

Abstract
Thermal electron transfer through hydrogen bonds remains largely unexplored. Here we report the study of electron transfer through amide-amide hydrogen bonded interfaces in mixed-valence complexes with covalently bonded Mo2 units as the electron donor and acceptor. The rate constants for electron transfer through the dual hydrogen bonds across a distance of 12.5 Å are on the order of ∼ 1010 s−1, as determined by optical analysis based on Marcus–Hush theory and simulation of ν(NH) vibrational band broadening, with the electron transfer efficiencies comparable to that of π conjugated bridges. This work demonstrates that electron transfer across a hydrogen bond may proceed via the known proton-coupled pathway, as well as an overlooked proton-uncoupled pathway that does not involve proton transfer. A mechanistic switch between the two pathways can be achieved by manipulation of the strengths of electronic coupling and hydrogen bonding. The knowledge of the non-proton coupled pathway has shed light on charge and energy transport in biological systems.
Similar content being viewed by others

Introduction
Understanding electronic coupling (EC) and electron transfer (ET) across hydrogen bonds (HBs) is of fundamental importance in elucidating important biochemical processes in a diverse number of biological systems, including enzymes, proteins, and DNA1. Unlike covalent bonds, hydrogen-bonding interactions (X‒H···Y) are predominantly electrostatic in nature, comprising of a single-electron owned H atom in the middle of three linearly connected atoms, which makes an HB relatively weak, flexible, and dynamic. The process of transferring electrons across such an intermolecular interface has long inspired explorations of theoretical and experimental chemists2,3. In this context, the two major questions are (i) how efficiently are H bonds able to transport electrons and (ii) whether the ET process occurs with the help of the proton. The first quantitative evaluation of through-HB ET by comparison with covalent σ and π bonds employed an elegantly designed D–HB–A molecule with zinc(II) and iron(III) porphyrins as the electron donor (D) and acceptor (A), respectively. Charge transfer across this acid–acid HB interface in the photoexcited states was monitored by ultrafast transient spectroscopy4. The study led to a remarkable conclusion that the extent of EC through HBs is greater than via C–C σ bonds, but lower than across a π-conjugated bridge. However, significant discrepancies have arisen from subsequent experimental5 and theoretical6 studies, and thus the efficiency of EC mediation by an HB interface, relative to covalent bonds, remains controversial.
Donor–acceptor ET crossing an HB interface is often accompanied by a proton transfer reaction associated with two HB states, X–H···Y (initial) and X···H–Y (final); the overall ET reaction is referred to as proton-coupled electron transfer (PCET). Therefore, X–H bond breakage and Y–H bond formation are the prerequisites for a PCET reaction to be considered. Within the classical ET theoretical framework, contributions from Cukier and Nocera7 and Hammes–Schiffer and Stuchebrukhov8 have led to the development of PCET theories by reformulating the Fermi’s golden rule with a modified Frank–Condon factor. To account for the large displacement of the proton, the proton motions are further separated from other nuclear vibrations in the spirit of the Born-Oppenheimer approach9; by doing so, the EC element for the unperturbed diabatic states is redefined7,8. Nonadiabatic PCET theories treat ET and PT as separate particle tunneling events, which may occur sequentially or in concert. Unfortunately, employing the golden rule formalism to derive ET rate constants necessitates the knowledge of some physical parameters that are not always experimentally available, which prevents validation of the theories and their broad application. In a D–HB–A system, vibronic coupling plays an important role in determining the ET kinetics due to the dynamic nature of the bridge. To address this issue, one needs to scale the HB strength, and thus, topologically well-defined experimental models are in demand. Furthermore, up to now, experimental studies of PCET kinetics has been focused on photoinduced ET7, which feature nonadiabaticity and high exothermicity. However, a number of biochemical systems undergo PCET without light and under less exothermic conditions; typical examples include light-independent reactions in the Calvin cycle and cellular respiration catalyzed by cytochrome c oxidase 1,10.
Mixed-valence (MV) D–B–A molecules, which have identical D and A sites differing only in formal oxidation states, are valuable experimental models for study of electron self-exchange reaction with Marcus–Hush theory11,12, which has been successfully used to evaluate intramolecular ET through covalently bonded bridges13,14. Recent efforts have produced various examples of hydrogen bonded MV complexes, in which self-complementary HB interactions are used to bridge the electron donor and acceptor15,16,17, with the aim to evaluate the D–A EC optically upon analysis of the intervalence charge transfer (IVCT) absorbance. However, this optical feature has only been observed in few examples of hydrogen bonded MV compounds, and lack of a test-bed series of compounds has hindered the kinetic study of thermal ET in electron self-exchange reactions with zero driving force (−ΔG° = 0)5,16,17.
Herein we report the study of thermally induced ET across an HB bridge in symmetrical MV D–HB–A systems (−ΔG° = 0) with a quadruply bonded Mo2 unit as the donor, and a Mo2 unit with a bond order of 3.5 as the acceptor. The quadruply bonded Mo2 unit has an electronic configuration of σ2π4δ2 18, and in a Mo2 D–B–A system, the δ orbital in the Mo2 unit is discriminated from the σ and π orbitals by symmetry and energy. Therefore, only the δ electrons in the HOMO of the Mo2 core are involved in ET from the donor to acceptor, which makes the system unique and most desirable as an experimental model for study of EC and ET14,19. By analysis of IVCT absorptions for the Mo2 D–HB–A systems reported here, we were able to determine the EC matrix elements (Hab) from the Mulliken–Hush expression20 and study the ET kinetics by employing Marcus–Hush theory11,12,20, which provides results in excellent agreement with the data obtained from analysis of ν(NH) vibrational band broadening. Comparisons of ET kinetics with the data obtained from analogous Mo2 dimers having π (phenylene) and σ (cyclohexylene) bond bridges that span similar D–A distances show that the HB interface conducts EC and ET as well as the π-conjugated bridges. This study has validated the PCET theory for electron self-exchange reactions, and demonstrated a usually overlooked, proton uncoupled electron transfer (PUET) pathway of thermal electron transfer in HB systems.
Results
Characterization of the HB bridged Mo2 dimers
The three categories of Mo2 complexes studied are of a general formula [Mo2]–bridge–[Mo2], in which [Mo2] = [Mo2(DArF)3(O2C)] (DArF = N,N′- diarylformamidinate) and the bridges are an amide–amide HB (1), phenylene (2), or cyclohexylene (3), as shown in Fig. 1a. To fine-tune the EC of the system21, each series consists of four members which are differentiated by the para substituents X (X = N(CH3)2(a), CH(CH3)2 (b), OCH3 (c), or CH3 (d)) on the DArF ligands. The HB bridged Mo2 dimers (1a–d) are self-assembled in less polar solvents from two Mo2 paddlewheel monomers, [Mo2(DArF)3(O2CCONH2)], in which the fourth equatorial coordination position of the Mo2 center is occupied by an acetamide ligand.

a Molecular skeletons for [Mo2]–bridge–[Mo2] complexes under investigation. b X-ray crystal structures for the Mo2 dimers bridged by amide–amide dual hydrogen bonds (H atoms are omitted for clarity). Each series (1–3) consists of four Mo2 dimers that differ in the substituents (X, a–d) on the formamidinate auxiliary ligands. By varying the electron-donating ability of X, the donor–acceptor electronic coupling in the mixed-valence complexes is tuned (see text)
The four HB-assembled Mo2 dimers (1a–d) have been structurally characterized by X-ray diffraction. The crystal structures are presented in Fig. 1b and the selected bond parameters are listed in Table 1. The structures of 1a–d show that the Mo2 monomers are dimerized in solid state through amide–amide dual HBs, giving these dimers the same topological geometry as that of the covalent π (2a–d) and σ bond (3a–d) bridged analogs (Fig. 1a). The Mo–Mo bond distances are about 2.1 Å, which is comparable to quadruple bonds with similar supporting ligands21, and the Mo2···Mo2 separations of ∼12.5 Å are slightly longer than those for series 2 and 3 (∼11.25 Å)21. The HB bridge is built by a pair of intermolecular N–H···O hydrogen bonds with N···O distances of 2.865 Å (1b) < 2.878 Å (1d) < 2.885 Å (1a) < 2.976 Å (1c) and ∠N–H···O angles of 172.46° (1a) > 169.96° (1d) > 154.42° (1b) > 151.35° (1c). A sp2 N atom and N–H bond distance of 0.86 Å are assumed for the amide group in all these compounds, resulting in O···H distances of 2.025 Å (1d) < 2.031 Å (1a) < 2.066 Å (1b) < 2.174 Å (1c). Therefore, the structural parameters show that the HBs in 1a and 1d are appreciably stronger than those in 1b and 1c. It appears that 1a has strong HBs, which may be due to the strong electron donating of the N(CH3)2 groups, but there is no strict correlation between HB strength and the electronic property of the X substituents. The HBs in these adducts are generally weak, in comparison with distances of <2.65 Å expected for strong N–H···O bonds22. However, the dual HBs in the amide–amide linkage should strengthen the bonding between the two Mo2 units. The 1H NMR spectra of compounds 1a–d in CDCl3 exhibit two widely separated resonances for the amide protons (N–H) at ∼5.6 ppm for the free proton and at ∼7.2 ppm for the bonded proton, a small downfield chemical shift indicating relatively weak HBs22. In contrast, in deuterated dimethylsulfoxide (DMSO- d6), the two amide protons exhibit similar chemical shifts (∼8 ppm), corresponding to the monomeric precursor; therefore, the dimeric structures of 1a–d in solid state and in less polar solvents such as dichloromethane (DCM) are clearly established.
Compounds 1b–d show two closely separated redox couples in their cyclic voltammograms (CVs) in DCM (Supplementary Figure 2). The chemical potentials (E1/2) fall in the range of −0.3 to 0.1 V (vs. Fc+/0) and the potential separations (ΔE1/2) are estimated to be ∼100 mV from Richard–Taube methods (Supplementary Figure 31)23, as listed in Table 2. In DMF, these complexes show only one redox couple, as expected, for the corresponding monomers (Supplementary Figure 3)15. For 1a, 2a, and 3a, the N(CH3)2 groups on the DArF ancillary ligands are redox active (E1/2 = 0.1–0.5 V vs. Fc+/0), which makes the redox waves of the Mo2 centers weak and irreversible. For the other HB adducts (1b, 1c, and 1d), the redox processes are attributed to the two one-electron oxidations occurring on each of the Mo2 centers, and the E1/2 and ΔE1/2 values are comparable with the phenylene bridged dimers. It is interesting to note that the ΔE1/2 values for 1b–d are larger in most cases than those for the corresponding phenylene bridged analogs (2b–d) (Table 2 and Supplementary Figure 32). This is especially the case for 1c, which is notable given that the HB bridge separates the two Mo2 units even further than the phenylene bridge in 2c (Supplementary Figure 4). The electrochemical behaviors demonstrate that these Mo2 complexes exist as HB dimers in both neutral and the oxidized states in DCM solution. This is contrary to the Ru3O D–HB–A system in which the HB bridged dimer is formed upon reduction of one of the metal cluster to yield the mixed-valent species24. The cyclohexylene bridged complexes 3a–d present the smallest ΔE1/2 values (∼70 mV) (Supplementary Figures 5 and 33), showing very weak coupling due to the electrostatic effect. The increased magnitude of ΔE1/2 for 1a–d account for the large resonant effect on the donor–acceptor coupling (vide infra) and probably the dynamic properties of the HB adducts as well.
All the HB adducts in DCM exhibit two isolated IR bands at ∼3517 and at ∼3398 cm−1, labeled as ν(NH)A and ν(NH)B in Fig. 2a–d, respectively. Similar ν(NH) bands are observed at 3529 and 3319 cm−1 for the 2-aminopyridine dimer25,26, and at 3364 and 3166 cm−1 for the benzamide dimers27. Evidently, ν(NH)A and ν(NH)B result from stretching vibrations of the free and bonded N–H bonds in the amide–amide dual HB linkage, respectively. The appearance of only two ν(NH) bands in the spectrum signals the presence of an inversion center on the HB motif. In DMF, two ν(NH) bands with similar stretching frequencies, ∼3545 and ∼3485 cm−1, are observed, indicating the presence of Mo2 monomers (Supplementary Figure 1)25. It is important to note that 1a and 1d exhibit an extra band at 3613 cm−1(Fig. 2a, d) that is absent for 1b and 1c. This high frequency band is assigned to the O–H stretching mode and is a direct evidence for tautomerism in these two systems28. The appearance of ν(OH) band in the spectra of 1a and 1d is ascribable to the stronger HBs that lower the PT energy barrier22. Contrarily, the non-linear structure (Fig. 1b, c) of weaker HBs in 1b and 1c inhibits the proton transfer.

Infrared spectra and assignments of N‒H vibrations for 1, 1+, and 12+. a X = N(CH3)2. b X = CH(CH3)2. c X = OCH3. d X = CH3. e Assignments of the IR N‒H and O‒H stretching bands for 1a and 1d in different oxidation states (0, +1 and +2). f Assignments of the IR N‒H stretching bands for 1b and 1c in different oxidation states (0, +1 and +2). Note that a ν(OH) band at 3613 cm−1 is observed for systems 1a and 1d, but not for 1b and 1c. For the system with X = N(CH3)2, the 3425 cm−1 band (marked with an asterisk), which is not affected by the ET and PT processes, is a frequency double band for a mid-IR band (1713 cm−1)
The cationic mixed-valence complexes of the three series (1+, 2+, and 3+) were prepared by one-electron oxidation of the corresponding neutral precursors using a stoichiometric amount of the oxidizing reagent ferrocenium hexafluorophosphate. The electron paramagnetic resonant (EPR) and UV–Vis–NIR spectra are recorded in situ. In the EPR spectra, all the MV complexes exhibit a symmetric peak at g = ∼1.94 (Supplementary Figure 6), indicating that for all Mo2 the dimers, the unpaired electron is mainly localized in the δ orbital29,30. The striking optical feature observed in the spectra for the MV complexes 1+ (Supplementary Figures 34–37) and 2+ (Supplementary Figures 39–41), except for 2a+, is a broad, symmetrical absorption band in the near-IR region, which is attributed to IVCT between the two Mo2 centers, as shown in Fig. 3. For both of the systems, the IVCT band parameters, including transition energy (EIT), band intensity (εIT), and half bandwidth (Δν1/2), are comparable with those found for other Mo2 dimers with various π bridges differing in length14,31,32, symmetry19, conjugation33, and conformation34. As expected, the absorption spectra of the corresponding monomers, [Mo2(DArF)3(O2CCONH2)]0/+, obtained in DMF do not exhibit the characteristic IVCT transition, as shown in Supplementary Figure 38. Therefore, the presence of a well-defined IVCT band for each of the HB adducts confirms their dimeric structure and the mixed-valency of the singly oxidized complexes. By contrast, no IVCT absorbance was observed in the spectra of complexes 3a+‒d+ (Supplementary Figure 42), which indicates very weak EC between the two bridged Mo2 centers in this series, consistent with the electrochemical results.

Near- to mid-infrared spectra of the mixed-valence complexes showing the vibronic intervalence charge transfer absorption bands. a For the hydrogen bond bridged MV complexes (1a–d)+ and b for the phenylene bridged MV complexes (2b–d)+. For complexes (1a–d)+ and (2b–d)+ a symmetrical IVCT absorption band is observed. The same color code is used to show the full Gaussian-shaped IVCT band profiles for differerent MV systems with the same X substituent
Notably, for 1+, the intervalence transition energies (EIT) in the range of 4000–4500 cm−1 are considerably lower than those for 2+ (EIT = 4500–4850 cm−1), and the bandwidths (Δν1/2) are only about a half of the Δν1/2 values for 2+ (Fig. 3, Supplementary Figure 8 and Table 2). These results are consistent with the large ΔE1/2 values observed for 1a–d, as a low energy, narrow IVCT band indicates strong EC. As expected, the strong electron donating of the substituents in 1a+ and 2b+ gives rise to the lowest transition energy EIT in the series (Table 2). However, for 1+, the IVCT absorption intensities (εIT, M−1 cm−1) are generally lower than those for 2+, except for 1a+ that exhibits an exceptionally intense IVCT band (Fig. 3 and Table 2). Unlike other members of the 2+ series, 2a+ does not exhibit an IVCT band. This different optical behavior can be rationalized by the redox active N(CH3)2 groups, which act as electron donors and become the redox partner of the cationic Mo2 unit. Indeed, for 2a+, an extra charge transfer absorption band is observed at λ = 1250 nm, which is tentatively assigned to charge transfer from the dimethylamine N atom to the Mo2 center.
Determination of H ab and k ET data for the Mo2 dimers
The well-defined IVCT bands for 1+ and 2+ allow determination of the electronic coupling matrix elements Hab from the Mulliken–Hush expression (Eq. (1)) 12,20.
Considering that the δ electrons are delocalized over the coordination shell of the Mo2 center14,19, the effective ET distances (rab) were estimated from the C to C distance between the two linked carboxylic groups, and found to be ~7.11 Å for 1+ and ~5.85 Å for 2+. These effective ET distances are around half of the Mo2···Mo2 separations, close to 40% of the geometrical distance between donor and acceptor, in accordance with previous literature reports35. For the 1+ series, the largest coupling parameter, Hab = 410 cm−1, is found for 1a+; while for the other three adducts, the Hab values decrease from 194 to 130 cm−1 as the substituents become less electron donating (Table 2). For all the complexes in series 2+, except for 2a+, similar coupling constants, Hab ≈ 450 cm−1, are found. Therefore, the MV compounds in series 1+ and 2+ belong to Class II in Robin–Day’s scheme36. For both series, the magnitudes of Hab vary following the same trend, showing remarkable correlation to the remote substituents as observed in previous work21. Interestingly, it appears that EC through an HB interface is even more sensitive to the electronic property of the substituents than through a π bridge (Table 2).
The activation energy ΔG* for system crossing the transition state of ET reaction is calculated according to Marcus theory (Eq. (2)) 11,37,
where reorganization energy λ = EIT + ΔG° and thus, for the current ET systems with a free energy change of zero (ΔG° = 0), λ = EIT12,20,37. For 1a+ and 2+, the calculated ΔG* (Table 2) are substantially smaller than λ/4, the ΔG* introduced by Marcus38 for the nonadiabatic limit of ET reactions, but close to λ/4 for 1b+, 1c+, and 1d+. This means that 1a+ and the three phenylene bridged analogs (2+) should be treated adiabatically, whereas the three weakly coupled D–HB–A systems are essentially on the border of the adiabatic and nonadiabatic regimes. This is further justified by the magnitude of 2Hab, the separation between the adiabatic potential energy surfaces12. For 1a+ and 2+, 2Hab are 4–5 times larger than the thermal energy barrier, kBT (207 cm−1),9 while for 1b+, 1c+ and 1d+, 2Hab ∼ kBT. The electron hopping frequencies (νel) are calculated in the range 1013–1014 s−1, close to nuclear vibrational frequency (νn) of 1012–1013 s−1 37. For the HB systems, νn is reduced by the nonadiabaticity of PT. Therefore, for 1b+–d+, the ET kinetics can be described either adiabatically using the Arrhenius equation (Eq. (3)) with an pre-exponential factor (A) of 5 × 1012 s−1 37, or non-adiabatically, using the rate expression (Eq. (4)) developed by Levich39, while for 1a+ and 2+, adiabatic treatment is more appropriate.
For the three weakly coupled HB bridged dimers (1b+–d+), almost identical kadia and knonadia are obtained (Table 2), as expected, and the calculated rate constants in the range of 4–8 × 1010 s−1 show a general substituent-dependence of kET, that is, that electron-donating of the substituents (X) accelerates ET. For 1a+ and 2+, the two approaches give the rate constants of ∼1011 s−1, with kadia < knonadia by a factor of two (Table 2) and the rate constants are larger than those for 1b+–d+ by one order of magnitude or less.
For the D–HB–A systems, we are able to measure the ET rate constants by analysis of ν(NH) band broadening in the IR spectra, a method used for determination of ET rate constants in Ru3O–Ru3O MV systems24,40. This methodology has also been employed to evaluate thermally induced ET reactions in MV D–B–A systems with covalent bridges using NMR41 and EPR13,35,42 spectroscopy techniques. In the IR spectra of 1+, substantial changes of ν(NH) frequency and band shape are observed, in comparison with the spectra for 1. As shown in Fig. 2a–d, the MV species (1+) show partially coalesced bands, ν(NH)C, for the free N‒H bonds, while the bonded N‒H bonds exhibit multiple absorptions shifted towards lower energy, denoted as ν(NH)D. The manifolds of the N–H vibrations show directly the multiple nuclear degrees of freedom caused by the ET or PCET. For the dication complexes (1a2+–d2+) obtained by two-electron oxidation, the free N–H bonds present a broad and intense band at ∼3460 cm−1 (ν(NH)E) vs. 3517 cm−1 (ν(NH)A) for the neutral complexes 1a–d. It is worthwhile to note that the IR spectra of 1b2+ and 1c2+ exhibit a small shoulder at higher energy (∼3460 cm−1). This feature arises from disproportionation equilibrium of the complexes, indicating poor stabilization of the MV state in 1b+ and 1c+ due to weak EC43. Therefore, for the MV species (1+), the coalesced ν(NH)C band profile is reflective of the dynamics of the free N–H bonds caused by ET across the HB interface (Fig. 2a–d), and thus can be exploited to simulate the ET kinetics24,40. For each of the four adducts band simulations were performed using the published software Zoerbex44, as shown in Fig. 4, giving the observed rate constants (kobs) listed in Table 2. Remarkably, the kobs values are in excellent agreement with the calculated rate constants, except for 1a+. In the series, the largest rate constant, kobs = 6.0 × 1010 s−1, is obtained for 1a+ and the smallest rate, kobs = 2.0 × 1010 s−1 for 1b+, essentially following the same trend in the calculated rate constants. Recently, Kubiak’s group reported a rate constant of 3.8 × 1011 s−1 derived by broadening analysis of the ν(CO) band for an HB bridged Ru3O–Ru3O MV complex, which has shorter ET distance24. Comparison of ET rates for these two distinct systems verifies the magnitudes of kET in the Mo2–Mo2 system. It is noted that for 1a+ the deviation of kobs (6.0 × 1010 s−1) from kadia (2.4 × 1011 s−1) is relatively large. This disagreement can be rationalized by the involvement of two ET reaction channels, i.e. ET and PCET2. While thermal ET is controlled by the transition state through EC (Hab), nonadiabatic PCET is governed by vibronic coupling (S)42,45. For 1a+ with 2Hab ≫ kBT, it is inappropriate to calculate the rate constant using Hab for the PCET reaction in the nonadiabatic regime. From this point of view, the PCET rate constant, namely, kPCET, can be derived from Eq. (5) 2.
where kET = kadia and kPCET = knonadia, viewing the concerted PCET process as a single tunneling event that crosses a longer tunneling path over a higher energy barrier. Indeed, Eq. (5) gives kPCET = 8.0 × 1010 s−1 for 1a+, in good agreement with kobs of 6.0 × 1010 s−1 (Table 2). In other words, the nonadiabaticity caused by PT lowers the rate constant by less than one order of magnitude, which is in agreement with the consideration of nonadiabaticity in PCET theories7,8, The ET rate constants in these Mo2 MV systems are comparable with the data obtained for photoinduced ET across the acid–acid HB interface in metal porphyrin-based donor–acceptor systems, and thus verify the order of capability of the bridges in modulating EC and ET, that is, C–C π bonds > HBs > C–C σ bonds4. Moreover, this MV series provides an unprecedented example showing the transition of ET systems from the vibronically adiabatic regime (1a+) to the nonadiabatic regime (1b+, 1c+, and 1d+)45, modulated by EC. This study substantiates the semiclassical theory with excellent consistency between the calculated and experimental data, which is a long-term pursuit in the field of ET5. To our knowledge, this is the first example that compares optical and thermal ET pathways for systems on the adiabatic/nonadiabatic borderline45 and on the vibrational time scale (∼10−12 s), whereas earlier works have verified the related theories in the adiabatic13,42 and nonadiabatic41 limits on the time scales of NMR (∼10−6 s) and EPR (∼10−8 s).

Comparison of observed (left) and simulated (right) stretching frequencies for the free N–H bonds in the MV complexes 1a+ (a, X = N(CH3)2), 1b+ (b, X = CH(CH3)2), 1c+ (c, X = OCH3), and 1d+ (d, X = CH3). Note that in the simulated spectra (right), the degree of band coalesce varies as a function of the intermolecular electron transfer rate. For each of the complex systems, the rate constant (kobs) is determined by comparison of the spectrum with the simulated spectra (Supplementary Figures 43−46 and Supplementary Table 2)
Discussion
The agreement between the measured kobs and the calculated kET for these D–HB–A systems proves the accuracy of the optically determined rate constants for electron self-exchange in the phenylene bridged MV Mo2 dimers. It is found that the rate constants (kadia) and ET distances (Rab) of 1c+ and 2c+ fit well the linear relationship between ln(kET) and Rab for MV {[Mo2]–bridge–[Mo2]}+complexes with the same [Mo2] donor and acceptor but varying π conjugated bridges, in accordance with the decay law (Eq. (6)) in the frame of the McConnel superexchange mechanism 46,
where k0 is a kinetic prefactor and β the attenuation factor. The linear relationship of ln(kET) vs. Rab gives β = 1.25 Å−1 (Supplementary Figure 9). This result indicates that the HB bridge transports the electron from donor to acceptor equally well as π bridges do, in agreement with recent reports in photoinduced ET 47.
For the three D–B–A series with different bridges, DFT calculations on the simplified models, generated by replacing the Ar groups on the formamidinate ligand with a hydrogen atoms, show that the HOMO and HOMO-1 arise from the phase-out and phase-in combinations of the δ orbitals of the two Mo2 units, respectively (Supplementary Figure 10). The LUMO for 1 and 2 is the π* orbital of the bridging ligand, which permits metal(δ) to bridging ligand(π*) electronic transitions to occur29, while for 3, similar bridge-based LUMOs are not present. Interestingly for 1 and 2, not only are the two δ-based HOMOs very similar, but the LUMOs have the same symmetry and similar density distributions, showing the similarity of the amide–amide six-membered ring to the π-conjugated phenylene group48, despite the differences in the chemical constitution of the bridges. The HOMO–LUMO gap of 2.74 eV for 1 and 2.18 eV for 2 are in good agreement with the MLCT energies observed in the UV–Vis spectra, ca. 2.75 eV for 1c+ and 2.55 eV for 2c+. The δ–δ interactions in 1 and 2 are further manifested by the HOMO–HOMO-1 energy splitting of 0.09 eV (1) and 0.19 eV (2), appreciably larger than the 0.02 eV for 3. These results suggest that ET in the HB bridged Mo2 dimers (1+) may proceed via a superexchange mechanism, specifically for 1b+ and 1c+ for which PT is not involved, as occurring in the π conjugated analogs (2+)14. However, simple DFT calculations show only a stationary ground state that does not take into account the fluctuational behavior of the HB bridge47. The small HOMO−HOMO-1 splitting for the adducts, relative to those for 2, does not fully account for the efficient mediation that the HBs have on EC and ET 26.
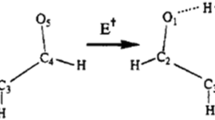
Significantly, this work illustrates that the electron self-exchange crossing HB interfaces can be mechanistically different in terms of PCET. For 1a+ and 1d +, the PCET pathway is directly evidenced by the ν(OH) stretches. In the existing PCET theories, while ET is generally described in the vibronically nonadiabatic regime, the PT process can be electronically nonadiabatic or adiabatic8. For the strongly coupled 1a+, kobs < kadia can be explained by slow PT that drags the ET process. This is the case of electronically nonadiabatic PT which takes place on a different time scale from that of ET8. For the strongly H-bonded 1d+, the weak EC (Hab = 130 cm−1) lowers down the adiabatic ET to the PT time scale, being a concerted PCET process7,8, with kobs ≈ knonadia. However, it appears that ET in 1b+ and 1c+ proceeds via a completely different ET mechanism, as evidenced by the IR spectral characteristics, specifically for the amide–amide HB moiety. While a large red-shift of the free N–H stretch is found for 1a+ (7 cm−1) and 1d+ (12 cm−1) upon one-electron oxidation (Fig. 2a, d), which signals the breakage of the bonded N–H bonds, the weak HB systems, 1b+ and 1c+, exhibit a small ν(NH) displacement (2 cm−1) of the free N–H bonds (Fig. 2b, c). For all the neutral complexes, the C = O stretching band of the amide group, ν(CO), appears at 1735 cm−1 (Supplementary Figure 11). Upon two-electron oxidation, this band is red-shifted to 1687 cm−1 for dication 1c2+, as expected for a C = O group with reduced bonding electron density. By contrast, for 1a2+, which has the strongest hydrogen bonding and strongest EC, the ν(CO) band disappears completely, as a result of conversion of C = O to C–OH, induced by proton transfer, in agreement with the observation of the ν(OH) band at 3613 cm−1 (Fig. 2a). For the two intermediate species 1b2+ and 1d2+, the C = O vibrational features are attenuated from 1b2+ to 1d2+ as the HB strength increases. All MV adducts display a weak, low energy (1687 cm−1) C = O band, as expected. For 1a+ and 1d+, simultaneous appearance of ν(OH) and ν(CO) stretches in the IR spectra visualize the PCET process, as described by Fig. 2e. On the other hand, together with the absence of ν(OH), it is evidenced that in 1b+ and 1c+, proton transfer does not occur during the course of ET. The amide–amide central moiety functions as a delocalized six-membered ring analogous to phenylene (Fig. 2f)48, conducting ET through the superexchange mechanism, as modeled by the DFT calculations (Supplementary Figure 10). In this case, the through-HB ET may be referred to as PUET. It is also worthwhile to note that for systems 1a and 1d, the O−H stretching bands appear in the spectra for the neutral, MV, and dicationic species (Fig. 2a, d), while all the oxidation states of 1b and 1c do not exhibit this band (Fig. 2b, c). This means that HB strength plays a critical role in the control of proton transfer, which affects the ET mechanism through interplay with the degree of EC.
To date, through-HB ET in both synthetic and naturally occurring systems has generally been treated as a PCET process1,49,50. This study demonstrates that ET across an HB interface may proceed via a proton-uncoupled pathway that can be more efficient than the PCET pathway because proton transfer dynamics can suppress the ET rate in PCET, as predicted by previous theories. Our results show that the PUET pathway predominates in weakly coupled systems that have weak HBs, but the mechanistic choice is subject to subtle variation of the structural and electronic factors of the system, as shown by the four HB adducts. These findings help to gain a deep and detailed understanding of ET reactions in which the redox partners are weakly electronic coupled through weak HBs, which is often the cases in biological systems involving enzymes, proteins, and DNA1. For instance, in the photoactivation of DNA photolyase, sequential ET among amino-acid residues is followed by proton transfer, and thus, charge compensating simultaneous proton transfer is not a prerequisite for intraprotein radical transfer51. Moreover, the unprecedented results in this study should direct additional theoretical attention, from a different perspective and in a broad sense, onto ET through HBs. While PCET theories are well developed to interpret ET dynamics and kinetics through the interplay between PT and ET, efficient EC and fast ET via the PUET channel should be further elucidated in terms of semiclassical formalism, quantum mechanics, and molecular dynamics.
Collectively, we have shown that ET across an HB interface may not only proceed via a PCET pathway, as predicted by existing theories, but also by a proton-uncoupled pathway (PUET) without proton translocation, which has not been addressed theoretically or observed experimentally before. Our results indicate that ET through HBs via both PCET and PUET pathways can be as equally efficient as ET through π bonds, and superior to σ bonds. A mechanistic switch from one pathway to the other can be realized by manipulating the strengths of the HB bridge and/or the donor–acceptor EC. The findings in this study have implications for long-distance, less exothermic ET across HBs in biological systems; thus, knowledge of the proton-uncoupled pathway may lead to better understanding of biochemical processes of charge and energy transport.
Methods
Synthesis
All manipulations were performed in a nitrogen-filled glove box or by using standard Schlenk-line techniques. All solvents were purified using a Vacuum Atmosphere (VAC) solvent purification system or freshly distilled over appropriate drying agents under nitrogen.
Preparation of 1a−d
The synthetic route is described by Supplementary Figure 12. A solution of acetamide (0.25 mmol) in 10 mL ethanol was transferred to a solution of Mo2(ArNCHNAr)3(O2CCH3) (0.2 mmol) in 10 mL tetrahydrofuran(THF). After stirring at room temperature for several minutes, the volatiles were removed under reduced pressure, producing an orange yellow solid. The solid product was washed with ethanol (3 × 10 mL) and collected by filtration. The dimeric structures of these compounds are confirmed by 1H NMR spectra in CDCl3 (Supplementary Figures 13, 15, 17 and 19) and the monomeric structures by 1H NMR spectroscopy in DMSO−d6 (Supplementary Figure 14, 16, 18 and 20).
Preparation of 2a−d
The synthetic route is described in Supplementary Figure 21. A solution of sodium ethoxide (0.20 mmol) in 10 mL ethanol was transferred to a solution of Mo2(ArNCHNAr)3(O2CCH3) (0.20 mmol) in 20 mL THF. After stirring at room temperature for 2 h, the mixture was transferred to the terephthalic acid (0.12 mmol). After stirring for 3 h the solvents were removed at room temperature; the residue was dissolved in 15 mL of DCM and filtered through a Celite-packed funnel. The solvent was evaporated under reduced pressure. The residue was washed with ethanol (3 × 20 mL) and then collected by filtration. The purity of the complexes was confirmed by 1H NMR spectroscopy in CDCl3 (Supplementary Figures 22−25).
Preparation of 3a−d
The synthetic route is described in Supplementary Figure 26. A solution of sodium ethoxide (0.25 mmol) in 10 mL ethanol was transferred to a solution of Mo2(ArNCHNAr)3(O2CCCH3) (0.2 mmol) in 10 mL THF. The solution was stirred at room temperature for about half an hour before the solvents were removed under vacuum. The residue was dissolved using 15 mL of DCM and filtered through a Celite-packed funnel. The DCM was removed, then 20 mL THF was added, and the solution was mixed with trans-1,4-cyclohexyldicarboxylic acid (0.12 mmol) in 10 mL ethanol. After stirring for 3 h at room temperature, the solvents were removed under vacuum. The residue was dissolved using 15 mL of DCM and filtered through a Celite-packed funnel before removing the solvent under reduced pressure. The residue was washed with ethanol (3 × 20 mL) and then collected by filtration. The purity of the complexes was confirmed by 1H NMR spectroscopy in CDCl3 (Supplementary Figures 27−30).
X-ray structural determination
For compounds 1a, 1c, and 1d, single crystals for X-ray structure determination were obtained by diffusion of hexane into the corresponding DCM solution and for 1b, by the solvent (DCM) evolution method. Single-crystal data for 1a·7CH2Cl2, 1b·7CH2Cl2, and 1d·CH2Cl2 were collected on a Rigaku XtaLAB Pro diffractometer (λ = 1.54178 Å) at 150 K and 1c were collected on an Agilent Xcalibur Nova diffractometer with Cu-Kα radiation (λ = 1.54178 Å) at 173 K. The empirical absorption corrections were applied using spherical harmonics, implemented in the SCALE3 ABSPACK scaling algorithm52. The structures were solved using direct methods, which yielded the positions of all non-hydrogen atoms. Hydrogen atoms were placed in calculated positions in the final structure refinement. Structure determination and refinement were carried out using the SHELXS-2014 and SHELXL-2014 programs, respectively53. The solvent molecules are disordered in multiple orientations, which were refined isotropically. All non-hydrogen atoms were refined with anisotropic displacement parameters (Supplementary Table 1).
Electrochemical studies
Electrochemical measurements on the neutral compounds were carried out in 0.1 M nBu4NPF6/DCM and DMF solutions. The CVs and differential pulse voltammograms were obtained using a CH Instruments model CHI660D electrochemical analyzer with Pt working and auxiliary electrodes, an Ag/AgCl reference electrode, and a scan rate of 100 mV/s. Under these conditions, the redox potential for ferrocene, E1/2(Fc+/0), is 0.52 V. The measured potentials for the complexes are referenced to E1/2(Fc+/0).
Spectroscopic measurements
UV–vis–NIR and mid-infrared spectra were measured on a Shimadzu UV-3600 UV–Vis–NIR and Thermo Electron Corporation Nicolet 6700 spectrophotometer, respectively. The UV–Vis–NIR spectra were measured using an IR quartz cell with a light path length of 2 mm. The IR measurements were carried out using a thin-layer CaF2 cell. The analyte concentration for the spectroscopic measurements is 1 × 10−3 mol/L in DCM and DMF.
Simulation of ET kinetics
Based on the Bloch equations formalisms54, published software Zoerbex44 was used to simulate the coalesced free N–H vibrational bands in the spectra to derive the ET rate constants for (1a+–d+). Since the band shapes of the spectra are not in accord with the simple Lorentzian function, the Voigt line shapes separated into Lorentzian and Gaussian components were used to simulate the IR peaks in each spectrum (Supplementary Figures 43–46)55. The input parameters for simulation include the full-width at half-maximum values for each contribution to the individual peaks as well as their center frequencies and relative populations. Each N–H vibrational band in (1a+–d+) was analyzed by curve fitting before the Voigt simulation.
DFT calculations
The ORCA 2.9.1 software packages56 were used for all DFT computations assuming an S = 0 spin state. DFT calculations were performed on the simplified models derived by replacing the p-anisyl groups in the DAniF ligands with hydrogen atoms. The geometry of the model complexes was optimized in the gas phase, employing the Becke−Perdew (BP86) functional57,58 and RI/J approximation59 without imposing any symmetry constraints. Geometry optimizations for the complexes were converged with the def2-SV(P) basis set60 and def2-SVP/J auxiliary basis set61,62 for C and H atoms, def2-TZVP(-f) basis set63 and def2-TZVP/J auxiliary basis set56 for N and O atoms. For Mo atoms, def2-TZVPP basis set57 and def2-TZVPP/J auxiliary basis set56 were used together with the ZORA approximation64. Tight optimization and tight self-consistent field convergence were employed along with a dense integration grid (ORCA Grid 5) for all geometry optimization calculations. Single-point calculations on optimized geometries were performed using the B3LYP functional65,66,67 and the COSMO methodology68 (using ε = 9.08 for DCM solvent). Isosurface plots of molecular orbitals were generated using the gOpenMol 3.00 program69,70 with isodensity values of 0.04.
Data availability
The X-ray crystallographic data (1a–d) reported in this study have been deposited at the Cambridge Crystallographic Data Centre (CCDC), under deposition number CCDC 1899032–1899035. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif. The data that support the findings of this study are available from the corresponding authors upon reasonable request.
Change history
04 June 2019
An amendment to this paper has been published and can be accessed via a link at the top of the paper.
02 May 2019
The original version of this Article contained errors in the symbols displayed in the eighteenth sentence of the third paragraph of the ‘Determination of Hab and kET data for the Mo2 dimers’ section of the Results, and the third sentence of the Discussion. This has been corrected in both the PDF and HTML versions of the Article.
References
Migliore, A., Polizzi, N. F., Therien, M. J. & Beratan, D. N. Biochemistry and theory of proton-coupled electron transfer. Chem. Rev. 114, 3381–3465 (2014).
Chang, C. J., Brown, J. D., Chang, K. M. C. Y., Baker, E. A. & Nocera, D. G. Electron Transfer in Chemistry (ed. Balzani, V.) Vol. 4, 409–461, Wiley-VCH, Weinheim, (2001).
Huynh, M. H. V. & Meyer, T. J. Proton-coupled electron transfer. Chem. Rev. 107, 5004–5064 (2007).
de Rege, P. J. F., Williams, S. A. & Therien, M. J. Direct evaluation of electronic coupling mediated by hydrogen bonds: implications for biological electron transfer. Science 269, 1409–1413 (1995).
Yang, J., Seneviratne, D., Arbatin, G., Andersson, A. M. & Curtis, J. C. Spectroscopic and electrochemical evidence for significant electronic coupling in mixed- valence hydrogen-bonded adducts of ruthenium cyano and ethylenediamine complexes. J. Am. Chem. Soc. 119, 5329–5336 (1997).
Newton, M. D. Medium reorganization and electronic coupling in long-range electron transfer. J. Electroanal. Chem. 438, 3–10 (1997).
Cukier, R. I. & Nocera, D. G. Proton-coupled electron transfer. Annu. Rev. Phys. Chem. 49, 337–369 (1998).
Hammes-Schiffer, S. & Stuchebrukhov, A. A. Theory of coupled electron and proton transfer reactions. Chem. Rev. 110, 6939–6960 (2010).
Newton, M. D. & Sutin, N. Electron transfer reactions in condensed phases. Ann. Rev. Phys. Chem. 35, 437–480 (1984).
Kaila, V. R. I., Verkhovsky, I. M. & Wikström, M. Proton-coupled electron transfer in cytochrome oxidase. Chem. Rev. 110, 7062–7081 (2010).
Marcus, R. A. & Sutin, N. Electron transfers in chemistry and biology. Biochim. Biophys. Acta 811, 265–322 (1985).
Hush, N. S. Homogeneous and heterogeneous optical and thermal electron transfer. Electrctrochim. Acta 13, 1005–1023 (1968).
Sun, D., Rosokha, S. V. & Kochi, J. K. Donor-acceptor (electronic) coupling in the precursor complex to organic electron transfer: intermolecular and intramolecular self-exchange between phenothiazine redox centers. J. Am. Chem. Soc. 126, 1388–1401 (2004).
Liu, C. Y., Xiao, X., Meng, M., Zhang, Y. & Han, M. J. Spectroscopic study of δ electron transfer between two covalently bonded dimolybdenum units via a conjugated bridge: adequate complex models to test the existing theories for electronic coupling. J. Phys. Chem. C 117, 19859–19865 (2013).
Wilkinson, L. A., McNeill, L., Meijer, A. J. H. M. & Patmore, N. J. Mixed valency in hydrogen bonded ‘dimers of dimers’. J. Am. Chem. Soc. 135, 1723–1726 (2013).
Sun, H., Steeb, J. & Kaifer, A. E. Efficient electronic communication between two identical ferrocene centers in a hydrogen-bonded dimer. J. Am. Chem. Soc. 128, 2820–2821 (2006).
Canzi, G. et al. On the observation of intervalence charge transfer bands in hydrogen-bonded mixed-valence complexes. J. Am. Chem. Soc. 136, 1710–1713 (2014).
Cotton, F. A. & Nocera, D. G. The whole story of the two-electron bond, with the δ bond as a paradigm. Acc. Chem. Res. 33, 483–490 (2000).
Yu, W. Y., Meng, M., Lei, H., He, X. D. & Liu, C. Y. Optical determination of electron transfer dynamics and kinetics for asymmetrical [Mo2]-ph-[Mo2] systems. J. Phys. Chem. C 120, 12411–12422 (2016).
Hush, N. S. Intervalence-transfer absorption. Part 2. Theoretical considerations and spectroscopic data. Prog. Inorg. Chem. 8, 391–444 (1967).
Cheng, T., Meng, M., Lei, H. & Liu, C. Y. Perturbation of the charge density between two bridged Mo2 centers: the remote substituent effects. Inorg. Chem. 53, 9213–9221 (2014).
Frey, P. A., Whitt, S. A. & Tobin, J. B. A low-barrier hydrogen bond in the catalytic triad of serine proteases. Science 264, 1927–1930 (1994).
Richardson, D. E. & Taube, H. Determination of E 2°- E 1° in multistep charge transfer by stationary-electrode pulse and cyclic voltammetry: application to binuclear ruthenium ammines. Inorg. Chem. 20, 1278–1285 (1981).
Porter, T. M., Heim, G. P. & Kubiak, C. P. Stable mixed-valent complexes formed by electron delocalization across hydrogen bonds of pyrimidinone-linked metal clusters. J. Am. Chem. Soc. 140, 12756–12759 (2018).
Wu, R. & Brutschy, B. Infrared depletion spectroscopy and structure of the 2-Aminopyridine dimer. J. Phys. Chem. A 108, 9715–9720 (2004).
Yamada, Y., Mikami, N. & Ebata, T. Relaxation dynamics of NH stretching vibrations of 2-aminopyridine and its dimer in a supersonic beam. Proc. Natl. Acad.Sci. USA 105, 12690–12695 (2008).
Saha, S. & Desiraju, G. R. Acid amide supramolecular synthon in cocrystals: from spectroscopic detection to property engineering. J. Am. Chem. Soc. 140, 6361–6373 (2018).
Nowak, M. J., Lapinski, L., Fulara, J., Les, A. & Adamowicz, L. Matrix isolation IR spectroscopy of tautomeric systems and its theoretical interpretation: 2-hydroxypyridine/2 (1H)-pyridinone. J. Phys. Chem. 96, 1562–1569 (1992).
Xiao, X. et al. Control of the charge distribution and modulation of the class II–III transition in weakly coupled Mo2–Mo2 systems. Inorg. Chem. 52, 12624–12633 (2013).
Chisholm, M. H., Pate, B. D., Wilson, P. J. & Zaleski, J. M. On the electron delocalization in the radical cations formed by oxidation of MM quadruple bonds linked by oxalate and perfluoroterephthalate bridges. Chem. Commun. 0, 1084–1085 (2002).
Xiao, X., Meng, M., Lei, H. & Liu, C. Y. Electronic coupling and electron transfer between two dimolybdenum units spaced by a biphenylene group. J. Phys. Chem. C 118, 8308–8315 (2014).
Zhu, G. Y., Meng, M., Tan, Y. N., Xiao, X. & Liu, C. Y. Electronic coupling between two covalently bonded dimolybdenum units bridged by a naphthalene group. Inorg. Chem. 55, 6315–6322 (2016).
Gao, H. et al. Electronic coupling and electron transfer between two Mo2 units through meta- and para-phenylene bridges. Chem. Eur. J. 25, 3930–3938(2019).
Kang, M. T., Meng, M., Tan, Y. N., Cheng, T. & Liu, C. Y. Tuning the electronic coupling and electron transfer in Mo2 donor–acceptor systems by variation of the bridge conformation. Chem. Eur. J. 22, 3115–3126 (2016).
Lancaster, K. et al. Intramolecular electron-transfer rates in mixed-valence triarylamines: measurement by variable-temperature ESR spectroscopy and comparison with optical data. J. Am. Chem. Soc. 131, 1717–1723 (2009).
Robin, M. B. & Day, P. Mixed valence chemistry-a study and classification. Adv. Inorg. Chem. Radiochem 9, 247–422 (1967).
Sutin, N. Theory of electron transfer reactions: insights and hindsights. Prog. Inorg. Chem. 30, 441–444 (1983).
Marcus, R. A. On the theory of oxidation-reduction reactions involving electron transfer. I. J. Chem. Phys. 24, 966–978 (1956).
Levich, V. G. Present state of the theory of oxidation-reduction in solution (bulk and electrode reactions). Adv. Electrochem. Electrochem. Eng. 4, 249–371 (1966).
Ito, T. et al. Effects of rapid intramolecular electron transfer on vibrational spectra. Science 277, 660–663 (1997).
Elliott, C. M., Derr, D. L., Matyushov, D. V. & Newton, M. D. Direct experimental comparison of the theories of thermal and optical electron-transfer: studies of a mixed-valence dinuclear iron polypyridyl complex. J. Am. Chem. Soc. 120, 11714–11726 (1998).
Nelsen, S. F., Ismagilov, R. F. & Trieber, D. A. II Adiabatic electron transfer: comparison of modified theory with experiment. Science 278, 846–849 (1997).
Creutz, C. Mixed valence complexes of d 5-d 6 metal centers. Prog. Inorg. Chem. 30, 1–73 (1983).
Zoerb, M. C. & Harris, C. B. A simulation program for dynamic infrared (IR) spectra. J. Chem. Educ. 90, 506–507 (2013).
Barbara, P. F., Meyer, T. J. & Ratner, M. A. Contemporary issues in electron transfer research. J. Phys. Chem. 100, 13148–13168 (1996).
McConnel, H. M. Intramolecular charge transfer in aromatic free radicals. J. Chem. Phys. 35, 508–515 (1961).
Warren, J. J. et al. Long-range proton-coupled electron-transfer reactions of bis (imidazole) iron tetraphenylporphyrins linked to benzoates. J. Phys. Chem. Lett. 4, 519–523 (2013).
Grabowski, S. J. Covalent character of hydrogen bonds enhanced by π-electron delocalization. Croat. Chem. Acta 82, 185–192 (2009).
Pannwitz, A. & Wenger, O. S. Recent advances in bioinspired proton-coupled electron transfer. Dalton Trans. https://doi.org/10.1039/C8DT04373F(2019).
Hammes-Schiffer., S. Proton-coupled electron transfer: moving together and charging forward. J. Am. Chem. Soc. 137, 8860–8871 (2015).
Aubert, C., Vos, M. H., Mathis, P., Eker, A. P. M. & Brettel, K. Intraprotein radical transfer during photoactivation of DNA photolyase. Nature 405, 589–590 (2000).
Crys Alis RED. Version 1.171.31.7 (Oxford Diffraction Ltd, Abington, UK, 2006).
Sheldrick, G. M. SHELXTL, Version 6.12 (Bruker Analytical X-ray Systems, Inc., Madison, WI, 2000).
Grevels, F.-W. & Jacke, J. Dynamic processes on the IR time scale: coalescence of CO stretching vibrational bands in [(ƞ4-Diene)Fe(CO)3] complexes. Angew. Chem. Int. Ed. Engl. 26, 885–887 (1987).
Nishiumi, T., Nomura, Y., Chimoto, Y., Higuchi, M. & Yamamoto, K. The class II/III transition electron transfer on an infrared vibrational time scale for N,N-Diphenyl-1,4-phenylenediamine structures. J. Phys. Chem. B 108, 7992–8000 (2004).
Neese, F. The ORCA program system. WIREs Comput. Mol. Sci. 2, 73–78 (2012).
Perdew, J. P. Density-functional approximation for the correlation energy of the inhomogeneous electron gas. Phys. Rev. B Condens. Matter Mater. Phys. 33, 8822–8824 (1986).
Becke, A. D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A At. Mol. Opt. Phys. 38, 3098–3100 (1988).
Neese, F. An improvement of the resolution of the identity approximation for the formation of the coulomb matrix. J. Comput. Chem. 24, 1740–1747 (2013).
Schäfer, A., Horn, H. & Ahlrichs, R. Fully optimized contracted Gaussian basis sets for atoms Li to Kr. J. Chem. Phys. 97, 2571–2577 (1992).
Eichkorn, K., Treutler, O., Öhm, H., Häser, M. & Ahlrichs, R. Auxiliary basis sets to approximate Coulomb potentials. Chem. Phys. Lett. 240, 283–290 (1995).
Eichkorn, K., Weigend, F., Treutler, O. & Ahlrichs, R. Auxiliary basis sets for main row atoms and transition metals and their use to approximate Coulomb potentials. Theor. Chem. Acc. 97, 119–124 (1997).
Weigend, F. & Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: design and assessment of accuracy. Phys. Chem. Chem. Phys. 7, 3297–3305 (2005).
Pantazis, D. A., Chen, X.-Y., Landis, C. R. & Neese, F. All-electron scalar relativistic basis sets for third-row transition metal atoms. J. Chem. Theory Comput. 4, 908–919 (2008).
Becke, A. D. A new mixing of Hartree–Fock and local density-functional theories. J. Chem. Phys. 98, 1372–1377 (1993).
Becke, A. D. Density-functional thermochemistry. III. The role of exact exchange.. J. Chem. Phys. 98, 5648–5652 (1993).
Lee, C., Yang, W. & Parr, R. G. Development of the Colic-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B Condens. Matter Mater. Phys. 37, 785–789 (1988).
Klamt, A. & Schüürmann, G. COSMO: a new approach to dielectric screening in solvents with explicit expressions for the screening energy and its gradient. J. Chem. Soc. Perkin Trans. 2. 5, 799–805 (1993).
Laaksonen, L. A graphics program for the analysis and display of molecular dynamics trajectories. J. Mol. Graph. 10, 33–34 (1992).
Bergman, D. L., Laaksonen, L. & Laaksonen, A. Visualization of solvation structures in liquid mixtures. J. Mol. Graph. Modell. 15, 301–306 (1997).
Acknowledgements
We acknowledge primary financial support from the National Natural Science Foundation of China (21371074), Natural Science Foundation of Guangdong Province (2018A030313894), Jinan University, and the Fundamental Research Funds for the Central Universities. We are grateful to Prof. Julia A. Weinstein (University of Sheffield) for comments and suggestions on the manuscript.
Author information
Authors and Affiliations
Contributions
C.Y.L. conceived and designed the experiments and worked on the manuscript. T.C. carried out the major experimental work and data analysis. M.M. completed the data collection and analysis of the X-ray crystal structures and S.M. performed DFT theoretical calculations. N.J.P. participated in discussion of the results and refinement of the manuscript. D.X.S., L.C., H.L.Z., S.F.Z., H.W.C., Y.Q., and Y.Y.W were involved in compound syntheses and electrochemical and spectroscopic measurements.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Journal peer review information: Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. Peer reviewer reports are available.
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Cheng, T., Shen, D.X., Meng, M. et al. Efficient electron transfer across hydrogen bond interfaces by proton-coupled and -uncoupled pathways. Nat Commun 10, 1531 (2019). https://doi.org/10.1038/s41467-019-09392-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-019-09392-7
- Springer Nature Limited
This article is cited by
-
From molecular to supramolecular electronics
Nature Reviews Materials (2021)
-
Crossover between the adiabatic and nonadiabatic electron transfer limits in the Landau-Zener model
Nature Communications (2021)
-
New insight into hydrogen bond-mediated electron transfer
Science China Chemistry (2019)