cell invasion of the basement membrane. Nat. Commun. 8, 1–13 (2017).
Friedl, P. & Wolf, K. Proteolytic interstitial cell migration: a five-step process. Cancer Metastasis Rev. 28, 129–135 (2009).
Wolf, K. et al. Physical limits of cell migration: control by ECM space and nuclear deformation and tuning by proteolysis and traction force. J. Cell Biol. 201, 1069–1084 (2013).
Lämmermann, T. et al. Rapid leukocyte migration by integrin-independent flowing and squeezing. Nature 453, 51–55 (2008).
Friedl, P. & Wolf, K. Proteolytic and non-proteolytic migration of tumour cells and leucocytes. Biochem. Soc. Symp. 70, 277–285 (2003).
Salmon, H. et al. Matrix architecture defines the preferential localization and migration of T cells into the stroma of human lung tumors. J. Clin. Investig. 122, 899–910 (2012).
Moreau, J.-F. et al. The emerging role of ECM crosslinking in T cell mobility as a hallmark of immunosenescence in humans. Ageing Res. Rev. 35, 322–335 (2017).
Owyong, M. et al. Overcoming barriers of age to enhance efficacy of cancer immunotherapy: the clout of the extracellular matrix. Front. Cell Dev. Biol. 6, 1–9 (2018).
Li, X. et al. Infiltration of CD8 + T cells into tumor cell clusters in triple-negative breast cancer. Proc. Natl Acad. Sci. USA 116, 3678–3687 (2019).
Wolf, K. et al. Multi-step pericellular proteolysis controls the transition from individual to collective cancer cell invasion. Nat. Cell Biol. 9, 893–904 (2007).
Sabeh, F. et al. Tumor cell traffic through the extracellular matrix is controlled by the membrane-anchored collagenase MT1-MMP. J. Cell Biol. 167, 769–781 (2004).
Feinberg, T. Y. et al. Divergent matrix-remodeling strategies distinguish developmental from neoplastic mammary epithelial cell invasion programs. Dev. Cell 47, 145–160.e146 (2018).
Artym, V. V., Zhang, Y., Seillier-Moiseiwitsch, F., Yamada, K. M. & Mueller, S. C. Dynamic interactions of cortactin and membrane type 1 matrix metalloproteinase at invadopodia: defining the stages of invadopodia formation and function. Cancer Res. 66, 3034–3043 (2006).
Hoshino, D. et al. Exosome secretion is enhanced by invadopodia and drives invasive behavior. Cell Rep. 5, 1159–1168 (2013).
Sung, B. H., Ketova, T., Hoshino, D., Zijlstra, A. & Weaver, A. M. Directional cell movement through tissues is controlled by exosome secretion. Nat. Commun. 6, 7164 (2015).
Gaggioli, C. et al. Fibroblast-led collective invasion of carcinoma cells with differing roles for RhoGTPases in leading and following cells. Nat. Cell Biol. 9, 1392–1400 (2007).
Goetz, J. G. et al. Biomechanical remodeling of the microenvironment by stromal caveolin-1 favors tumor invasion and metastasis. Cell 146, 148–163 (2011).
Attieh, Y. et al. Cancer-associated fibroblasts lead tumor invasion through integrin-β3-dependent fibronectin asse. J. Cell Biol. 216, 3509–3520 (2017).
Jia, H. et al. The tumor cell-secreted matricellular protein WISP1 drives pro-metastatic collagen linearization. EMBO J. 38, e101302 (2019).
Labernadie, A. et al. A mechanically active heterotypic E-cadherin/N-cadherin adhesion enables fibroblasts to drive cancer cell invasion. Nat. Cell Biol. 19, 224–237 (2017).
Ray, A., Slama, Z. M., Morford, R. K., Madden, S. A. & Provenzano, P. P. Enhanced directional migration of cancer stem cells in 3D aligned collagen matrices. Biophys. J. 112, 1023–1036 (2017).
Riching, K. M. et al. 3D collagen alignment limits protrusions to enhance breast cancer cell persistence. Biophys. J. 107, 2546–2558 (2014).
Fraley, S. I. et al. Three-dimensional matrix fiber alignment modulates cell migration and MT1-MMP utility by spatially and temporally directing protrusions. Sci. Rep. 5, 14580 (2015).
Semenza, G. L. The hypoxic tumor microenvironment: a driving force for breast cancer progression. Biochim. Biophys. Acta 1863, 382–391 (2016).
Muñoz-Nájar, U. M., Neurath, K. M., Vumbaca, F. & Claffey, K. P. Hypoxia stimulates breast carcinoma cell invasion through MT1-MMP and MMP-2 activation. Oncogene 25, 2379–2392 (2006).
Choi, J. Y., Jang, Y. S., Min, S. Y. & Song, J. Y. Overexpression of MMP-9 and HIF-1α in breast cancer cells under hypoxic conditions. J. Breast Cancer 14, 88–95 (2011).
Erler, J. T. et al. Lysyl oxidase is essential for hypoxia-induced metastasis. Nature 440, 1222–1226 (2006).
Gilkes, D. M., Bajpai, S., Chaturvedi, P., Wirtz, D. & Semenza, G. L. Hypoxia-inducible factor 1 (HIF-1) promotes extracellular matrix remodeling under hypoxic conditions by inducing P4HA1, P4HA2, and PLOD2 expression in fibroblasts. J. Biol. Chem. 288, 10819–10829 (2013).
Hofbauer, K.-H. et al. Oxygen tension regulates the expression of a group of procollagen hydroxylases. Eur. J. Biochem. 270, 4515–4522 (2003).
Ju, J. A. et al. Hypoxia selectively enhances integrin α 5 β 1 receptor expression in breast cancer to promote metastasis. Mol. Cancer Res. 15, 723–734 (2017).
Natarajan, S. et al. Collagen remodeling in the hypoxic tumor-mesothelial niche promotes ovarian cancer metastasis. Cancer Res. 79, 2271–2284 (2019).
Bergers, G. & Benjamin, L. E. Tumorigenesis and the angiogenic switch. Nat. Rev. Cancer 3, 401–410 (2003).
Goveia, J. et al. An integrated gene expression landscape profiling approach to identify lung tumor endothelial cell heterogeneity and angiogenic candidates. Cancer Cell 37, 21–36.e13 (2020).
Houck, K. A., Leung, D. W., Rowland, A. M., Winer, J. & Ferrara, N. Dual regulation of vascular endothelial growth factor bioavailability by genetic and proteolytic mechanisms. J. Biol. Chem. 267, 26031–26037 (1992).
Belotti, D. et al. Matrix metalloproteinases (MMP9 and MMP2) induce the release of vascular endothelial growth factor (VEGF) by ovarian carcinoma cells: implications for ascites formation. Cancer Res. 63, 5224–5229 (2003).
Deryugina, E. I. & Quigley, J. P. Tumor angiogenesis: MMP-mediated induction of intravasation- and metastasis-sustaining neovasculature. Matrix Biol. 44–46, 94–112 (2015).
Shao, R. et al. Acquired expression of periostin by human breast cancers promotes tumor angiogenesis through up-regulation of vascular endothelial growth factor receptor 2 expression. Mol. Cell Biol. 24, 3992–4003 (2004).
Rupp, T. et al. Tenascin-C orchestrates glioblastoma angiogenesis by modulation of pro- and anti-angiogenic signaling. Cell Rep. 17, 2607–2619 (2016).
Van Obberghen-Schilling, E. et al. Fibronectin and tenascin-C: accomplices in vascular morphogenesis during development and tumor growth. Int. J. Dev. Biol. 55, 511–525 (2011).
Rodriguez-Manzaneque, J. C. et al. Thrombospondin-1 suppresses spontaneous tumor growth and inhibits activation of matrix metalloproteinase-9 and mobilization of vascular endothelial growth factor. Proc. Natl Acad. Sci. USA 98, 12485–12490 (2001).
Lawler, P. R. & Lawler, J. Molecular basis for the regulation of angiogenesis by thrombospondin-1 and -2. Cold Spring Harb. Perspect. Med. 2, a006627 (2012).
Wagenblast, E. et al. A model of breast cancer heterogeneity reveals vascular mimicry as a driver of metastasis. Nature 520, 358–362 (2015). This study describes that highly metastatic tumour cells form vascular like structures and that his process is accompanied by the upregulation of two proteases inhibitors Serpin E2 and SLPI that serve as anticoagulant and support blood perfusion.
Seftor, R. E. et al. Cooperative interactions of laminin 5 gamma2 chain, matrix metalloproteinase-2, and membrane type-1-matrix/metalloproteinase are required for mimicry of embryonic vasculogenesis by aggressive melanoma. Cancer Res. 61, 6322–6327 (2001).
Barnett, F. H. et al. Macrophages form functional vascular mimicry channels in vivo. Sci. Rep. 6, 36659 (2016).
Ting, D. T. et al. Single-cell RNA sequencing identifies extracellular matrix gene expression by pancreatic circulating tumor cells. Cell Rep. 8, 1905–1918 (2014).
Yu, M. et al. Circulating breast tumor cells exhibit dynamic changes in epithelial and mesenchymal composition. Science 339, 580–584 (2013).
Nieswandt, B., Hafner, M., Echtenacher, B. & Männel, D. N. Lysis of tumor cells by natural killer cells in mice is impeded by platelets. Cancer Res. 59, 1295–1300 (1999).
Cheung, K. J. et al. Polyclonal breast cancer metastases arise from collective dissemination of keratin 14-expressing tumor cell clusters. Proc. Natl Acad. Sci. USA 113, E854–863 (2016).
Najmeh, S. et al. Neutrophil extracellular traps sequester circulating tumor cells via β1-integrin mediated interactions. Int. J. Cancer 140, 2321–2330 (2017).
Liu, Y. & Cao, X. Characteristics and significance of the pre-metastatic niche. Cancer Cell 30, 668–681 (2016).
Weigelt, B. et al. No common denominator for breast cancer lymph node metastasis. Br. J. Cancer 93, 924–932 (2005).
Naba, A. et al. Extracellular matrix signatures of human primary metastatic colon cancers and their metastases to liver. BMC Cancer 14, 518 (2014).
Mayorca-Guiliani, A. E. et al. ISDoT: in situ decellularization of tissues for high-resolution imaging and proteomic analysis of native extracellular matrix. Nat. Med. 23, 890–898 (2017).
Høye, A. M. & Erler, J. T. Structural ECM components in the premetastatic and metastatic niche. Am. J. Physiol. Cell Physiol. 310, C955–967 (2016).
Zeltz, C. & Gullberg, D. Post-translational modifications of integrin ligands as pathogenic mechanisms in disease. Matrix Biol. 40, 5–9 (2014).
Song, S. & Yu, Y. Progression on citrullination of proteins in gastrointestinal cancers. Front. Oncol. 9, 15 (2019).
Rohrbach, A. S., Slade, D. J., Thompson, P. R. & Mowen, K. A. Activation of PAD4 in NET formation. Front. Immunol. 3, 360 (2012).
Yuzhalin, A. E. et al. Colorectal cancer liver metastatic growth depends on PAD4-driven citrullination of the extracellular matrix. Nat. Commun. 9, 4783 (2018).
Cox, T. R. et al. The hypoxic cancer secretome induces pre-metastatic bone lesions through lysyl oxidase. Nature 522, 106–110 (2015).
Lynch, C. C. et al. MMP-7 promotes prostate cancer-induced osteolysis via the solubilization of RANKL. Cancer Cell 7, 485–496 (2005).
Ara, T. et al. Interleukin-6 in the bone marrow microenvironment promotes the growth and survival of neuroblastoma cells. Cancer Res. 69, 329–337 (2009).
Lu, X. et al. ADAMTS1 and MMP1 proteolytically engage EGF-like ligands in an osteolytic signaling cascade for bone metastasis. Genes Dev. 23, 1882–1894 (2009).
Leeming, D. J. et al. The relative use of eight collagenous and noncollagenous markers for diagnosis of skeletal metastases in breast, prostate, or lung cancer patients. Cancer Epidemiol. Biomark. Prev. 15, 32–38 (2006).
Dai, J. et al. Vascular endothelial growth factor contributes to the prostate cancer-induced osteoblast differentiation mediated by bone morphogenetic protein. Cancer Res. 64, 994–999 (2004).
Logothetis, C. J. & Lin, S.-H. Osteoblasts in prostate cancer metastasis to bone. Nat. Rev. Cancer 5, 21–28 (2005).
Casbon, A.-J. et al. Invasive breast cancer reprograms early myeloid differentiation in the bone marrow to generate immunosuppressive neutrophils. Proc. Natl Acad. Sci. USA 112, E566–575 (2015).
Peinado, H. et al. Pre-metastatic niches: organ-specific homes for metastases. Nat. Rev. Cancer 17, 302–317 (2017).
Erler, J. T. et al. Hypoxia-induced lysyl oxidase is a critical mediator of bone marrow cell recruitment to form the premetastatic niche. Cancer Cell 15, 35–44 (2009).
Kaplan, R. N. et al. VEGFR1-positive haematopoietic bone marrow progenitors initiate the pre-metastatic niche. Nature 438, 820–827 (2005). This was one of the first studies showing that tumour cells initiate remodelling of the ECM in sites distant to the primary tumour creating a pre-metastatic niche in order to support metastatic colonization.
Sangaletti, S. et al. Osteopontin shapes immunosuppression in the metastatic niche. Cancer Res. 74, 4706–4719 (2014).
Seubert, B. et al. Tissue inhibitor of metalloproteinases (TIMP)-1 creates a premetastatic niche in the liver through SDF-1/CXCR4-dependent neutrophil recruitment in mice. Hepatology 61, 238–248 (2015).
Barbazán, J. et al. Molecular characterization of circulating tumor cells in human metastatic colorectal cancer. PLoS ONE 7, e40476 (2012).
Huang, Y. et al. Pulmonary vascular destabilization in the premetastatic phase facilitates lung metastasis. Cancer Res. 69, 7529–7537 (2009).
Ghajar, C. M. et al. The perivascular niche regulates breast tumour dormancy. Nat. Cell Biol. 15, 807–817 (2013).
Shimoda, M. & Khokha, R. Metalloproteinases in extracellular vesicles. Biochim. Biophys. Acta Mol. Cell Res. 1864, 1989–2000 (2017).
Becker, A. et al. Extracellular vesicles in cancer: cell-to-cell mediators of metastasis. Cancer Cell 30, 836–848 (2016).
Budczies, J. et al. The landscape of metastatic progression patterns across major human cancers. Oncotarget 6, 570–583 (2015).
Barney, L. E. et al. A cell–ECM screening method to predict breast cancer metastasis. Integr. Biol. 7, 198–212 (2015).
Hoshino, A. et al. Tumour exosome integrins determine organotropic metastasis. Nature 527, 329–335 (2015). Landmark study which shows that tumour-derived exosomes activate stroma cells in distant organs to remodel the ECM creating a pre-metastatic niche. Organtropism is thereby dictated by specific integrin expression on the exosome surface.
Costa-Silva, B. et al. Pancreatic cancer exosomes initiate pre-metastatic niche formation in the liver. Nat. Cell Biol. 17, 816–826 (2015).
Barbazán, J. et al. Liver metastasis is facilitated by the adherence of circulating tumor cells to vascular fibronectin deposits. Cancer Res. 77, 3431–3441 (2017). This study describes how endothelial cells are triggered by tumour cells to deposit fibronectin that enhances tumour cell adhesion, extravasation and consequently metastasis.
Hebert, J. D. et al. Proteomic profiling of the ECM of xenograft breast cancer metastases in different organs reveals distinct metastatic niches. Cancer Res. 80, 1475–1485 (2020). This study determined the ECM proteins present at brain, lung, liver, and bone metastatic sites in a breast cancer mouse model. The matrisome composition was unique at each metastatic site and differed from healthy tissue.
Oskarsson, T. et al. Breast cancer cells produce tenascin C as a metastatic niche component to colonize the lungs. Nat. Med. 17, 867–874 (2011).
Malanchi, I. et al. Interactions between cancer stem cells and their niche govern metastatic colonization. Nature 481, 85–89 (2011).
Thiery, J. P., Acloque, H., Huang, R. Y. J. & Nieto, A. M. Epithelial-mesenchymal transitions in development and disease. Cell 139, 871–890 (2009).
Insua-Rodríguez, J. et al. Stress signaling in breast cancer cells induces matrix components that promote chemoresistant metastasis. EMBO Mol. Med. 10, e9003 (2018).
Gao, D. et al. Myeloid progenitor cells in the premetastatic lung promote metastases by inducing mesenchymal to epithelial transition. Cancer Res. 72, 1384–1394 (2012).
Albrengues, J. et al. Neutrophil extracellular traps produced during inflammation awaken dormant cancer cells in mice. Science 361, eaao4227 (2018). First study which shows that ECM remodelling a mechanism how dormant cancer cells awaken to form metastasis and highlights the importance of bioactive ECM components in that process.
Jailkhani, N. et al. Noninvasive imaging of tumor progression, metastasis, and fibrosis using a nanobody targeting the extracellular matrix. Proc. Natl Acad. Sci. USA 116, 14181–14190 (2019).
Ishihara, J. et al. Matrix-binding checkpoint immunotherapies enhance antitumor efficacy and reduce adverse events. Sci. Transl. Med. 9, eaan0401 (2017).
Ishihara, J. et al. Improving efficacy and safety of agonistic anti-cd40 antibody through extracellular matrix affinity. Mol. Cancer Ther. 17, 2399–2411 (2018).
Ishihara, J. et al. Targeted antibody and cytokine cancer immunotherapies through collagen affinity. Sci. Transl. Med. 11, eaau3259 (2019).
Momin, N. et al. Anchoring of intratumorally administered cytokines to collagen safely potentiates systemic cancer immunotherapy. Sci. Transl. Med. 11, eaaw2614 (2019).
Sachs, N. et al. A living biobank of breast cancer organoids captures disease heterogeneity. Cell 172, 373–386.e310 (2018).
Naba, A. et al. Characterization of the extracellular matrix of normal and diseased tissues using proteomics. J. Proteome Res. 16, 3083–3091 (2017).
Keren, L. et al. A structured tumor-immune microenvironment in triple negative breast cancer revealed by multiplexed ion beam imaging. Cell 174, 1373–1387.e1319 (2018).
Miyamoto, K. et al. Tumour-suppressive miRNA-26a-5p and miR-26b-5p inhibit cell aggressiveness by regulating PLOD2 in bladder cancer. Br. J. Cancer 115, 354–363 (2016).
Chen, Y. et al. Lysyl hydroxylase 2 induces a collagen cross-link switch in tumor stroma. J. Clin. Invest. 125, 1147–1162 (2015).
Gjaltema, R. A., de Rond, S., Rots, M. G. & Bank, R. A. Procollagen lysyl hydroxylase 2 expression is regulated by an alternative downstream transforming growth factor beta-1 activation mechanism. J. Biol. Chem. 290, 28465–28476 (2015).
Kurozumi, A. et al. Regulation of the collagen cross-linking enzymes LOXL2 and PLOD2 by tumor-suppressive microRNA-26a/b in renal cell carcinoma. Int. J. Oncol. 48, 1837–1846 (2016).
Elia, I. et al. Breast cancer cells rely on environmental pyruvate to shape the metastatic niche. Nature 568, 117–121 (2019).
Barker, H. E., Cox, T. R. & Erler, J. T. The rationale for targeting the LOX family in cancer. Nat. Rev. Cancer 12, 540–552 (2012).
Siegel, M. & Khosla, C. Transglutaminase 2 inhibitors and their therapeutic role in disease states. Pharmacol. Ther. 115, 232–245 (2007).
Mehta, K., Fok, J., Miller, F. R., Koul, D. & Sahin, A. A. Prognostic significance of tissue transglutaminase in drug resistant and metastatic breast cancer. Clin. Cancer Res. 10, 8068–8076 (2004).
Mangala, L. S. & Mehta, K. Tissue transglutaminase (TG2) in cancer biology. Prog. Exp. Tumor Res. 38, 125–138 (2005).
Richter, P. et al. IIICS de novo glycosylated fibronectin as a marker for invasiveness in urothelial carcinoma of the urinary bladder (UBC). J. Cancer Res. Clin. Oncol. 134, 1059–1065 (2008).
Freire-de-Lima, L. et al. Involvement of O-glycosylation defining oncofetal fibronectin in epithelial-mesenchymal transition process. Proc. Natl Acad. Sci. USA 108, 17690–17695 (2011).
Singh, C. et al. Integrin expression and glycosylation patterns regulate cell-matrix adhesion and alter with breast cancer progression. Biochem. Biophys. Res. Commun. 499, 374–380 (2018).
Yalak, G. & Vogel, V. Ectokinases as novel cancer markers and drug targets in cancer therapy. Cancer Med. 4, 404–414 (2015).
Yalak, G., Shiu, J. Y., Schoen, I., Mitsi, M. & Vogel, V. Phosphorylated fibronectin enhances cell attachment and upregulates mechanical cell functions. PLoS ONE 14, e0218893 (2019).
Skhinas, J. N. & Cox, T. R. The interplay between extracellular matrix remodelling and kinase signalling in cancer progression and metastasis. Cell Adh. Migr. 12, 529–537 (2018).
Bordoli, M. R. et al. A secreted tyrosine kinase acts in the extracellular environment. Cell 158, 1033–1044 (2014).
Soares da Costa, D., Reis, R. L. & Pashkuleva, I. Sulfation of glycosaminoglycans and its implications in human health and disorders. Annu. Rev. Biomed. Eng. 19, 1–26 (2017).
Escobar Galvis, M. L. et al. Transgenic or tumor-induced expression of heparanase upregulates sulfation of heparan sulfate. Nat. Chem. Biol. 3, 773–778 (2007).
Vlodavsky, I., Gross-Cohen, M., Weissmann, M., Ilan, N. & Sanderson, R. D. Opposing functions of heparanase-1 and heparanase-2 in cancer progression. Trends Biochem. Sci. 43, 18–31 (2018).
Jaisson, S. et al. Impact of carbamylation on type I collagen conformational structure and its ability to activate human polymorphonuclear neutrophils. Chem. Biol. 13, 149–159 (2006).
Said, G. et al. Impact of carbamylation and glycation of collagen type I on migration of HT1080 human fibrosarcoma cells. Int. J. Oncol. 40, 1797–1804 (2012).
Chan, N. et al. Influencing the tumor microenvironment: a phase II study of copper depletion using tetrathiomolybdate in patients with breast cancer at high risk for recurrence and in preclinical models of lung metastases. Clin. Cancer Res. 23, 666–676 (2017).
Nazari, S. S. & Mukherjee, P. An overview of mammographic density and its association with breast cancer. Breast Cancer 25, 259–267 (2018).
Huo, C. W. et al. High mammographic density is associated with an increase in stromal collagen and immune cells within the mammary epithelium. Breast Cancer Res. 17, 79 (2015).
Sherratt, M. J., McConnell, J. C. & Streuli, C. H. Raised mammographic density: causative mechanisms and biological consequences. Breast Cancer Res. 18, 45 (2016).
Schedin, P., O’Brien, J., Rudolph, M., Stein, T. & Borges, V. Microenvironment of the involuting mammary gland mediates mammary cancer progression. J. Mammary Gland Biol. Neoplasia 12, 71–82 (2007).
Schedin, P. Pregnancy-associated breast cancer and metastasis. Nat. Rev. Cancer 6, 281–291 (2006).
Russo, J., Rivera, R. & Russo, I. H. Influence of age and parity on the development of the human breast. Breast Cancer Res. Treat. 23, 211–218 (1992).
Murrow, L. M. et al. Pregnancy and obesity modify the epithelial composition and hormone signaling state of the human breast. bioRxiv https://doi.org/10.1101/430611 (2020).
Goddard, E. T. et al. Quantitative extracellular matrix proteomics to study mammary and liver tissue microenvironments. Int. J. Biochem. Cell Biol. 81, 223–232 (2016).
Martinson, H. A., Jindal, S., Durand-Rougely, C., Borges, V. F. & Schedin, P. Wound healing-like immune program facilitates postpartum mammary gland involution and tumor progression. Int. J. Cancer 136, 1803–1813 (2015).
Guo, Q., Betts, C., Pennock, N., Mitchell, E. & Schedin, P. Mammary gland involution provides a unique model to study the TGF-β cancer paradox. J. Clin. Med. 6, 10 (2017).
Senthebane, D. A. et al. The role of tumor microenvironment in chemoresistance: to survive, keep your enemies closer. Int. J. Mol. Sci. 18, 1586 (2017).
Joyce, M. H. et al. Phenotypic basis for matrix stiffness-dependent chemoresistance of breast cancer cells to doxorubicin. Front. Oncol. 8, 337 (2018).
Najafi, M., Farhood, B. & Mortezaee, K. Extracellular matrix (ECM) stiffness and degradation as cancer drivers. J. Cell. Biochem. 120, 2782–2790 (2019).
Roma-Rodrigues, C., Mendes, R., Baptista, P. V. & Fernandes, A. R. Targeting tumor microenvironment for cancer therapy. Int. J. Mol. Sci. 20, 840 (2019).
Neuzillet, C. et al. Targeting the TGFβ pathway for cancer therapy. Pharmacol. Ther. 147, 22–31 (2015).
Winer, A., Adams, S. & Mignatti, P. Matrix metalloproteinase inhibitors in cancer therapy: turning past failures into future successes. Mol. Cancer Ther. 17, 1147–1155 (2018).
Isaacson, K. J., Martin Jensen, M., Subrahmanyam, N. B. & Ghandehari, H. Matrix-metalloproteinases as targets for controlled delivery in cancer: an analysis of upregulation and expression. J. Control Release 259, 62–75 (2017).
Acknowledgements
Zena Werb’s outstanding mentorship made this work possible. She will be dearly missed. We apologise to all colleagues whose work could not be mentioned due to space restrictions. We thank N. Ataii, M. Owyong and C. Diadhiou for their support. This study was supported by funds from the National Institutes of Health (R01 CA190851, R01 CA201537 and U01 CA199315), MetaVIVOR, an EMBO long-term post-doctoral fellowship (EMBO ALTF 159-2017 to J.W.), an NIH postdoctoral traineeship (T32 CA108462 to A.A.-O.), and an American Cancer Society–2017 Seattle Gala Paddle Raise Postdoctoral Fellowship (PF-18-118-01-CDD to K.J.M.).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Winkler, J., Abisoye-Ogunniyan, A., Metcalf, K.J. et al. Concepts of extracellular matrix remodelling in tumour progression and metastasis. Nat Commun 11, 5120 (2020). https://doi.org/10.1038/s41467-020-18794-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-020-18794-x
- Springer Nature Limited
This article is cited by
-
Reconstructed colorectal cancer model to dissect the anti-tumor effect of mesenchymal stromal cells derived extracellular vesicles
Experimental Hematology & Oncology (2024)
-
CDCA5 promoted cell invasion and migration by activating TGF-β1 pathway in human ovarian cancer cells
Journal of Ovarian Research (2024)
-
Effects of mechanical ventilation on the interstitial extracellular matrix in healthy lungs and lungs affected by acute respiratory distress syndrome: a narrative review
Critical Care (2024)
-
Deciphering the performance of macrophages in tumour microenvironment: a call for precision immunotherapy
Journal of Hematology & Oncology (2024)
-
An overview of CCN4 (WISP1) role in human diseases
Journal of Translational Medicine (2024)