

Abstract
Facet engineering of oxide nanocrystals represents a powerful method for generating diverse properties for practical and innovative applications. Therefore, it is crucial to determine the nature of the exposed facets of oxides in order to develop the facet/morphology–property relationships and rationally design nanostructures with desired properties. Despite the extensive applications of electron microscopy for visualizing the facet structure of nanocrystals, the volumes sampled by such techniques are very small and may not be representative of the whole sample. Here, we develop a convenient 17O nuclear magnetic resonance (NMR) strategy to distinguish oxide nanocrystals exposing different facets. In combination with density functional theory calculations, we show that the oxygen ions on the exposed (001) and (101) facets of anatase titania nanocrystals have distinct 17O NMR shifts, which are sensitive to surface reconstruction and the nature of the steps on the surface. The results presented here open up methods for characterizing faceted nanocrystalline oxides and related materials.
Similar content being viewed by others

Introduction
Faceted oxide nanocrystals have attracted much research attention in a variety of fields, including catalysis1,2,3,4, photocatalysis5,6,7,8, solar hydrogen generation9, photoelectrochemical application10, gas sensoring11, and energy storage12, owing to their specific surface structures. Identification of the exposed facets is thus fundamental to the preparation and applications of oxide nanomaterials. Current characterization tools for studying the surface structure of nanocrystals are mostly based on electron microscopy13,14,15,16,17,18. At a resolution that the exposed facet can be determined, however, the field of view of microscopy techniques is often so small, or the particles may show considerable aggregation that it is possible that the region investigated is not representative of the whole sample19. Therefore, the development of complementary characterization methods that can give detailed structural information concerning the nature of the exposed facets of nanocrystals is urgently required.
Solid-state NMR spectroscopy is a powerful technique that has been widely used in studying the local environments of solids20. 17O NMR spectra, e.g., can give detailed structural and dynamic information of important functional oxygen-containing materials21,22,23,24,25,26, benefiting from the large 17O chemical shift range (>1000 ppm). However, few publications are available on the 17O NMR studies of nanosized oxides, in spite of their widespread applications, largely owing to the high cost of 17O and structure change during isotopic labeling. Recently, Wang et al.27 developed a surface-selective labeling method for oxide nanomaterials at low temperatures and revealed that the 17O species on the first few layers of ceria nanomaterials are associated with different 17O chemical shifts. However, direct experimental evidence is still missing concerning the relationship between the 17O chemical shifts and the nature of the exposed facets.
Here, we demonstrate a new approach based on NMR and surface-selective 17O labeling to determine the structures of the exposed facets on the technologically important anatase titania nanocrystals6, 9, 10, 28,29,30. With the help of density functional theory (DFT) calculations, oxygen species on different facets can be distinguished by their NMR shifts. The nature of surface steps and reconstructions of these surfaces, particularly on reaction with water, are also revealed.
Results
Morphology of anatase TiO2 nanosheets and nano-octahedra
Two types of anatase TiO2 nanocrystals with different tailored facets were examined, i.e., anatase TiO2 nanosheets with dominant exposed (001) facets (NS001-TiO2), and nano-octahedra preferentially exposing (101) facets (NO101-TiO2)31. Their crystal forms were confirmed with X-ray diffraction (XRD) (Supplementary Fig. 1). High-resolution transmission electron microscopy (HRTEM) results show that NS001-TiO2 (Supplementary Fig. 2) are nanosheets with a thickness of 6–7 nm, while NS101-TiO2 (Supplementary Fig. 3) are nano-octahedra with an average size of 14 nm. According to the statistical analysis of the data (Supplementary Figs. 2, 3), an average of 77% of the exposed surfaces of NS001-TiO2 are (001) facets, while 96% of the exposed surfaces of NO101-TiO2 are (101) (see Supplementary Table 1, Supplementary Fig. 4 and additional discussion in Supplementary Note 1). X-ray photoelectron spectroscopy (XPS) spectra (Supplementary Fig. 5) suggest that there is no evidence for the existence of F− or Cl− on the surface of either sample, while the concentrations of carbon (C) or nitrogen (N) impurities in both samples are also very small according to the elemental analysis (Supplementary Table 2).
17O NMR spectra of the surface-selectively labeled samples
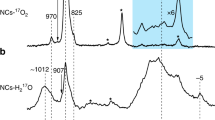
After exposing to 17O-water for surface-selective labeling27, the anatase nanocrystals were characterized with 17O magic angle spinning (MAS) NMR spectroscopy and were compared to a non-faceted anatase TiO2 sample with a smaller surface area (denoted as NF1-TiO2, see Supplementary Fig. 6) labeled nonselectively with 17O2 at 500 °C, as shown in Fig. 1. Bulk anatase TiO2 consists of TiO6 octahedra that share 4 O–O edges (Supplementary Fig. 7a and Supplementary Table 3) and all of the O ions are 3-coordinated (OTi3, denoted as O3c) with an average Ti–O bond length of 0.195 nm32. Therefore, 17O NMR spectrum of the anatase TiO2 sample enriched with 17O2 at high temperature show a single sharp peak at 558 ppm (Fig. 1), corresponding to O3c species in the “bulk” part, consistent with previous reports33. It is clear that the 17O NMR spectra of surface-labeled NS001-TiO2 and NO101-TiO2 differ significantly and are also distinct from the spectrum of the nonselectively labeled anatase TiO2 (Fig. 1), reflecting their different local structures (see Supplementary Fig. 7 and Supplementary Table 3, and further discussions below), suggesting that 17O NMR spectroscopy can be a new method to distinguish faceted oxide nanocrystals.

17O NMR spectra of faceted anatase titania nanocrystals compared to the non-faceted sample. Anatase TiO2 nanosheets with dominant exposed (001) facets (NS001-TiO2), and nano-octahedra preferentially exposing (101) facets (NO101-TiO2) were surface-selectively 17O-labeled and vacuum dried for 2 and 12 h, respectively. The other sample, NF1-TiO2, was nonselectively 17O-labeled. All data were obtained at 9.4 T under a MAS frequency of 14 kHz. A rotor synchronized Hahn-echo sequence (π/6−τ−π/3−τ—acquisition) and optimized recycle delays (0.5 s for NS001-TiO2 and NO101-TiO2, and 50 s for NF1-TiO2, see Supplementary Fig. 8), with 1H decoupling, were used to obtain the NMR data. 120,000, 110,000, and 1200 scans were collected for NS001-TiO2, NO101-TiO2, and NF1-TiO2, respectively. The spectra are normalized according to the sample mass and the number of scans (Supplementary Table 4). Asterisks denote sidebands. The dependence of the 17O MAS NMR spectra of the two faceted samples on the vacuum-drying time is shown in Supplementary Figs. 9 and 10, and discussed in the Supplementary Notes 2 and 3. Comparison of the 17O NMR spectra of the two faceted nanocrystalline samples to a surface-selectively labeled, non-faceted anatase TiO2 nanoparticle sample with comparable surface area (denoted as NF2-TiO2 and the TEM image of the sample is shown in Supplementary Fig. 11) can be found in Supplementary Fig. 12 and Supplementary Note 4
The signals observed in the 17O NMR spectra of NS001-TiO2 and NO101-TiO2 can be categorized into three types. The resonances at 480–570 ppm should arise from O3c species on the surface of titania, since their chemical shifts are close to that of bulk O3c. The peaks at higher frequencies (600–750 ppm) can be assigned to O species with lower coordination numbers (e.g., O2c) on the surface of titania nanostructure27. The broad signals at much lower frequencies (−150 to 300 ppm) can be attributed to hydroxyl groups in surface hydroxyls and/or water environments27, 34, 35. The peak centered at 150 ppm in the spectrum of NS001-TiO2 can also be observed in 1H→17O cross-polarization (CP) MAS NMR spectra (Supplementary Fig. 13 and Supplementary Note 5), confirming that this signal arises from oxygen ions in close proximity to proton. Such signal is very weak in the spectrum of NO101-TiO2 while an additional peak can be found centered at −75 ppm (Fig. 1 and Supplementary Figs. 10, 14). According to the shift, this lower-frequency resonance is assigned to adsorbed water molecules (see Supplementary Fig. 10 and Supplementary Note 3). The observation of surface OH species on the (001) facet while only water on the (101) surface, on the vacuum-dried samples, agrees with the previous DFT calculations that water prefers to dissociate on anatase TiO2 (001) facet to form surface OHs36, while it tends to adsorb molecularly on (101) surface37, 38.
Surface reconstruction of anatase TiO2(001)
In order to help the spectral assignment, DFT calculations were performed on anatase titania structures with different exposed facets. Since water molecules prefer to dissociate on the high-energy (001) facets36, and surface reconstructions are likely to occur on (001)39, four possible surface models were constructed for NS001-TiO2, including the un-reconstructed clean TiO2(001) (CL), hydrated TiO2(001) at a water coverage of 1/2 ML (dissociative adsorption, DA), 1 × 4-reconstructed clean TiO2(001) (RC-CL), and hydrated 1 × 4-reconstructed TiO2(001) (RC-DA) (see Fig. 2 and Supplementary Figs. 15–18 for details). 1/2 ML means that every two surface Ti5c take one water molecule, and it also corresponds to a fully hydrated surface state36. The calculated isotropic chemical shifts of each oxygen sites (δ iso), quadrupolar coupling constant (C Q), asymmetry parameter (η), and center of gravity of the NMR signals (δ CG) are given in Supplementary Tables 5–8. In all the models investigated, the calculated chemical shifts (δ CG) of oxygen ions in the “bulk” part (middle layers) of the anatase structures are close to 558 ppm, which is the observed chemical shift of O3c in the nonselectively labeled anatase TiO2. The chemical shifts of the oxygen species in the first few layers, however, deviate noticeably from the “bulk” values and depend on the specific local structure.

Experimental and simulated 17O NMR spectra of NS001-TiO2. NS001-TiO2 (Exp) was surface selectively 17O-labeled and vacuum-dried for 2 h. The simulated spectra are based on DFT calculations on different structures, i.e., un-reconstructed clean anatase TiO2(001) (CL), hydrated anatase TiO2(001) at a water coverage of 1/2 molecular layer (DA), 1 × 4-reconstructed clean anatase TiO2(001) (RC-CL), and hydrated 1 × 4-reconstructed anatase TiO2(001) (RC-DA). Insets are models of corresponding surface structures, where gray, white, blue, and red spheres represent Ti, H, O in surface hydroxyl groups, and other O species, respectively. Full view of all four structural models, isotropic chemical shifts of each oxygen sites and their quadrupolar parameters are presented in Supplementary Figs. 15–18 and Supplementary Tables 5–8. Asterisks denote sidebands. Ampersand denotes sideband overlapping with the OH signal. The arrows show the calculated isotropic chemical shift values of surface oxygen species. The centers of masses of the resonances owing to OH appear at lower frequencies due to the significant second-order quadrupolar induced shifts
The calculated results were used to simulate the 17O NMR spectra at different external magnetic fields (Fig. 2 and Supplementary Fig. 19) by considering the surface oxygen species only, whose isotropic chemical shifts have been marked in the structural models in Fig. 2. The simulated signals arising from the OH species generated in the DA and RC-DA structures give a fair match with the experimental data (450–0 ppm), further supporting that water dissociates on the (001) facets. Furthermore, they also allow us to assign a weak peak centered at approximately 400 ppm that overlaps with the sidebands from the surface oxygen sites to another OH environment. The calculation results also show that the majority species that give rise to the signals at 600–760 ppm in the experimental data are actually the O2c environments, rather than the O3c site, and that these species can only be ascribed to reconstructed surfaces (i.e., contributions from the RC-CL and/or RC-DA structures). Therefore, these results provide compelling evidence that structure reconstruction does indeed occurs on the (001) surface. On the basis of the 1H NMR results (Supplementary Fig. 20 and Supplementary Table 9), the water coverage on this sample is 0.3 ML, indicating that a reconstructed surface is energetically favored at this state36, and both RC-CL and RC-DA surface conditions should exist, due to the insufficient water coverage. Therefore, it can be concluded that, at this specific water coverage (0.3 ML), surface reconstruction occurs on (001) surface of anatase titania, and water dissociates on this surface.
Step edges of anatase TiO2(101)
For NO101-TiO2, three defect-free structure models, including clean anatase TiO2(101) (CL), hydrated anatase TiO2(101) under a water (molecular adsorption) coverage of 1/2 ML (MA), and hydrated anatase TiO2(101) with dissociatively adsorbed water under the coverage of 1/2 ML (DA, which is energetically less favorable37, 38), were constructed first to calculate the NMR parameters (Supplementary Figs. 21–23 and Supplementary Tables 10–12). However, the simulated spectra do not match the experimental data (for surface O2c sites in particular) (Supplementary Fig. 24). Surface defects, however, often occur on the (101) facets according to scanning tunneling microscopy investigations40, 41 as well as first-principles calculations41. Particularly, “step edges”, associated with higher reactivity41, are considered as the most common defects on this surface. Gong et al. have proposed several types of step-edge defects42 with monoatomic height along trapezoidal or triangular islands on (101) surface40, 43. The so-called type-D steps occur along two nonparallel sides of the trapezoidal islands (or two sides of the triangular ones), and they are also the most prevalent ones among all the steps. Accordingly, in the current work, an anatase TiO2(134) vicinal surface with such type-D steps and (101) planes (see Fig. 3a) was constructed for the chemical shift calculations.

The structure model and 17O NMR spectra of NO101-TiO2. a The structure model of the TiO2(134) vicinal surface for DFT calculations, which contains type-D steps and (101) planes. b The experimental 17O spin-echo NMR spectrum of the fully dried surface-selectively 17O-labeled NO101-TiO2 (black line) and the simulated spectra (colored lines and peaks) by using parameters obtained from DFT calculations. Water molecules are adsorbed in two orientations (OA and OB). The contributions of both adsorption orientations are also shown in b (dark yellow line for OB and blue line for OA). Other colored peaks denote the individual components of OA, which correspond to the oxygen atoms labeled with the same numbers in the structural model in a. The parameters adopted in the simulation are listed in Supplementary Table 16. Full views of the models with the two adsorption orientations are presented in Supplementary Figs. 25–26. Asterisks denote spinning sidebands, while ampersands denote sidebands that overlap with the signal of the adsorbed water
According to our calculations, water are molecularly adsorbed at the Ti5c sites (TiO5) of type-D step-edges and have two different orientations (denoted as OA and OB) with similar adsorption strength, distinguished by the lengths of the hydrogen bonds formed with the adjacent oxygen ions at the edge (Supplementary Figs. 25–26 and Supplementary Tables 13–14). In both adsorption modes, water has higher adsorption energies than that found at flat (101) surface (Supplementary Table 15). Since the adsorbed water molecules in two orientations have similar adsorption energies, each orientation is weighted the same and only 14 different surface/subsurface oxygen species are considered in the spectral simulation. The calculated structures, NMR parameters, and simulated spectra, along with the experimental data, are shown in Fig. 3, Supplementary Fig. 27, and Supplementary Table 16. For clarity, the simulated spectrum of the 14 oxygen species in OA is also presented as colored and shaded peaks in Fig. 3b.
The simulated spectra agree remarkably well with the experimental data at different external magnetic fields (Fig. 3b and Supplementary Fig. 27b), except for the center of gravity of the NMR signal for the adsorbed water species (Fig. 3b, peak 1). The experimental line width of this peak is smaller than the calculated one, which can be attributed to the motion of the adsorbed water molecules (see Supplementary Fig. 28 and Supplementary Note 6). Other signals from surface sites probably originate from the dissociation of H2 17O at oxygen vacancies generated in the vacuum-drying pretreatment at 100 °C (see Supplementary Fig. 29 and Supplementary Note 7) and possible subsequent migration of oxygen ions within the structure of TiO2, since water molecules are not expected to dissociate on type-D step edges37, 44. The major resonance at 730 ppm (peak 3) arises from O2c species at the step edges (Fig. 3). In comparison, peak 2, corresponding to O2c species at the middle of (101) plane, has much smaller intensity. Considering the fact that there is only a small fraction of oxygen ions at step edges (4 ± 1.5%)42, the much stronger intensity of peak 3 implies that O2c at the step edge has higher activity in the initial labeling process than the species on (101) plane. The other relatively strong peak owing to O2c ions occurs at 640 ppm (peak 5). Such oxygen species is at flat terraces below the adjacent step edge and is attached with the adsorbed water through hydrogen bond. The signals at 480–560 ppm can be assigned to surface and subsurface O3c species. The much stronger intensity of the O2c species compared to the O3c ones confirms that the 17O-enrichment method adopted in this work does achieve an effective surface-selective labeling.
Discussion
17O solid-state NMR spectroscopy, in combination with DFT calculations, can be used to distinguish two anatase TiO2 nanocrystals with different exposed facets and explore the details of their unique surface local environments. The 17O NMR spectra provide definitive evidence that surface reconstruction occurs when (001) faceted anatase TiO2 nanosheets adsorb a small amount of water, while “step edges” are the main defects present on the anatase TiO2(101) surface. The results indicate that 17O solid-state NMR spectroscopy is a sensitive method to probe the local environments of the exposed facets of oxide nanocrystals, the structures of these facets playing a vital role in determining their properties. Further studies based on this approach can be readily envisaged to study possible changes that may occur on the faceted oxide nanocrystals in catalytic processes and other related applications.
Methods
Sample preparation
The anatase TiO2 nanosheets, mainly dominated by exposed (001) facets, i.e. NS001-TiO2, were prepared according to Han’s work45. (101) facets dominated anatase nano-octahedra (NO101-TiO2), and non-faceted anatase TiO2 nanoparticles (NF2-TiO2) were prepared hydrothermally according to Liu’s work31. The obtained materials were washed thoroughly with NaOH aqueous solution and water to remove F− or Cl− on the surface, which were introduced in the preparation. Experiment details are given in the Supplementary Methods. Another non-faceted anatase TiO2 sample with smaller surface area, NF1-TiO2, was purchased from Sigma-Aldrich Corporation, and used as received.
Characterization
The powder XRD analysis was carried out on a Philips X’Pro X-ray diffractometer using Cu Kα irradiation (λ = 1.54184 Å) operated at 40 kV and 40 mA at 25 °C. High-resolution TEM images were obtained on an FEI Titan 80/300 S/TEM with an acceleration voltage of 200 kV. Electron paramagnetic resonance (EPR) spectra were recorded on the samples with the same mass (50 mg) by a Bruker EMX-10/12 spectrometer at room temperature. The Brunauer–Emmett–Teller specific surface areas of the samples were measured by nitrogen adsorption at 77 K using a Micromeritics tristar ASAP 2020 instrument. The contents of C and N impurities of the samples were analyzed using a Heraeus CHN-0-Rapid analyzer. XPS spectra of both faceted samples were obtained on an Ulvac-PHI PHI 5000 VersaProbe instrument.
17O enrichment
Faceted NS001-TiO2, NO101-TiO2, and non-faceted NF2-TiO2 nanocrystalline samples were surface-selectively 17O-labeled through a vacuum line using 90% 17O-enriched H2O (Cambridge Isotope Laboratories). The sample (typically 300 mg) was first activated in a glass tube by vacuum drying at 100 °C for 1.5 h. After the sample was cooled down to room temperature, it was exposed to the saturated vapor of 17O-enriched H2O for 10 min for adequate adsorption. Then the sample was sealed in the glass tube, heated to 40 °C and kept at this temperature for 5 h to achieve an optimized 17O labeling of the surface oxygen species. The other non-faceted anatase TiO2 sample NF1-TiO2, with a smaller surface area, was 17O-labeled nonselectively by calcining in 17O2 (70% 17O, Cambridge Isotope Laboratories) within a sealed glass tube at 500 °C for 12 h.
Solid-state NMR measurement
17O MAS NMR spectra were measured on 9.4 and 14.1 T Bruker Avance III spectrometers using 4.0 mm MAS probes doubly tuned to 17O at 54.2 and 81.3 MHz, and 1H at 400.0 and 600.0 MHz, respectively. All samples were packed into rotors in a N2 glove box. 17O chemical shift is referenced to H2O at 0.0 ppm.
DFT calculations
Spin-polarized DFT calculations were performed with the Perdew–Burke–Ernzerhof functional46 by using the Vienna Ab initio Simulation Package (VASP)47. The 17O chemical shifts were calculated by using the linear response method. We used the project-augmented wave method48 to describe the core-valence electron interactions in structure optimization, chemical shift, and electric field gradients (EFGs) calculations at a kinetic energy cutoff of 500 eV with Ti (3s, 3p, 3d, 4s), O (2s, 2p), and H (1s) electrons being treated as valence electrons. All of the atoms were allowed to relax during structure optimization with a force stopping-criterion of 0.02 eV/Å on each relaxed ion. During electronic minimization, we used an extremely high stopping criterion of 10−8 eV for all the calculations27. With a 3 × 3 × 3 k-point mesh, we obtained optimized lattice parameters of a = 3.80 Å and c = 9.51 Å for bulk anatase TiO2, which is very close to the experimental values (a = 3.78 Å and c = 9.50 Å)49. It should be noted that the on-site Coulomb interaction of localized d electrons was also considered by using the DFT+U approach with an optimum Hubbard U value of 4.0 eV50, and lattice parameters of a = 3.86 Å and c = 9.53 Å was obtained. This indicates pristine DFT method can give reliable structural information. Since correct structural information is crucial to chemical shift calculations, we then used the pristine DFT method to do all the calculations.
The anatase TiO2 structures were modeled by surface slabs that are thick enough to maintain trivial fluctuations of chemical shift values in their middle layers (see Supplementary Figs. 15–18 and 21–23 for details). For un-reconstructed TiO2(001) surface, 1 × 4-reconstructed TiO2(001) surface39, TiO2(101) surface, and TiO2(134) vicinal surface consisting of type-D steps and (101) planes42, we used a 1 × 2, 2 × 4, 1 × 2, and 1 × 1 surface cell, respectively, with a corresponding 4 × 2 × 1, 2 × 1 × 1, 2 × 2 × 1, and 2 × 3 × 1 k-point mesh, respectively, for the Brillouin zone integration. All the slabs also contain a large vacuum gap (~12 Å for un-reconstructed anatase TiO2(001), 1 × 4-reconstructed anatase TiO2(001), anatase TiO2(101) surfaces, and ~13 Å for anatase TiO2(134) vicinal surfaces) to remove the slab–slab interactions.
The isotropic chemical shift (δ iso) can be computed as δ iso = δ cal + δ ref 27, where δ cal is the chemical shift obtained in VASP, δ ref is the reference chemical shift. Considering the fact that bulk oxygens have more regular arrangements than those near the surfaces, all the δ ref for each model (except TiO2(134) vicinal surface) were determined by aligning the average δ cal of middle four layers to the experimental δ iso of bulk O3c (561 ppm, Supplementary Figs. 15–18, 21–23). For the anatase TiO2(134) vicinal surface consisting of type-D steps and (101) planes, shown in Supplementary Figs. 25 and 26, the average δ iso of atom 35–39, which are in the middle layer, is set as δ iso of bulk O3c. All determined δ ref is in the range of 50–60 ppm (given in caption of Supplementary Figs. 15–18, 21–23, 25, 26), which is close to the reported value of 52 ppm for CeO2 27.
To calculate the quadrupole coupling constant (C Q) and asymmetry parameter (η), we used the following equations:
where h is the Planck constant, e is the absolute value of the electron charge, and V ii (ii = XX, YY, or ZZ) are the eigenvalues of the EFG tensor with |V ZZ| > |V YY| > |V XX|. We used the experimental quadrupole moment (Q) of −0.02558 barns51 for 17O.
The adsorption energy of H2O (E ads) was calculated as follows:
where \(E_{{{\rm{H}}_2}{\rm{O}}}\), E sub, and \(E_{{{{\rm{H}}_2}{\rm{O}}}/{\rm{sub}}}\) are the DFT total energies of the gas phase H2O, the TiO2 substrate, and the adsorption complex, respectively.
17O NMR spectra simulation
Wsolids package developed by Dr. K. Eichele was used to simulate the 17O NMR spectra using the NMR parameters obtained with DFT calculations, as shown in Figs. 2, 3b and Supplementary Figs. 19b, 24, 27b, 28.
For the simulated spectra from the models of (001) facet (Fig. 2 and Supplementary Fig. 19b), only surface oxygen sites were considered, whose isotropic chemical shifts have been marked in the structural models in Fig. 2. The O2c and O3c sites have been given the same weight of peak area in the calculated spectra. Twice of the weight has been given to the hydroxyl groups centered around 420 ppm, and four times of the weight has been given to the hydroxyl groups centered around 150 ppm, for the sake of presentation. For simulating the NMR spectra of the defect-free (101) facet (as shown in Supplementary Fig. 24), a similar approach was used. Surface sites, i.e., sites 1–3 in Supplementary Fig. 21 and sites 1–7 in Supplementary Figs. 22 and 23, respectively, were considered. Twice the weight of the peak areas have been given to the signals of hydroxyl groups and adsorbed water, in comparison to those of the surface O2c and O3c sites. For simulating the spectra of (101) facet with type-D steps (Fig. 3b and Supplementary Fig. 27b), NMR parameters of surface and subsurface oxygen sites (1–14) in Supplementary Tables 13 and 14 were adopted, only with their percentage adjustable to achieve the best fitting. Furthermore, in Supplementary Fig. 28, C Qs of the adsorbed water in both adsorption orientations were also allowed to change in the simulation, in order to examine the influence of the motion of the adsorbed water on its NMR signal.
Data availability
All relevant data are available from the authors.
References
Xie, X. W., Li, Y., Liu, Z. Q., Haruta, M. & Shen, W. J. Low-temperature oxidation of CO catalysed by Co3O4 nanorods. Nature 458, 746–749 (2009).
Zhou, K. & Li, Y. D. Catalysis based on nanocrystals with well-defined facets. Angew. Chem. Int. Ed. 51, 602–613 (2012).
Yang, P. Surface chemistry: crystal cuts on the nanoscale. Nature 482, 41–42 (2012).
Li, Y. & Shen, W. Morphology-dependent nanocatalysts: rod-shaped oxides. Chem. Soc. Rev. 43, 1543–1574 (2014).
Liu, G., Yu, J. C., Lu, G. Q. & Cheng, H.-M. Crystal facet engineering of semiconductor photocatalysts: motivations, advances and unique properties. Chem. Commun. 47, 6763–6783 (2011).
Gordon, T. R. et al. Nonaqueous synthesis of TiO2 nanocrystals using TiF4 to engineer morphology, oxygen vacancy concentration, and photocatalytic activity. J. Am. Chem. Soc. 134, 6751-6761 (2012).
Huang, W.-C., Lyu, L.-M., Yang, Y.-C. & Huang, M. H. Synthesis of Cu2O nanocrystals from cubic to rhombic dodecahedral structures and their comparative photocatalytic activity. J. Am. Chem. Soc. 134, 1261–1267 (2012).
Zhu, J. et al. Direct imaging of highly anisotropic photogenerated charge separations on different facets of a single BiVO4 photocatalyst. Angew. Chem. Int. Ed. 54, 9111–9114 (2015).
Yuan, Y. J. et al. Constructing anatase TiO2 nanosheets with exposed (001) facets/layered MoS2 two-dimensional nanojunctions for enhanced solar hydrogen generation. ACS Catal. 6, 532–541 (2016).
Li, C. et al. Facet-dependent photoelectrochemical performance of TiO2 nanostructures: an experimental and computational study. J. Am. Chem. Soc. 137, 1520–1529 (2015).
Wang, H. K. et al. Engineering of facets, band structure, and gas-sensing properties of hierarchical Sn2+-doped SnO2 nanostructures. Adv. Funct. Mater. 23, 4847–4853 (2013).
Cheng, X. L., Jiang, J. S., Jiang, D. M. & Zhao, Z. J. Synthesis of rhombic dodecahedral Fe3O4 nanocrystals with exposed high-energy {110} facets and their peroxidase-like activity and lithium storage properties. J. Phys. Chem. C 118, 12588–12598 (2014).
Yang, H. G. et al. Anatase TiO2 single crystals with a large percentage of reactive facets. Nature 453, 638–641 (2008).
Pan, J., Liu, G., Lu, G. M. & Cheng, H. M. On the true photoreactivity order of {001}, {010}, and {101} facets of anatase TiO2 crystals. Angew. Chem. Int. Ed. 50, 2133–2137 (2011).
Chen, C. D. et al. Synthesis of [111]- and {010}-faceted anatase TiO2 nanocrystals from tri-titanate nanosheets and their photocatalytic and DSSC performances. Nanoscale 7, 7980–7991 (2015).
Mao, J. et al. Pt-loading reverses the photocatalytic activity order of anatase TiO2 {001} and {010} facets for photoreduction of CO2 to CH4. Appl. Catal. B Environ. 144, 855–862 (2014).
Lauritsen, J. V. & Reichling, M. Atomic resolution non-contact atomic force microscopy of clean metal oxide surfaces. J. Phys. Condens. Matter 22, 263001 (2010).
Boneschanscher, M. P. et al. Electron tomography resolves a novel crystal structure in a binary nanocrystal superlattice. Nano Lett. 13, 1312–1316 (2013).
Browning, N. D. et al. in Modeling Nanoscale Imaging in Electron Microscopy (eds. Vogt, T., Dahmen, W., & Binev, P.) 11–40 (Springer, 2012).
Peng, Y.-K. et al. Trimethylphosphine-assisted surface fingerprinting of metal oxide nanoparticle by P-31 solid-state NMR: a zinc oxide case study. J. Am. Chem. Soc. 138, 2225–2234 (2016).
Kim, N. & Grey, C. P. Probing oxygen motion in disordered anionic conductors with O-17 and V-51 MAS NMR spectroscopy. Science 297, 1317–1320 (2002).
Peng, L. M., Liu, Y., Kim, N. J., Readman, J. E. & Grey, C. P. Detection of Bronsted acid sites in zeolite HY with high-field 17O MAS NMR techniques. Nat. Mater. 4, 216–219 (2005).
Halat, D. M. et al. Probing oxide-ion mobility in the mixed ionic-electronic conductor La2NiO4+δ by solid-state O-17 MAS NMR spectroscopy. J. Am. Chem. Soc. 138, 11958–11969 (2016).
Seymour, I. D. et al. Characterizing oxygen local environments in paramagnetic battery materials via O-17 NMR and DFT calculations. J. Am. Chem. Soc. 138, 9405–9408 (2016).
Griffin, J. M. et al. Ionothermal O-17 enrichment of oxides using microlitre quantities of labelled water. Chem. Sci. 3, 2293–2300 (2012).
Merle, N. et al. O-17 NMR gives unprecedented insights into the structure of supported catalysts and their interaction with the silica carrier. J. Am. Chem. Soc. 134, 9263–9275 (2012).
Wang, M. et al. Identification of different oxygen species in oxide nanostructures with 17O solid-state NMR spectroscopy. Sci. Adv. 1, e1400133 (2015).
Liu, G. et al. Titanium dioxide crystals with tailored facets. Chem. Rev. 114, 9559–9612 (2014).
Hagfeldt, A., Boschloo, G., Sun, L. C., Kloo, L. & Pettersson, H. Dye-sensitized solar cells. Chem. Rev. 110, 6595–6663 (2010).
Wu, B., Guo, C., Zheng, N., Xie, Z. & Stucky, G. D. Nonaqueous production of nanostructured anatase with high-energy facets. J. Am. Chem. Soc. 130, 17563–17567 (2008).
Liu, L. C. et al. Anion-assisted synthesis of TiO2 nanocrystals with tunable crystal forms and crystal facets and their photocatalytic redox activities in organic reactions. J. Phys. Chem. C 117, 18578–18587 (2013).
Sun, X. M. et al. Solid-state NMR investigation of the 16/17O isotope exchange of oxygen species in pure-anatase and mixed-phase TiO2. Chem. Phys. Lett. 594, 34–40 (2014).
Bastow, T. J., Moodie, A. F., Smith, M. E. & Whitfield, H. J. Characterisation of titania gels by O-17 nuclear magnetic resonance and electron diffraction. J. Mater. Chem. 3, 697–702 (1993).
Zhao, L. et al. Investigating local structure in layered double hydroxides with 17O NMR spectroscopy. Adv. Funct. Mater. 24, 1696–1702 (2014).
Blanchard, J., Bonhomme, C., Maquet, J. & Sanchez, C. Characterisation of sol-gel derived titanium oxopolymers: first evidence of Ti-OH groups through 1H-17O CP NMR experiments. J. Mater. Chem. 8, 985–989 (1998).
Gong, X. Q. & Selloni, A. Reactivity of anatase TiO2 nanoparticles: the role of the minority (001) surface. J. Phys. Chem. B 109, 19560–19562 (2005).
He, Y. B., Tilocca, A., Dulub, O., Selloni, A. & Diebold, U. Local ordering and electronic signatures of submonolayer water on anatase TiO2(101). Nat. Mater. 8, 585–589 (2009).
Tilocca, A. & Selloni, A. Structure and reactivity of water layers on defect-free and defective anatase TiO2(101) surfaces. J. Phys. Chem. B 108, 4743–4751 (2004).
Lazzeri, M. & Selloni, A. Stress-driven reconstruction of an oxide surface: the anatase TiO2(001)−(1 × 4) surface. Phys. Rev. Lett. 87, 266105 (2001).
Diebold, U., Ruzycki, N., Herman, G. S. & Selloni, A. One step towards bridging the materials gap: surface studies of TiO2 anatase. Catal. Today 85, 93–100 (2003).
Setvin, M. et al. Charge trapping at the step edges of TiO2 anatase (101). Angew. Chem. Int. Ed. 53, 4714–4716 (2014).
Gong, X. Q., Selloni, A., Batzill, M. & Diebold, U. Steps on anatase TiO2(101). Nat. Mater. 5, 665–670 (2006).
Hebenstreit, W., Ruzycki, N., Herman, G. S., Gao, Y. & Diebold, U. Scanning tunneling microscopy investigation of the TiO2 anatase (101) surface. Phys. Rev. B 62, R16334–R16336 (2000).
Gong, X. Q. & Selloni, A. Role of steps in the reactivity of the anatase TiO2(101) surface. J. Catal. 249, 134–139 (2007).
Han, X., Kuang, Q., Jin, M., Xie, Z. & Zheng, L. Synthesis of titania nanosheets with a high percentage of exposed (001) facets and related photocatalytic properties. J. Am. Chem. Soc. 131, 3152–3153 (2009).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865 (1996).
Kresse, G. & Hafner, J. Ab initio molecular-dynamics simulation of the liquid-metal-amorphous-semiconductor transition in germanium. Phys. Rev. B Condens. Matter 49, 14251–14269 (1994).
Blöchl, P. E. Projector augmented-wave method. Phys. Rev. B Condens. Matter 50, 17953–17979 (1994).
Burdett, J. K., Hughbanks, T., Miller, G. J., Richardson, J. W. Jr & Smith, J. V. Structural-electronic relationships in inorganic solids: powder neutron diffraction studies of the rutile and anatase polymorphs of titanium dioxide at 15 and 295 K. J. Am. Chem. Soc. 109, 3639–3646 (1987).
Finazzi, E., Valentin, C. D., Pacchioni, G. & Selloni, A. Excess electron states in reduced bulk anatase TiO2: comparison of standard GGA, GGA+U, and hybrid DFT calculations. J. Chem. Phys. 129, 154113 (2008).
Sundholm, D. & Olsen, J. Finite element multiconfiguration Hartree-Fock calculations on carbon, oxygen, and neon: the nuclear quadrupole moments of 11C, 17O, and 21Ne. J. Phys. Chem. 96, 627–630 (1992).
Acknowledgements
This work was supported by the National Basic Research Program of China (2013CB934800), the National Natural Science Foundation of China (NSFC) (21573103, 21421004, 21222302, and 20903056), NSFC—Royal Society Joint Program (21661130149 and 21111130201), Program for New Century Excellent Talents in University (NCET-10-0483), the Fundamental Research Funds for the Central Universities (1124020512), and National Science Fund for Talent Training in Basic Science (J1103310). The ECUST group also thanks the Programme of Introducing Talents of Discipline to Universities (B16017) and National Super Computing Centre in Jinan for computing time. L.P. thanks Royal Society and Newton Fund for Royal Society—Newton Advanced Fellowship. C.P.G. thanks the European Research Council for an Advanced Fellowship. This work was also supported by a Project Funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions.
Author information
Authors and Affiliations
Contributions
N.J., F.G. and L.D. carried out the synthesis of anatase nanostructures. Y.L., L.S., H.S., Y.W., M.W., X.G., W.H. and W.D. carried out XRD, EPR, C and N element analyzing, XPS and surface area measurement; M.L. performed HRTEM; Y.L., L.S., H.S., M.W., X.K. and L.P. performed 17O isotope enrichment, collected, and analyzed the NMR spectra; Y.L., Z.Y., X.K. and L.P. collected and analyzed the high-field NMR spectra; X.-P.W. and X.-Q.G. conducted the DFT calculations; Y.L., X.-P.W., M.L., L.D., W.H., W.D., C.P.G., X.-Q.G. and L.P. wrote the manuscript, and all authors discussed the experiments and final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Li, Y., Wu, XP., Jiang, N. et al. Distinguishing faceted oxide nanocrystals with 17O solid-state NMR spectroscopy. Nat Commun 8, 581 (2017). https://doi.org/10.1038/s41467-017-00603-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-017-00603-7
- Springer Nature Limited